
Imonoglobulin G4 asociované onemocnění v gastroenterologii
Immunoglobulin G4-related disease in gastroenterology
IgG4-related disease is a recently defined clinical entity that can manifest itself in any organ. The most common gastrointestinal manifestations are diseases of the pancreas (autoimmune pancreatitis type 1) and biliary tree (IgG4-associated cholangitis); involvement of liver parenchyma is uncommon and the affection of tubular organs is very rare. IgG4-related pancreatitis and cholangitis can mimic malignancies in their clinical presentation. Diagnosis is often difficult and requires careful evaluation of the combination of symptoms, serology and imaging findings, while adhering to the established diagnostic criteria. The first line of treatment is the administration of corticoids and the remission is achieved in the vast majority of patients. In case of contraindication, intolerance or failure of corticotherapy, patients should receive B cell depletion therapy (rituximab). Based on the available knowledge, monotherapy with other immunosuppressants is not considered to be sufficiently effective. Some patients may benefit from maintenance treatment to prevent relapse, which is otherwise common in both IgG4-related pancreatitis and cholangitis. Recognized IgG4-related disease has a good prognosis, but some patients develop irreversible fibrotic changes in the affected organ with consequent dysfunction; the possible association of the disease with a higher risk of malignancy has not yet been reliably elucidated.
Keywords:
IgG4-related disease – IgG4-related pancreatitis – autoimmune pancreatitis – IgG4-related cholangitis
Authors:
Peter Mačinga; Jana Jarošová
![]() ; Julius Špičák; Tomáš Hucl
; Julius Špičák; Tomáš Hucl
Authors‘ workplace:
Klinika hepatogastroenterologie, Institut klinické a experimentální medicíny, Praha
Published in:
Vnitř Lék 2021; 67(2): 76-83
Category:
Main Topic
Overview
IgG4 asociované onemocnění je recentně definovaná klinická jednotka, která se může projevit postižením jakéhokoliv orgánu. Z gastrointestinální manifestace je nejčastější onemocnění pankreatu (autoimunitní pankreatitida typ 1) a žlučového stromu (IgG4 asociovaná cholangitida), méně časté je postižení jaterního parenchymu, onemocnění orgánů trávicí trubice je velmi vzácné. IgG4 asociovaná pankreatitida i cholangitida svým klinickým obrazem často napodobují maligní onemocnění, stanovení diagnózy bývá obtížné, vyžaduje pečlivé zhodnocení kombinace symptomů, sérologie a nálezu zobrazovacích metod, a to v adherenci k zavedeným diagnostickým kritériím. První linií léčby je podávání kortikoidů, remisi dosáhne naprostá většina pacientů. Při kontraindikaci, intoleranci nebo selhání kortikoterapie lze u pacientů podávat depleční léčbu proti B lymfocytům (rituximab). Monoterapie jinými imunosupresivy se na základě dostupných znalostí nejeví být dostatečně účinná. Část pacientů může profitovat z udržovací léčby k prevenci relapsu, který je jinak v případě IgG4 asociované pankreatitidy i cholangitidy častý. Rozpoznané IgG4 asociované onemocnění má dobrou prognózu, u části pacientů ale dochází k přestavbě postiženého orgánu s projevy dysfunkce, možná asociace onemocnění s vyšším rizikem malignity doposud nebyla spolehlivě objasněna.
Klíčová slova:
IgG4 asociované onemocnění – IgG4 asociovaná pankreatitida – autoimunitní pankreatitida – IgG4 asociovaná cholangitida
Úvod
Imunoglobulin G4 (IgG4) asociované onemocnění (anglicky IgG4-Related Disease; IgG4-RD) je systémový chronický zánětlivý syndrom definovaný multiorgánovým postižením, relabujícím průběhem, typickým histolopatologickým a radiologickým nálezem, elevací sérové hladiny IgG4 a dobrou odpovědí na léčbu kortikoidy a na depleční terapii proti B lymfocytům.
Terminologie
IgG4-RD se jako klinická jednotka dostala do povědomí odborné veřejnosti zejména v první dekádě tohoto století, a to pod variabilním pojmenováním. Sjednocující termín IgG4-related disease byl navržen japonskými odborníky v roce 2010 a poté konsenzuálně přijat na mezinárodním sympóziu v roce 2011 (1, 2). Před termínem „IgG4-associated“ bylo upřednostněno označení „IgG4-related“ disease, pro které ale v českém jazyce není analogický ekvivalent, v české odborné literatuře se tudíž obvykle užívá termín IgG4 asociované onemocnění.
Epidemiologie
IgG4-RD se řadí mezi vzácná onemocnění, přesná prevalence a incidence ale není známa. Dostupné epidemiologické údaje pochází zejména z asijských zemí a pravděpodobně nejsou platné univerzálně v celosvětovém měřítku. V japonské dotazníkové studii z roku 2009 byla stanovena incidence onemocnění na 0,28 až 1,08 na 100 000 obyvatelů, přičemž počet nemocných v celém Japonsku byl odhadnut na 8 000 případů (62 pacientů na milion obyvatel) (3). Tento údaj je již ale s jistotou překonán, v roce 2016 byl v Japonsku počet pacientů pouze s AIP odhadnut na více než 13 000 (4).
Onemocnění bývá častěji diagnostikováno u mužů (v poměru 1 : 0,77 až 4 : 1 k ženám), obvykle v pátém až sedmém decéniu, může se ale manifestovat i v dětském věku (3, 5).
IgG4-RD může postihnout prakticky kterýkoliv orgán, až v 85 % případů se manifestuje postižením některého z těchto sedmi – pankreas, žlučové cesty, slinné žlázy, slzné žlázy, ledviny, retroperitoneum a plíce (6). Recentně byly v mezinárodní studii čítající téměř 800 pacientů metodou analýzy latentních tříd definovány čtyři klinické fenotypy IgG4-RD, každý s charakteristickými klinickými, epidemiologickými a sérologickými rysy (7). Tyto fenotypy jsou (I.) pankreatobiliární postižení (31 %), (II.) retroperitoneální fibróza s/bez aortitidy (24 %), (III.) postižení omezené na oblast hlavy a krku (24 %) a (IV.) klasický Mikuliczův syndrom (IgG4 asociovaná dakryoadenitida a sialoadenitida) se systémovými projevy (22 %). Postižení více orgánů bylo zaznamenáno u více než tří čtvrtin pacientů (75,6 %), onemocnění pankreatu a žlučových cest bylo nejvíce zastoupené v první skupině (87 a 55 %), nebylo ale pro tento fenotyp exkluzivní. V menší míře se pankreatobiliární postižení manifestovalo i u pacientů v II. (12 a 1 %), III. (15 a 1 %) a IV. (46 a 27 %) fenotypu. Využití této klasifikace v klinické praxi bylo následně potvrzeno v italské kohortě pacientů s IgG4-RD (8).
Etiologie
IgG4-RD bylo při detekci zvýšeného titru řady autoprotilátek a dobré odpovědi na terapii kortikoidy historicky definováno jako autoimunitní onemocnění. Současné znalosti o onemocnění ale naznačují existenci vícero antigenů (nejenom auto-), které pravděpodobné indukují obdobnou imunitní reakci v různých orgánech u geneticky predisponovaných jedinců; polymorfismy zvyšující susceptibilitu k nemoci byly identifikovány v několika genech včetně HLA (9).
Jako potenciální autoantigeny byly identifikovány galectin-3, annexin‑A11 nebo laminin-511, jejich role v patogenezi onemocnění ale prozatím nebyla potvrzena (10–12). Předpokládaným mechanismem indukce onemocnění by byla ztráta imunologické tolerance, příčina tohoto fenoménu ale zůstává neznámá. IgG4-RD pravděpodobně může být vyvoláno i chronickou expozicí „zevním“ antigenům, jako jsou mikroorganismy nebo chemická agens (rozpouštědla, kovový prach) (6). U pacientů s IgG4 asociovanou cholangitidou bylo výrazně vyšší zastoupení dělnických profesí, než u pacientů s PSC (88 % vs. 14 %)(13).
Postižení gastrointestinálního traktu v rámci IgG4-RD
Nejčastější gastrointestinální manifestací IgG4-RD je IgG4 asociovaná pankreatitida (AIP) a IgG4 asociovaná cholangitida (IAC) - přítomny u 45 % a 20 % pacientů s IgG4-RD, které budou detailně popsány níže (6). V rámci IgG4-RD bylo referováno i postižení žlučníku, jater a orgánů trávicí trubice (2). IgG4 asociována cholecystitida byla zaznamenána u 25–32 % pacientů s AIP, klinická relevance tohoto pozorování je ale nejasná (14). V kontextu primární IgG4 asociované hepatopatie (IAH) byly popsány IgG4 asociované pseudotumory, tzn. ložiskové léze m.j. napodobující intrahepatální cholangiocelulární karcinom, obvykle spojené s přítomností IAC. Návrh IgG4 asociované autoimunitní hepatitidy jako primární IAH vznikl na základě analýzy tří malých kohort a prozatím nebyl plně přijat (15). V trávicí trubici se může IgG4-RD manifestovat jako ulcerace, polypoidní a submukózní léze anebo ztluštěním stěny. Koncept IgG4 asociované kolitidy, enteritidy, ezofagitidy nebo pouchitidy je postaven na malých sériích případů nebo dokonce jednotlivých kazuistikách, postižení žaludku (IgG4 asociovaná gastritida a gastrické léze) bylo ale v rámci IgG‑RD akceptováno (6, 16).
Histopatologie
Typický histopatologický obraz je pro AIP i IAC patognomický a sdílí stejné rysy s ostatními onemocněními v rámci IgG4-RD syndromu (26), tzn. přítomnost (I.) difúzní nebo fokální lymfoplazmocytární infiltrace, (II.) storiformní fibrózy, (III.) obliterativní flebitidy a (IV.) zvýšené množství IgG4+ plazmocytů. U AIP by měl počet těchto elementů dosáhnout 10/HPF (high power field) v případě hodnocení bioptického vzorku nebo 50/HPF při hodnocení resekátu, pro IAC je diagnostický počet 10/HPF. U obou nemocí mělo být zastoupení IgG4+ plazmocytů na celkovém počtu alespoň 40%.
IgG4-asociovaná pankreatitida
Terminologie
V rámci IgG4-RD je pankreatická orgánová manifestace (rovněž označovaná jako autoimunitní pankreatitida 1. typu) prototypem a pravděpodobně je i nejvíce studovaná. Koncept autoimunitní pankreatitidy (AIP) vytvořil v roce 1995 Yoshida, který popsal AIP jako formu chronické pankreatitidy autoimunitní etiologie (17). V roce 2001 pozoroval Hamano souvislost mezi AIP a elevací sérové hladiny IgG4, načež v roce 2003 navrhl Kamisawa koncept AIP jako součásti spektra IgG4-RD (18, 19). Studie z Evropy a USA ale upozornily na dvojí histopatologickou morfologii pankreatitidy referované jako AIP. Jednou byla lymfoplazmocytární sklerozující pankreatitida (LPSP), která se shodovala s popisem AIP japonských autorů (tzn. IgG4 asociována), druhou idiopatická duktocentrická pankreatitida (IDCP) (20, 21). V roce 2009 tudíž byly akceptovány dva podtypy: (I.) AIP typ 1 – LPSP a (II.) AIP typ 2 – IDCP. Společný název byl ponechán z důvodu podobné manifestace obou onemocnění, byť se jedná o rozdílné klinické jednotky s unikátním patologickým nálezem a odlišnou patogenezí i klinickým profilem. Někteří experti z výše uvedených důvodů navrhují používat pojem AIP výhradně pro LPSP a AIP 2. typu označit pouze jako IDCP (22); tento přístup ale nebyl univerzálně přijat. V klinické praxi rozlišujeme ještě AIP‑NOS (not otherwise specified), a to u pacientů, které nelze na základě dostupných vyšetření přesně klasifikovat ani do jednoho z podtypů (23).
Za účelem zjednodušení textu bude dále zkratka AIP referovat pouze typ 1 autoimunitní pankreatitidy.
Epidemiologie
Recentně byla stanovena prevalence onemocnění v roce 2016 v Japonsku na 10,1 na 100 000 obyvatel a incidence na 3,1 případů na 100 000 obyvatel ročně; průměrný věk v době diagnózy byl 64,8 let a poměr můžu a žen byl 3 : 1 (4). Ve srovnání s předchozí studií z roku 2011 se za pět let počet pacientů více než zdvojnásobil (z 5 745 případů narostl na 13 436) (24). Epidemiologická data z Evropy jsou velice sporá. Roční incidence onemocnění v Rhine‑Neckar regiónu v jihozápadním Německu byla odhadnuta na 0,29 na 100 000 obyvatelů (25). V regionu jižní Moravy byla AIP zjištěna u 4,7 % pacientů sledovaných s diagnózou idiopatické chronické pankreatitidy (26).
Klinický obraz
Symptomatologie
Klinická manifestace AIP je dána povahou nemoci a anatomií, z příznaků je nejčastější ikterus (35–70 %), který je podmíněn buď fokálním (v hlavě) nebo difuzním zvětšením pankreatu, popřípadě může být manifestací přidruženého postižení žlučového stromu v rámci IgG4-RD, které je přítomno u 40–80 % pacientů s AIP a je nejčastějším extrapankreatickým postižením u pacientů s AIP (24, 27–29). Mezi další symptomy patří bolest břicha nebo bolesti zad, popřípadě váhový úbytek (přítomný až u 25 % pacientů); manifestace onemocnění obrazem akutní pankreatitidy je považována za vzácnou, v některých kohortách ale dosahuje až 22 % (27, 29). Mezi další projevy patří funkční postižení žlázy (diabetes nebo steatorea), až třetina pacientů ale může být asymptomatických (4).
Sérologie
Zvýšení sérové hladiny IgG4 nad 1,35 g/l je přítomno u přibližně dvou třetin pacientů, až 20 % nemocných s AIP ale může mít hodnoty v mezích normy (30). Z dalších laboratorních nálezů byla u pacientů s AIP zaznamenána pozitivita autoprotilátek proti karbonanhydráze, laktoferinu, mitochondriím (AMA), hladkému svalu (ASMA), tyreoglobulinu nebo proteinu vážícího plasminogen (PBP) (29, 31). U řady z nich ale následující studie nedokázaly replikovat jejích diagnostický potenciál a v klinické praxi se rutinně nevyužívají (32, 33). U pacientů s AIP mohou být rovněž detekovány antinukleární protilátky (ANA) a revmatoidní faktor (RF) (34).
Zobrazovací metody a endoskopie
Pro stanovení diagnózy AIP je stěžejní zhodnocení morfologie parenchymu i pankreatického vývodu. Sonografie je levná a dostupná metoda, kterou lze zaznamenat zvětšený, difuzně hypoechogenní pankreas, většinou ale neumožňuje řádné zobrazení pankreatického vývodu.
Tomografické metody jako CT a MRI umožňují lepší posouzení přítomnosti parenchymatózních změn sugestivních pro AIP, kterými jsou (I.) difuzní nebo (multi)fokální zvětšení pankreatu se ztrátou lobulizace (sausage‑like pankreas), (II.) obdelníkový tvar kaudy pankreatu (cut‑tail příznak), (III.) přítomnost diskrétního peripankreatického lemu nebo sytícího se pouzdra (capsule sign) a nakonec MR obraz (IV.) difuzně snížené intenzity signálu na T1 vážených obrazech, lehce zvýšené na T2 obrazech a postkontrastní nepravidelné tečkované sycení v pozdní arteriální fázi (15).
Změny na pankreatickém vývodu pozorované při MR nebo endoskopické cholangiografii svědčící pro AIP jsou (I.) zúžení dlouhého segmentu (≥ 1/3 délky) vývodu bez proximální dilatace, (II.) postižení vícero segmentů pankreatického vývodu s interponovaným normálním vývodem (tzv. skip lesions), (III.) viditelné lumen wirsungu nebo choledochu procházejícího fokální expanzí pankreatu (tzv. duct‑penetrating sign) a (IV.) hladké pozvolné zvětšení průsvitu pankreatického vývodu postupující proximálně od léze (icicle sign) (15).
Důležitou roli v diagnostice AIP má i endosonografie (EUS), která umožní zhodnocení jak parenchymatózních, tak i duktálních změn a rovněž umožňuje odběr biologického materiálu k histopatologickému vyšetření (35).
Diagnostická kritéria
Výše uvedené typické nálezy na zobrazovacích metodách nejsou přítomny u všech pacientů, stanovení diagnózy AIP je často obtížné a vyžaduje komplexní přístup. Od rozpoznání nemoci byla publikována řada diagnostických kritérií, v západních zemích jsou nejčastěji používaná HISORt kritéria (název je v anglickém jazyku mnemonický pro sledované parametry) a mezinárodní konsenzuální diagnostická kritéria (ICDC) (36, 37). Oba systémy hodnotí histologický nález (H), morfologii pankreatického parenchymu a vývodu na zobrazovacích metodách (I), serologický nález (S), přítomnost postižení jiných orgánů (OOI) a odpověď na kortikoterapii (Rt). V ICDC kritériích existují pro každý sledovaný parametr dvě úrovně průkaznosti, jejíchž kombinací je možné určit pravděpodobnou či definitivní diagnózu AIP 1. typu (viz Tab. 1. a – b). Doporučený postup pro diagnostiku AIP v České republice byl vypracován v roce 2015 (38).


Diferenciální diagnostika
Klinickým obrazem (starší pacient s bezbolestným ikterem a expanzí pankreatu) AIP sugestivně napodobuje karcinom pankreatu (KP), odlišení těchto onemocnění bývá často velmi obtížné (Tab. 2) (39). Zvýšení sérové hladiny IgG4 není exkluzivní pro AIP, hodnota nad 1,4 g/l byla zaznamenána i u 10 % pacientů s KP, u 1 % i nad dvojnásobek normy (40). Zavedeným a levným laboratorním vyšetřením při podezření na KP je stanovení hladiny Ca19-9, která ale může být zvýšená i u (ikterických) pacientů s AIP. Recentně bylo publikováno několik prací s mezní hladinou Ca19-9 od 74 do 306 U/ml, která dokázala odlišit KP od AIP se senzitivitou 73–56 % a specificitou 74–96 % (41, 42), tato zjištění ale nebyla prozatím validována dalšími studiemi.

Důležité je pečlivé zhodnocení nálezů zobrazovacích metod, kde lze pozorovat řadu odlišností, v případě fokální expanze je pravidlem provedení EUS s odběrem biopsie. Nápomocné v odlišení karcinomu a AIP je posouzení odpovědi na kortikoterapii. Pacient, u kterého nelze na základě provedených vyšetření s velkou pravděpodobností vyloučit KP, je indikován k chirurgické léčbě, a to i za cenu „zbytečného výkonu“, prokáže‑li se v resekátu AIP.
Prognóza a komplikace onemocnění
V průběhu onemocnění může dojít k rozvoji funkčního postižení žlázy, diabetes mellitus byl v průběhu sledování zaznamenán u 39–57 % pacientů, 34–82 % pacientů vyvinulo exokrinní pankreatickou insuficienci (43). U řady pacientů lze pozorovat morfologické změny pankreatu. Atrofie žlázy, pravděpodobně odpovídající „vyhořelé“ fázi onemocnění, je přítomna přibližně u 30 % z nich, litiáza byla detekována u 5–40 % nemocných a zobrazovací kritéria diagnózy chronické pankreatitidy splní až 22 % pacientů s AIP (44–46).
Aktuálním tématem je výskyt maligních onemocnění u pacientů s IgG4-RD, řada studií totiž ukázala vyšší incidenci zhoubných nádorů ve srovnání s obecnou populací (15). Pro toto pozorování není jasné vysvětlení, jedna z hypotéz přepokládá IgG4-RD jako paraneoplastický fenomén a je založena na nálezu zvýšeného výskytu nádoru zejména v prvním roce od diagnózy IgG4-RD a absenci relapsu onemocnění po úspěšné léčbě malignity (47). Jiným vysvětlením ale může být pečlivé sledování pacientů s diagnózou IgG4-RD a tudíž časnější a četnější záchyt malignity. Obdobné analýzy v kohortách pacientů s AIP mají rozporuplné výsledky, tři z pěti retrospektivních analýz zvýšené riziko nepotvrdily (15). V kontextu AIP je pak studovaným fenoménem výskyt KP, a to i vzhledem k známému zvýšenému riziku této malignity u pacientů s chronickou pankreatitidou (48). V literatuře byla publikována řada případů koincidence AIP a KP (43, 49). V retrospektivní analýze z našeho pracoviště byla AIP zjištěna u 15 (5,1 %) z 295 pacientů po resekci pankreatu pro podezření nádor, u šesti z nich (40 %) byl v resekátu nalezen synchronní KP (50). V longitudinálních kohortách byla zaznamenána incidence 0–4,8 % (51, 52), zvýšené riziko KP u pacientů s AIP ale prozatím nebylo prokázáno. Zajímavý je nález recentní národní japonské studie, kdy bylo popsáno 18 případů KP v 1370 analyzovaných případech AIP (tzn. 1,3 %) a KP byl příčinou úmrtí u čtvrtiny ze všech zemřelých pacientů (4).
Navzdory výše uvedenému je prognóza onemocnění dobrá a dlouhodobé přežití pacientů s AIP je srovnatelné s obecnou populací (51).
IgG4-asociovaná cholangitida
Terminologie
IgG4-asociovaná cholangitida (IAC) byla rozpoznána jako součást IgG4-RD syndromu teprve v roce 2004 (53, 54). Dle recentních evropských doporučení je vhodné pro onemocnění používat termín „IgG4-related cholangitis“, bez adjektiva „sclerosing“. Tato změna má odrážet obvykle benigní průběh onemocnění a absenci pokročilých změn u léčených pacientů (15).
Epidemiologie
V současnosti není dostupná žádná epidemiologická studie zabývající se cíleně IAC, nicméně extrapolací dat z kohort pacientů s AIP byla prevalence v Japonsku odhadnuta na 2 případy na 100 000 obyvatel (55). IAC je onemocnění výrazně častěji postihující muže (s poměrem 4 : 1 až 8 : 1 k ženám), diagnostikováno pouze u dospělých, nejčastěji v šesté a sedmé dekádě, a je velmi často (90 %) asociováno s AIP 1. typu (56, 57). Pro případy IAC bez AIP bylo zavedeno označení „izolovaná IAC“ (58).
Klinický obraz
Symptomatologie
Onemocnění se nejčastěji manifestuje ikterem, mezi další projevy patří pruritus, bolesti břicha, váhový úbytek nebo bakteriální cholangitida. Frekvence příznaků a symptomů je v publikovaných pracích značně variabilní. V retrospektivní (japonské) kohortě 527 pacientů byl ikterus zaznamenán v 35 % případů, pruritus u 13 % a bolest břicha u 11 % pacientů. Onemocnění bylo asymptomatické až u čtvrtiny pacientů – 95 % z nich ale mělo postižení jiného orgánu v rámci IgG4-RD (59). Naproti tomu v recentní studii ze Spojených států bylo z 89 pacientů asymptomatických pouze 6 % a ikterus byl přítomen až u 88 % z nich (60).
Sérologie
Zvýšení sérové hladiny IgG4 je přítomno u 90 % pacientů s IAC (61), z dalších laboratorních parametrů může být detekována hypergamaglobulinemie (50 %), zvýšení celkového IgG (6–70 %), pozitivita ANA (40–50%) nebo RF (20 %) a periferní eozinofilie (15–25 %) (62).
Zobrazovací metody a endoskopie
Sonograficky lze u pacientů s IAC pozorovat koncentrické zesílení stěny žlučovodů (intra - a/nebo extrahepatálních), obvykle s průchodným lumen (63). Při časté asociaci onemocnění s AIP je možné touto metodou zaznamenat pankreatopatii, pro zhodnocení nálezu na pankreatu (a k případné detekci jiných intraabdominálních manifestací IgG4-RD) je ale vhodnější počítačová tomografie. Na CT svědčí pro IAC zvýšené sycení v pozdní arteriální fázi, homogenně zvýšené sycení v postkontrastní fázi, multifokální biliární stenózy s nápadným zesílením stěny žlučovodů (s hladkými okraji) a zúženým, ale viditelným lumen, a to současně se zesílením stěny žlučníku a absencí vaskulární invaze (64).
Pro diagnózu IAC je stěžejní cholangiografie, od které se odvíjí i klasifikace IAC do čtyř typů (viz Obr. 1) (65).
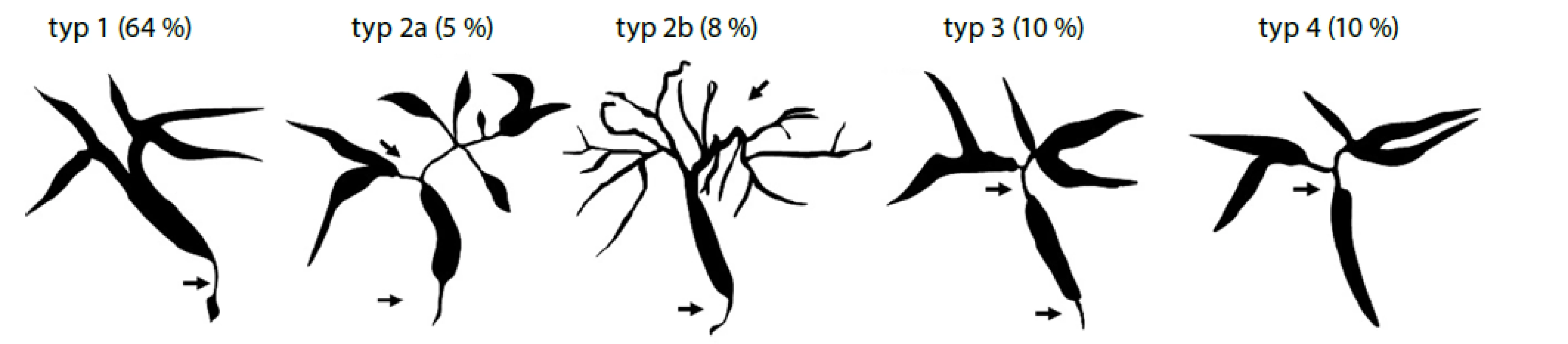
* frekvence jednotlivých typů platí pro případy IgG4 asociované cholangitidy s koincidencí s autoimunitní pankreatitidou
Diagnostická kritéria
K stanovení diagnózy IAC jsou v současnosti užívány dvě diagnostická kritéria. HISORt kritéria obsahují podobné parametry, jako je tomu u AIP (67). Japonská kritéria (Tab. 3) jsou v hodnocených parametrech podobná, na rozdíl od HISORt ale umožňují kategorizaci pacientů ve smyslu definitivní, pravděpodobné nebo možné diagnózy (68).

Diferenciální diagnostika
Svou manifestací napodobuje IAC jiné sklerozující cholangitidy, u druhého typu je obtížné zejména odlišení od PSC, u sekundárních cholangitid bývá často příčina sklerozujícího postižení známá (iatrogenní, ischemická, HIV‑asociovaná etc.). Klinický obraz u typů 3 a 4 často imponuje jako cholangiogenní karcinom (CCC). Diferenciálně diagnostický proces je obtížný, vyžaduje kombinaci vícero metod a měl by tudíž být proveden na pracovištích se zkušeností s IAC, obvykle terciárních.
Obdobně jako je tomu u AIP, je i pro IAC zvýšení sérové hladiny IgG4 typické, ale ne patognomické; elevace sérové IgG4 je přítomna i u 9–22 % pacientů s PSC, 8–14 % pacientů s CCC a 5 % zdravé populace (55, 69). Senzitivita a specifita toho markeru v diagnostice IAC se tudíž odvíjí od zvolené mezní hodnoty. Při hladině 1,4 g/l byla referována senzitivita 90 % a specificita 85 % (70), jako optimální mezní hodnota k odlišení IAC a PSC byla doporučena hladina 2,5 g/l se senzitivitou 67–89 % a specificitou 95%, k odlišení IAC a CCC pak 2 g/l (61). K odlišení IAC od CCC a jiných cholangiopatií může být nápomocné stanovení poměru IgG4 k celkovému IgG nebo IgG1; pro IAC pak svědčí poměr IgG4/IgG > 0,1 a IgG4/IgG1 > 0,24 (70, 71).
Při diferenciaci IAC (typ 2) od PSC může být užitečné stanovení protilátek proti cytoplazmě neutrofilů (ANCA), které bývají pozitivní až u 84 % pacientů s PSC, zatímco u IAC zcela výjimečně (72, 73). Z invazivních výkonů je pak v rámci diagnostiky možné provést necílenou jaterní biopsii, onemocnění mají odlišný histopatologický nález, nicméně vzhledem k fokální distribuci změn při IAC bývá ve vzorku zachycen charakteristický nález pouze v 25 % (74, 75). Z dalších metod je možné provést koloskopii k eventuálnímu průkazu IBD, typicky asociovanému s PSC.
Při diagnostickém procesu typů IAC napodobujících karcinom (1, 3 a 4) je vyjma obvyklých vyšetřovacích metod (CT, ERCP s biopsií) často prováděna biopsie z Vaterské papily, endosonografie s FNAB, IDUS nebo cholangioskopie (76, 77).
Stručný přehled diferenciální diagnostiky IAC je shrnut v tabulce 4.

Prognóza a komplikace onemocnění
Rozpoznaná a léčená IAC má dobrou prognózu, u pacientů s IAC byl v recentní studii z Mayo Clinic ve srovnání s pacienty s PSC prokázán významně nižší výskyt hepatobiliárních komplikací (cirhóza, CCC) a rovněž byla zaznamenána tendence k lepšímu desetiletému přežití (79 vs. 68 %, p = 0,11) (60). V západních zemích byla pozorována progrese onemocnění do cirhózy u 4,4 až 7,5 % pacientů (60, 78), celoživotní riziko rozvoje CCC se u pacientů s IAC pohybovalo na úrovni 3,4 až 10 %, což v jedné ze studií znamenalo až 130násobně vyšší incidenci této malignity ve srovnání s obecnou populací (59, 60). Navzdory těmto číslům ale prozatím není dostatečná evidence k označení IAC jako rizikového faktoru CCC (55).
Terapie gastrointestinální manifestace IgG4-RD
Léčba pacientů s AIP a IAC je totožná a vzhledem k mládí konceptu IgG4-RD v současnosti stojí na expertních doporučeních (15, 79, 80), obvykle založených na nízké úrovni důkazů a čerpající zejména z retrospektivních analýz. Prospektivní studie stran terapie IgG4-RD, potažmo AIP, jsou nečetné, provedené na malém počtu pacientů a většinou nerandomizované.
Při terapii IgG4-RD je nezbytné zohlednit několik důležitých aspektů, a to: (I.) klinický profil pacientů, tzn. starší, často polymorbidní populace (s diabetem, hypertenzí nebo osteoporózou), u které je vhodné minimalizovat expozici kortikoidům, zároveň (II.) je ale nutno předcházet ireverzibilní změně orgánů s alterací jejích funkce (tzn. nezbytnost dosažení remise a pokud možno prevence relapsu u rizikových pacientů), a to vše v kontextu (III.) načasování léčby, tzn. její zahájení v zánětlivé fázi onemocnění, jelikož ve fibrotické („vyhořelé“) fázi již léčba může být zbytečná při absenci terapeutické odpovědi.
Při výše uvedených omezeních na straně znalostí o onemocnění a řady proměnných na straně pacientů nejsou obvykle tato doporučení rigidní, ale spíše tvoří obecný rámec, který je pak dále upraven dle zkušeností jednotlivých center. To nevyhnutelně dále vede při stále malém počtu pacientů s IgG4-RD a geografických odlišnostech k významné variaci léčebných přístupů a obtížnému srovnání jejích výstupů. Aktuálně probíhající mezinárodní studie (i za účasti českých center) – PrescrAIP, analyzující prozatím největší počet pacientů v evropských podmínkách a jeden z největších vůbec, si klade za cíl popsat právě léčebné režimy v Evropě a srovnat jejich efektivitu v terapii onemocnění (81).
Při hodnocení efektu léčby je nezbytné definovat stěžejní parametry. Remise je klasifikována do pěti kategorií – (I.) symptomatická, (II.) sérologická, (III.) radiologická, (IV.) histologická a (V.) funkční. Ve většině studií se za kompletní remisi považuje kombinace první, druhé a třetí kategorie, za inkompletní pak remisi alespoň v jedné z prvních tří kategorií při absenci odpovědi v (některé ze) zbylých dvou. Relaps je definován jako rekurence symptomů a sérologických, radiologických nebo histologických abnormalit typických pro IgG4-RD, a to po navození kompletní nebo inkompletní remise. Relaps se pak může projevit v původně postiženém orgánu, nebo v některém z jiných systémů (82).
Farmakoterapie
Indukční léčba
U nezanedbatelné části pacientů s IgG4-RD (10–25 %) může dojít k spontánní remisi onemocnění (79). Zahájení systémové terapie je tudíž indikováno pouze u symptomatických pacientů a lze ji ale zvážit u (I.) asymptomatických pacientů s AIP s perzistující fokální expanzí k vyloučení nádoru a (II.) anikterických pacientů s IAC s přetrvávající elevací jaterních testů. Pro preventivní léčbu asymptomatických pacientů s AIP s cílem předcházet funkčnímu postižení žlázy není relevantní evidence (15).
První linií léčby jsou kortikoidy (Prednison) v dáce 0,6–0,8 mg/kg/den (tzn. obvykle 30–40 mg/den) po dobu jednoho měsíce. V případě oligosymptomatických starších (polymorbidních) pacientů lze zvážit indukční léčbu 20 mg/den, nižší dávky již pravděpodobně snižují frekvenci dosažení remise (79). Odpověď na terapii je vhodné posoudit v odstupu dvou až čtyř týdnů, a to kombinací klinického, laboratorního a morfologického nálezu. Doporučeno je pozvolné snižování dávky rychlostí 5 mg/2 týdny. Adekvátní kortikoterapie vede k dosažení remise v 97–100 % případů (15).
Při kontraindikaci (nebo intoleranci) kortikoidů je lékem druhé volby rituximab, jehož efektivita v navození remise byla prokázána v několika studiích na pacientech s IgG4-RD nebo AIP (83, 84). Klinická odpověď obvykle nastává v odstupu čtyř týdnů od zahájení léčby a remise byla zaznamenána u 67–97 % léčených pacientů (15, 85). V terapii IgG4-RD bylo užito celé spektrum imunosupresiv (azathioprin, 6-merkaptopurin, mykofenolát, tacrolimus, metotrexát), současná evidence ale nesvědčí pro jejich účinnost při navození remise onemocnění v monoterapii. Lze ale zvážit jejích přidání do kombinace v případě suboptimální odpovědi na kortikoterapii, dle recentní metaanalýzy byla míra dosažení remise u kombinované léčby vyšší než při monoterapii kortikoidy (86).
Terapie relapsu a udržovací léčba
Relaps onemocnění po ukončení indukční léčby nastává u 26–70 % pacientů s AIP (87) a 35–71 % nemocných s IAP (67, 88). Racionální léčbou relapsu se jeví být u pacientů tolerujících předchozí kortikoterapii znovuzahájení léčby Prednisonem, ve vyšší než původní dávce a s prodloužením detrakce. Při takovém postupu lze dosáhnout remise až u 95 % pacientů (15).
Udržovací léčba Prednisonem v dávce 5–10 mg/den vede ke významnému snížení rizika relapsu, ale signifikantně zvyšuje kumulativní dávku (a toxicitu) kortikoterapie. Z toho důvodu je arbitrárně doporučena pouze japonskými experty (v dávce 5 mg/den po dobu tří let) (55), mezinárodní a evropský konsenzus doporučuje zvážení zahájení udržovací léčby u pacientů s vyšším rizikem relapsu (tzn. vysokou vstupní nebo postindukční hladinou sérového IgG4, multiorgánovým postižením, periferní eozinofilií nebo proximálním typem IAC) (15, 79, 85). K udržení remise lze pokračovat v terapii rituximabem (83), popřípadě v terapii kortikoidy v kombinaci s imunosupresivy, kde lze očekávat menší riziko relapsu ve srovnání s monoterapií kortikoidy (86). Pro doporučení udržovací léčby v monoterapii imunosupresivy není dostatečná evidence, v případě nutnosti takové léčby se pak jako nejvhodnější jeví azathioprin, v dávce ne nižší než 2–2,5 mg/kg/den (15).
Endoskopická léčba
Zavedení biliární drenáže je v současnosti doporučeno u všech ikterických pacientů s IAC (55). Toto doporučení je ale slabé při nízké úrovni důkazů a zohledňuje zejména nutnost provedení diagnostického ERCP u většiny pacientů s IAC. Ve dvou studiích na malém počtu ikterických pacientů s AIP 15 a IAC 19 byla na kortikoterapii pozorována remise onemocnění i bez nutnosti zavedení biliární drenáže u všech pacientů vyjma jednoho nemocného s IAC, přičemž nebyl zaznamenán ani jeden případ bakteriální cholangitidy navzdory imunosupresivní léčbě (89, 90). Zdá se tudíž, že ERCP (a biliární drenáž) nejsou nezbytné u pacientů s definitivní diagnózou IAC, u kterých není nutný odběr bioptického vzorku (kupříkladu typ 1 s AIP).
Shrnutí
IgG4 asociované onemocnění a jeho nejčastější manifestace v trávicím traktu – autoimunitní pankreatitida 1. typu a IgG4 asociovaná cholangitida jsou dnes již široce rozeznávanou jednotkou v diferenciální diagnostice onemocnění pankreatu a žlučových cest. Nadále se ale jedná o velmi vzácné onemocnění, s incidencí řádově nižší ve srovnání se zhoubnými nádory, které svou manifestací často napodobují. Diagnostický proces je obvykle náročný, a tudíž by měl být prováděn v centrech se zkušeností s IgG4-RD. Nezbytná je adherence k některému z etablovaných diagnostických kritérií, primární je nadále vyloučení malignity, důležité je ale také vyvarovat se falešně pozitivní diagnózy u jiných benigních onemocnění („overdiagnosis“), které pak vede k zbytečné zátěži pacientů imunosupresivní léčbou. V terapii onemocnění jsou lékem první volby kortikoidy s výbornou účinností, optimální indukční dávka, délka kortikoterapie nebo indikace (a trvání) udržovací léčby jsou nadále předmětem studia. Účinnost léčby jinými imunosupresivy (kupříkladu azathioprinu) v monoterapii nebyla spolehlivě potvrzena. Naopak byla v posledních letech prokázána efektivita depleční terapie proti B‑lymfocytům, rituximab je dnes akceptovaným lékem druhé volby. Podmínkou dobré prognózy pacientů s gastrointestinální manifestací IgG4-RD je pečlivé sledování, které umožní včas rozpoznat a léčit dlouhodobé komplikace onemocnění ve smyslu insuficience postiženého orgánu při jeho fibrotické přestavbě. Dispenzarizace je nezbytná i v kontextu možného vyššího rizika maligních onemocnění u pacientů s IgG4-RD, byť tato asociace nebyla prozatím jasně prokázána a zůstává předmětem studií.
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Peter Mačinga, Ph.D.
Klinika hepatogastroenterologie, Institut klinické a experimentální medicíny
Vídeňská 9
140 21 Praha
Cit. zkr: Vnitř Lék 2021; 67(2): 76–83
Článek přijat redakcí: 18. 12. 2020
Článek přijat po recenzích: 14. 1. 2021
Sources
1. Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012; 22(1): 1–14.
2. Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012; 64(10): 3061–3067.
3. Uchida K, Masamune A, Shimosegawa T, Okazaki K. Prevalence of IgG4-Related Disease in Japan Based on Nationwide Survey in 2009. Int J Rheumatol. 2012; 2012 : 358371.
4. Masamune A, Kikuta K, Hamada S, Tsuji I, Takeyama Y, Shimosegawa T et al. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2016. J Gastroenterol. 2020; 55(4): 462–470.
5. Karim F, Loeffen J, Bramer W, Westenberg L, Verdijk R, van Hagen M et al. IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatr Rheumatol Online J. 2016; 14(1): 18.
6. Miyabe K, Zen Y, Cornell LD, Rajagopalan G, Chowdhary VR, Roberts LR et al. Gastrointestinal and Extra ‑ Intestinal Manifestations of IgG4-Related Disease. Gastroenterology. 2018; 155(4): 990–1003 e1.
7. Wallace ZS, Zhang Y, Perugino CA, Naden R, Choi HK, Stone JH et al. Clinical phenotypes of IgG4-related disease: an analysis of two international cross‑sectional cohorts. Ann Rheum, DiS. 2019; 78(3): 406–412.
8. Lanzillotta M, Campochiaro C, Mancuso G, Ramirez GA, Capurso G, Falconi M, et al. Clinical phenotypes of IgG4-related disease reflect different prognostic outcomes. Rheumatology (Oxford). 2020; 59(9): 2435–2442.
9. Kawa S, Ota M, Yoshizawa K, Horiuchi A, Hamano H, Ochi Y et al. HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology. 2002; 122(5): 1264–1269.
10. Perugino CA, AlSalem SB, Mattoo H, Della‑Torre E, Mahajan V, Ganesh G et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol. 2019; 143(2): 736–745 e6.
11. Shiokawa M, Kodama Y, Sekiguchi K, Kuwada T, Tomono T, Kuriyama K et al. Laminin 511 is a target antigen in autoimmune pancreatitis. Sci Transl Med. 2018; 10(453).
12. Hubers LM, Vos H, Schuurman AR, Erken R, Oude Elferink RP, Burgering B et al. Annexin A11 is targeted by IgG4 and IgG1 autoantibodies in IgG4-related disease. Gut. 2018; 67(4): 728–735.
13. de Buy Wenniger LJ, Culver EL, Beuers U. Exposure to occupational antigens might predispose to IgG4-related disease. Hepatology. 2014; 60(4): 1453–1454.
14. Detlefsen S, Drewes AM. Autoimmune pancreatitis. Scand J Gastroenterol. 2009; 44(12): 1391–407.
15. Lohr JM, Beuers U, Vujasinovic M, Alvaro D, Frokjaer JB, Buttgereit F et al. European Guideline on IgG4-related digestive disease - UEG and SGF evidence‑based recommendations. United European Gastroenterol J. 2020; 8(6): 637–666.
16. Deshpande V. IgG4-Related Disease of the Gastrointestinal Tract: A 21st Century Chameleon. Arch Pathol Lab Med. 2015; 139(6): 742–749.
17. Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995; 40(7): 1561–1568.
18. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001; 344(10): 732–738.
19. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003; 38(10): 982–984.
20. Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol. 2003; 27(8): 1119–1127.
21. Zamboni G, Luttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004; 445(6): 552–563.
22. Hart PA, Zen Y, Chari ST. Recent Advances in Autoimmune Pancreatitis. Gastroenterology. 2015; 149(1): 39–51.
23. de Pretis N, Vieceli F, Brandolese A, Brozzi L, Amodio A, Frulloni L. Autoimmune pancreatitis not otherwise specified (NOS): Clinical features and outcomes of the forgotten type. Hepatobiliary Pancreat Dis Int. 2019; 18(6): 576–579.
24. Kanno A, Masamune A, Okazaki K, Kamisawa T, Kawa S, Nishimori I et al. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2011. Pancreas. 2015; 44(4): 535–539.
25. Schneider A, Michaely H, Weiss C, Hirth M, Ruckert F, Wilhelm TJ et al. Prevalence and Incidence of Autoimmune Pancreatitis in the Population Living in the Southwest of Germany. Digestion. 2017; 96(4): 187–198.
26. Dítě P, Ševčíková A, Novotný I et al. Autoimunní pankreatitida v České republice – region jižní Morava. Čes a Slov Gastroent a Hepatol 2007; 61(2): 82–85.
27. Vujasinovic M, Valente R, Maier P, von Beckerath V, Haas SL, Arnelo U et al. Diagnosis, treatment and long‑term outcome of autoimmune pancreatitis in Sweden. Pancreatology. 2018; 18(8): 900–904.
28. Blaho M, Dite P, Kunovsky L, Kianicka B. Autoimmune pancreatitis - An ongoing challenge. Adv Med Sci. 2020; 65(2): 403–408.
29. Nagpal SJS, Sharma A, Chari ST. Autoimmune Pancreatitis. Am J Gastroenterol. 2018; 113(9): 1301.
30. Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011; 23(1): 108–113.
31. Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C, Falconi M, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med. 2009; 361(22): 2135–2142.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2021 Issue 2
Most read in this issue
- Diagnostika a terapie chronické pankreatitidy dle UEG guidelines
- Glukagon v léčbě hypoglykemie – novinky
- Perindopril – dlouholetá jistota v léčbě hypertenze
- Mimostřevní komplikace idiopatických střevních zánětů