
Transformace IgM-MGUS do Waldenströmovy makroglobulinemie u 2 z 6 pacientů v průběhu léčby syndromu Schnitzlerové
Transformation of IgM-MGUS into Waldenström´s macroglobulinemia in two of six patients treated for Schnitzler´s syndrome
Transformation of IgM-MGUS into Waldenström´s macroglobulinemia in two of six patients treated for Schnitzler´s syndrome Schnitzler´s syndrome is a very rare, adult-onset, apparently acquired autoinflammatory disease. Chronic urticarial rash and symptoms of systemic inflammation including fever, arthralgia and bone pain with the presence of monoclonal immunoglobulin M (IgM), rarely IgG, are among hallmarks of the disease. We performed a retrospective study of 6 patients (5 men, 1 woman) diagnosed with Schnitzler´s syndrome fulfilling the Strasbourg criteria who had been treated at our centre in the University Hospital Brno from 2007 to 2021. Median age at diagnosis was 54 (45–67) years, median follow up was 8 (3–14) years. All 6 patients had IgM κ monoclonal gammopathy, increased CRP and/or erythrocyte sedimentation rate and arthralgia or bone pain, 4 patients suffered from fever, three had leucocytosis ≥ 10 × 109/L and lymphadenopathy was found in one patient. 18FDG-PET/CT scan with low-dose total body CT became a part of the initial baseline assessment in 5 patients with suspected Schnitzler´s syndrome, while Na18F-PET/CT was used in one patient to confirm the presence of osteosclerotic leasions as a criterion of the disease. All patients had osteosclerotic or hyperostotic bone lesions detected by low-dose CT examination, with increased 18FDG uptake in illiac and femoral bone marrow. The patient with Na18F-PET/CT scan revealed intensive abnormal tracer uptake with Na18F-PET/CT being more sensitive for detection of osteosclerotic lesions in Schnitzler´s syndrome than 18FDG-PET/CT. All patients were treated with daily subcutaneous anakinra without any adverse events, with excellent clinical results. We observed complete disappearance of urticaria and other symptoms persisting during years of anakinra administration.
IgM-MGUS transformed into Waldenström´s macroglobulinemia in two of six patients, but only one patient developed symptoms requiring RBD (Rituximab, Bendamustin, and Dexamethasone) treatment, which induced almost complete remission of the disease. Successful RBD therapy enabled to prolong intervals of maintenance anakinra from 24 to 48 hours with almost complete control of urticarial rash and other symptoms. We suggest close monitoring of patients with Schnitzler´s syndrome to early capture potential transformation into Waldenström´s macroglobulinemia with succesful treatment of both conditions.
Keywords:
Schnitzler´s syndrome – 18FDG-PET/CT – Na18F-PET/CT – anakinra – Waldenström´s disease
Authors:
Zdeněk Adam 1; Miroslav Tomíška 1; Zdeněk Řehák 2; Renata Koukalová 2; Marta Krejčí 1; Zdeněk Král 1; Zuzana Adamová 4; Sabina Ševčíková 3; Luděk Pour 1; Martin Štork 1; Martin Krejčí 1; Viera Sandecká 1
Authors‘ workplace:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Oddělení nukleární medicíny Masarykův onkologický ústav Brno
2; Ústav patologické fyziologie LF MU a FN Brno
3; Chirurgické oddělení nemocnice Vsetín
4
Published in:
Vnitř Lék 2021; 67(E-3): 15-23
Category:
Review Articles
Overview
Syndrom Schnitzlerové je velmi vzácná autoinflamatorní choroba manifestující se v dospělosti. Hlavními znaky je chronická urtika a příznaky systémové zánětlivé reakce (horečka, artralgie, bolesti kostí) a monoklonální imunoglobulin typu IgM (M-IgM) vzácně IgG. Do analýzy jsme zařadili našich šest pacientů se syndromem Schnitzlerové, kteří splnili Štrasburská kritéria nemoci a jsou léčeni od roku 2007 do 2021. Medián věku pacientů (5 mužů 1 žena) v době stanovení diagnózy byl 54 (45–67) let. Medián sledování je 8 (3–14) roků. Všech šest pacientů mělo M-IgM, bolesti kloubů anebo bolesti kostí, zvýšenou hodnotu sedimentace erytrocytů a CRP, čtyři měli horečky, tři měli leukocytózu ≥ 10 × 109/l, lymfadenopatii měl jen jeden pacient. Pro detekci osteosklerotických změn, jednoho z kritérií této nemoci, jsme použili u 5 pacientů18FDG-PET/CT, u jednoho pacienta Na18F-PET/CT vždy s low-dose CT vyšetřením celého těla. Všichni pacienti měli při zobrazení metodou low-dose CT zřetelná osteosklerotická neboli hyperostotická kostní ložiska. Akumulace 18FDG byla mírně zvýšená v kostní dřeni pánve a femurů, zatímco akumulace Na18F byla výrazná právě v osteosklerotických ložiscích. Zobrazení pomocí Na18F-PET/CT je více senzitivní pro detekci osteosklerotických ložisek než 18FDG-PET/CT. Všichni pacienti byli a jsou léčeni anakinrou bez jakýchkoliv nežádoucích účinků, s excelentními výsledky – kompletním vymizením urtiky a dalších symptomů, aniž by se po letech léčby její účinek zmenšoval.
IgM-MGUS se transformoval do Waldenströmovy makroglobulinemie (WM) u dvou pacientů, ale zatím pouze u jednoho se jednalo o symptomatickou WM, pro jejíž léčbu byla použita kombinace RBD (rituximab, bendamustin, dexametazon). Léčba docílila téměř úplné vymizení M-IgM a zároveň poklesla intenzita projevů syndromu Schnitzlerové, takže bylo možné protáhnout intervaly mezi aplikací anakinry z 24 na 48 hodin.
U pacientů se syndromem Schnitzlerové je nutné sledovat vývoj M-IgM a v případě transformace do symptomatické WM včas podat účinnou léčbu, obvykle kombinaci rituximabu a chemoterapii s cílem dosáhnout kompletní hematologické odpovědi a snížení intenzity příznaků syndromu Schnitzlerové.
Klíčová slova:
syndrom Schnitzlerové – 18FDG-PET/CT – Na18F-PET/CT – anakinra – Waldenströmova makroglobulinémie
Úvod
Syndrom Schnitzlerové je autoinflamatorní choroba manifestující se v dospělosti, která má tři hlavní znaky:
monoklonální imunoglobulin typu IgM (M-IgM), vzácně typu IgG,
opakované výsevy kopřivkových morf či chronickou urtiku (Obr. 1),
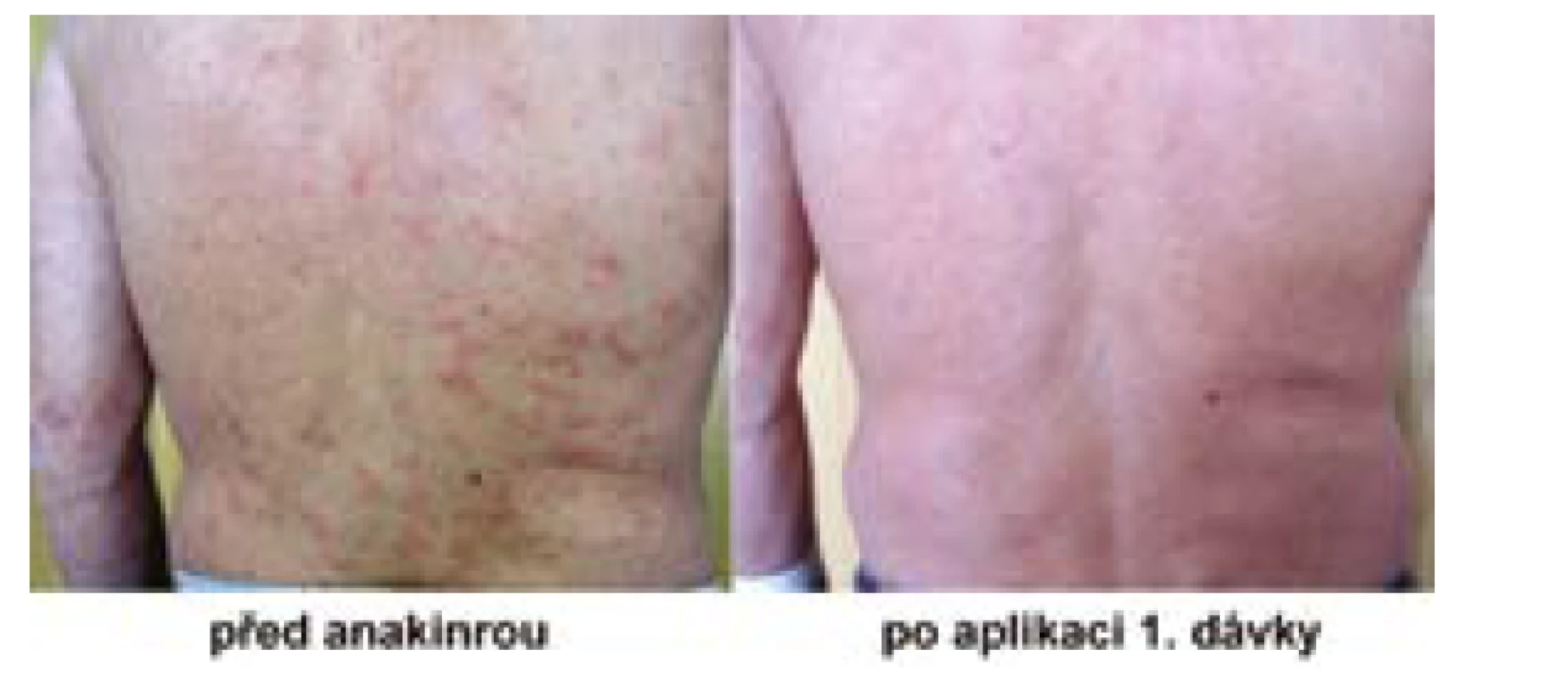
bolesti kostí a kloubů, obvykle dolních končetin.
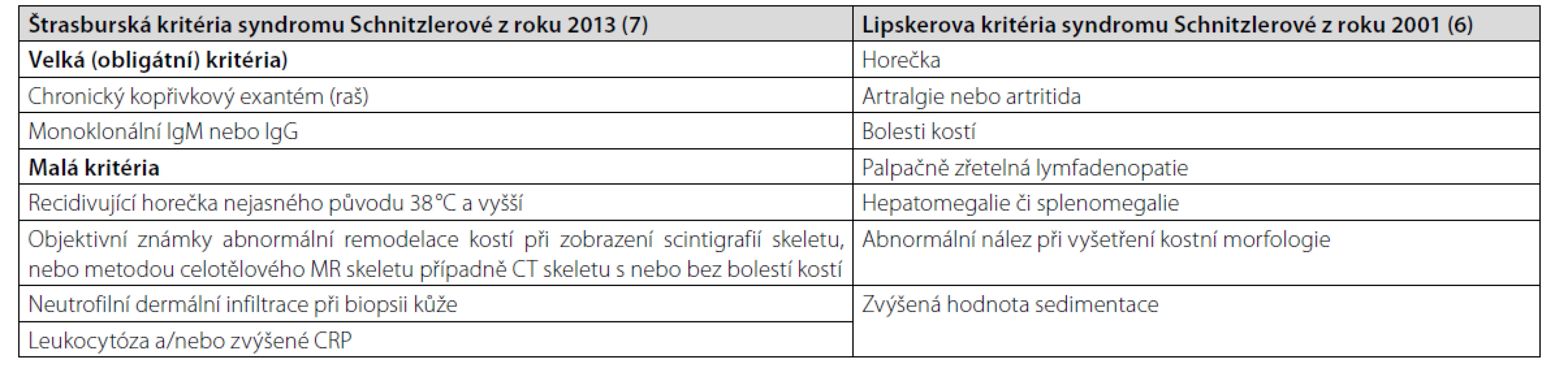
Malá diagnostická kritéria pak tvoří: rekurentní horečky, leukocytóza a/nebo zvýšená hodnota CRP, neutrofilní kožní dermatóza při biopsii kopřivkové morfy a známky remodelace kostí s nebo bez bolestí kostí (1–3). Mohou být přítomny i další příznaky, jako jsou bolesti hlavy, myalgie, artralgie, fatigue, periferní neuropatie, úbytek hmotnosti a lymfadenopatie, které tuto nemoc často provázejí, ale nejsou zahrnuty do diagnostických kritérií (1, 4, 5). První prezentace nemoci obvykle začíná v páté životní dekádě, obvykle bez familiární kumulace této nemoci (1). Asi u 15–20 % pacientů se v průběhu sledování vyvine symptomatická forma lymfoproliferativního onemocnění, obvykle Waldenströmova makroglobulinemie (1). Syndrom Schnitzlerové nemá jako četné jiné nemoci žádný jednoznačný definující znak, a proto se stále používají diagnostická kritéria uvedená v tabulce 1. Starší se nazývají po svém autorovi Lipskerova kritéria a novější dle místa konference, na níž byla dohodnuta, Štrasburská kritéria (6, 7).
Cílem publikace je připomenout tuto jednotku, která léta trápí nemocné, aniž by byla správně diagnostikována, shrnout zkušenosti s mnohaletou léčbou této nemoci preparátem anakinra (Kineret), poukázat na riziko transformace do symptomatické lymfoproliferace, která již vyžaduje cílenou léčbu. V závěru jsou připomenuty léčebné možnosti dostupné v roce 2021.
Soubor pacientů
Na našem pracovišti v roce 2021 léčíme celkem šest nemocných se syndromem Schnitzlerové preparátem anakinra (KINERET). Jedná se o pět mužů a jednu ženu, medián věku, ve kterém byla zahájena léčba anakinrou, je 54 (45–67) let. Medián trvání léčby je 8 (3–14) let. Po vyšetření na našem pracovišti splnili všichni pacienti Štrasburská kritéria (Tab. 1). U všech pacientů byly příznaky této nemoci přítomny již několik (3–6) let před stanovením diagnózy a tito pacienti prošli obvykle nejen ambulancemi kožních lékařů, ale bolesti kostí a kloubů je přivedly do ambulancí revmatologů a internistů. Někdy je nález zvýšené koncentrace imunoglobulinu IgM přivedl i do ambulance hematologů.
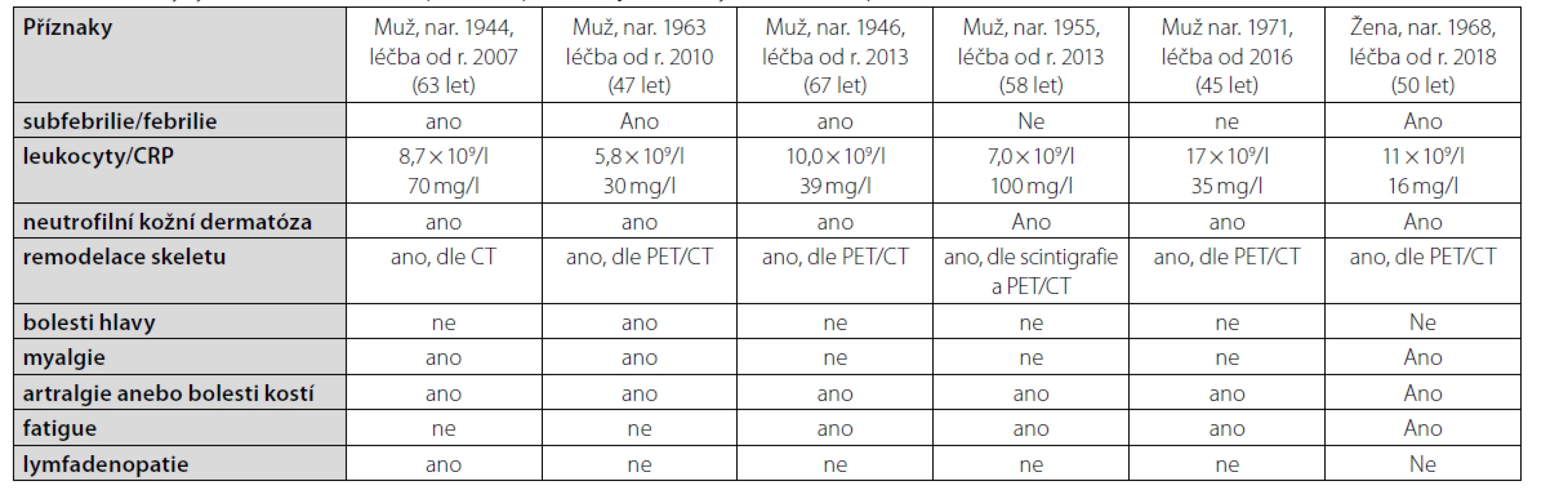
První pacient po četných předchozích neúspěšných léčebných pokusech zahájil léčbu anankinrou v březnu roku 2007, je tedy na léčbě 14 let a byl popsán již dříve. Poslední, šestá pacientka, je léčena anakinrou od března roku 2018, příznaky udávala již asi od roku 2013. Příznaky této nemoci u jednotlivých pacientů jsou shrnuty v tabulce 2.
Všichni pacienti při stanovení diagnózy měli provedenu trepanobiopsii s histologickým a flowcytometrickým hodnocením kostní dřeně. Toto vstupní vyšetření kostní dřeně neprokázalo infiltraci lymfoplazmocytárním lymfomem, a tak byla diagnóza uzavřena jako IgM–MGUS (monoklonální gamapatie nejistého významu typu IgM).
Všichni pacienti zahájili léčbu podkožní aplikací 1 ampulky ankinry (100 mg) denně. Tato léčba, s výjimkou druhého nemocného, odstranila kompletně veškeré příznaky nemoci již v průběhu prvního dne aplikace, tedy nejen kopřivku, ale také další projevy, subfebrilie, patologickou únavu, bolesti kloubů a kostí, případně svalů. Jen u jednoho pacienta ze šesti se potíže zmírnily, ale nevymizely zcela, takže léčebnou odpověď hodnotíme jako parciální remisi. Tento pacient si občas při nové atace symptomů musí výjimečně aplikovat i další ampulku denně. U jednoho ze šesti pacientů se brzy po zahájení léčby ukázalo, že je dostačující aplikace každý druhý den. Všiml si, že když si lék nepíchne, tak potíže se mu nevrátí po 24 hodinách, ale až po 48 hodinách. Tak si zkusil experimentálně lék podávat 1× za 2 dny a zjistil, že mu tento interval zcela vyhovuje. Při delším intervalu se již potíže začaly vracet. Ostatní pacienti mají formu onemocnění, která se připomene návratem symptomů po 24 hodinách od poslední injekce, pokud si zapomenou píchnout další dávku anakinry. U žádného nemocného v průběhu mnoholeté léčby nedošlo k poklesu či ztrátě účinnosti. Efekt léčby je pořád stejný.
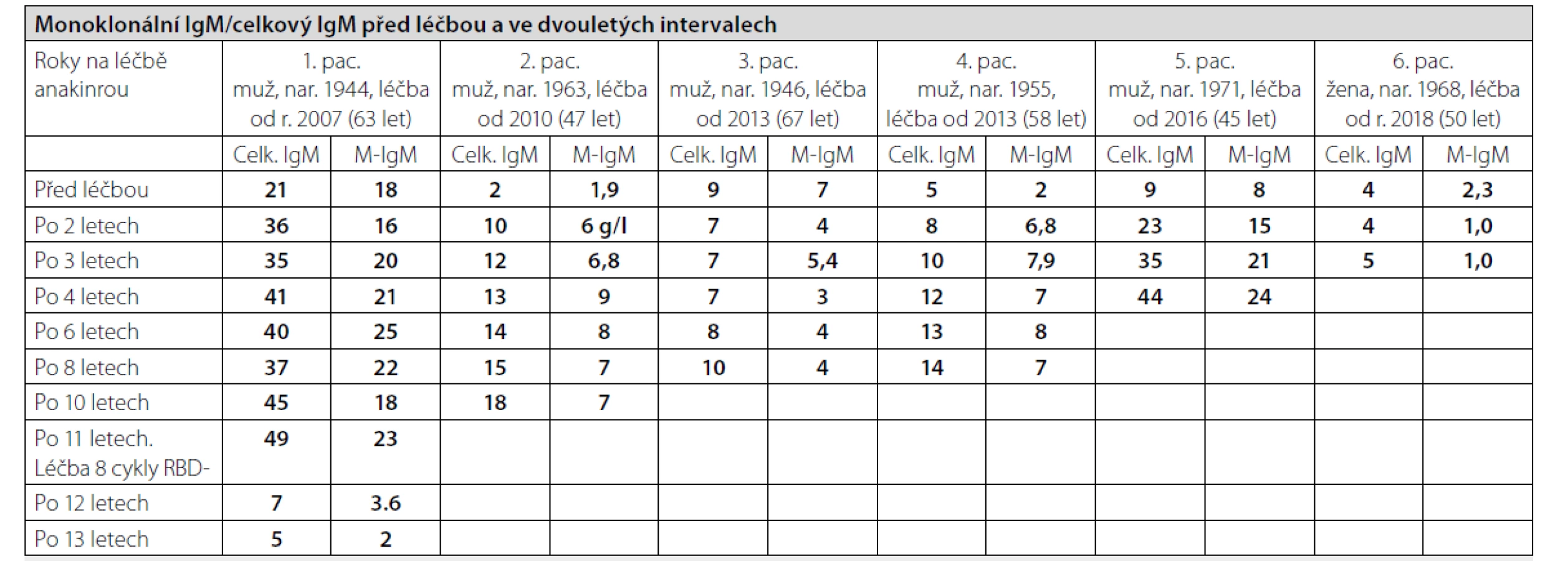
Pacienti k nám dojíždějí pravidelně ve tříměsíčních intervalech na kontrolu a pro předpis dalších balení anakinry (Kineretu). U pacientů sledujeme četné laboratorní parametry, do tabulky 2 jsme vložili vývoj celkového imunoglobulinu typu IgM a M-IgM. Sledovali jsme samozřejmě také volné lehké řetězce a polyklonální imunoglobuliny typu IgG, IgM IgA. Hodnoty celkové koncentrace imunoglobulinu typu IgM se zvyšují paralelně s hodnotu M-IgM. Informaci o přítomnosti osteosklerotických ložisek, která jsou jedním z malých kritérií této nemoci, jsme měli v 5 případech ze vstupního 18FDG-PET/CT vyšetření, v jednom případě Na18F-PET/PCT. Osteosklerotická ložiska byla popsána při zobrazení skeletu metodou low-dose CT. Akumulace 18FDG-byla jen mírně zvýšená v kostní dřeni femurů a v pánvi, zatímco akumulace Na18F byla výrazně zvýšená v místech, kde na low dose CT byly patrné osteosklerotické změny. Proto považujeme Na18F za vhodnější radiofarmakum k detekci osteosklerotických ložisek, pokud je jejich detekce potřebná ke splnění Štrasburských kritérií této nemoci. Pokud jsou pacienti po léčbě asymptomatičtí a jejich stav se nemění, tak neprovádíme kontrolní 18FDG-PET/CT, ani samotné low-dose CT, takže nemáme informace o vývoji těchto osteosklerotických ložisek. Měření kostní hustoty pomocí dexa jsme v průběhu léčby neprováděli.
U všech nemocných je vidět vývoj koncentrace M-IgM i celkového imunoglobulinu typu IgM, ale pouze u 2 z 6 se nemoc transformovala do Waldenströmovy makroglobulinemie (pacient 1 a pacient 5). Pouze u těchto dvou pacientů jsme pro výraznější vzestup koncentrace M-IgM provedli další trepanobiopsii, u ostatních jsme vyšetření kostní dřeně neopakovali. U obou pacientů histologické hodnocení válečku kostní dřeně mělo závěr: infiltrace kostní dřeně lymfoplazmocytárním lymfomem, což je morfologický podklad Waldenströmovy makroglobulinemie.
Ale pouze u jednoho (pacient č. 1) se Waldenströmova makroglobulinemie stala v roce 2018 symptomatickou (postupný pokles koncentrace hemoglobinu pod 100 g/l, výrazný vzestup celkového imunoglobulinu typu IgM na 49 g/l a M-IgM na 23 g/l. Koncentrace FLC kappa byla 224 mg/l, lambda 4,63 mg/l poměr K/L 48,3. Celková bílkovina se zvýšila jen na 89 g/l, na očním pozadí nebyly známky fundus paraproteinemicus, i když hodnocení očního pozadí bylo obtížnější při současné hypertenzi a diabetu. Každopádně jeho nemoc vyžadovala v roce 2018 léčbu potlačující infiltraci kostní dřeně lymfoplazmocytárním lymfomem.
U pacienta č. 1 bylo v roce 2018 podáno 8 cyklů léčby RBD (rituximab, bendamustin, dexametazon). Tato léčba u něj navodila výrazný pokles monoklonálního imunoglobulinu, který je zatím setrvalý, jak dokumentuje graf 1 a tabulka 3. Poslední vyšetření z února 2021 prokázalo M-IgM v koncentraci 1,5 g/l, volné lehké řetězce kappa 52, lambda 6 mg/l poměr K/L 8,8. Hodnota imunoglobulinu typu IgG zůstává také nízká, jen 3,1 g/l.

Po ukončení této léčby Waldenströmovy makroglobulinemie pacient zjistil, že zatímco dříve se mu při opomenutí aplikace anakinry příznaky vracely za 24 hodin od poslední injekce, tak nyní, po ukončení léčby RBD, mu stačí aplikace anakinry 1x za 2 dny, a to platí stále k datu 12. 4. 2021, více než dva roky od ukončení uvedené léčby RBD.
U pátého pacienta je vidět v tabulce 3 a na grafu 2 poměrně výrazný vzestup monoklonálního imunoglobulinu v průběhu sledování, v roce 2020 již byla dle nálezu v kostní dřeni splněna kritéria Waldenströmovy makroglobulinemie, v roce 2020 jsme jej hodnotili jako asymptomatickou formu Waldenströmovy makroglobulinemie. Ale vyšetření z března 2021 prokázalo další vzestup koncentrace celkové bílkoviny a M-IgM a změny dalších sledovaných parametrů, viz tabulka 3, a tak je zřejmé, že pacientovo onemocnění spěje k symptomatické formě Waldenströmovy makroglobulinemie. Zatím běží léčba anakinrou, ale pacient je pozván na dřívější kontrolu s cílem přešetření stavu nemoci včetně provedení FDG-PET/CT, vyšetření očního pozadí a laboratorní kontrolu. Pokud naplní kritéria symptomatické Waldenströmovy makroglobulinemie, zahájíme u něj léčbu RBD. O ostatních pacientů nemá koncentrace monoklonálního imunoglobulinu v průběhu léčby anakinrou tak jasný vzestup, jak ilustruje graf 3.
M-IgM = koncentrace monoklonálního imunoglobulinu typu IgM
Pos. I-fix = pozitivní imunizační průkaz přítomnosti monoklonálního imunoglobulinu


Diskuze
Syndrom Schnitzlerové je velmi vzácná choroba registrovaná v Mezinárodní klasifikaci nemocí MKN-10 pod kódem L50.8. Choroba byla poprvé popsána francouzskou dermatoložkou prof. Liliane Schnitzler v roce 1972. Hlavními znaky tohoto syndromu jsou chronická kopřivka a poté projevy systémové zánětlivé odpovědi (horečky, bolesti kostí a kloubů). Díky monoklonálnímu imunoglobulinu, obvykle IgM méně často IgG, je řazena do skupiny krevních chorob zvaných monoklonální gamapatie. Etiopatogeneze zánětlivých projevů však odpovídá autoinflamatorním chorobám, a proto je současně řazena do skupiny autoinflamatorních chorob manifestujících se v dospělosti, kam patří mimo jiné Stillova choroba. Obě choroby mají některé společné znaky. Skupina autoinflamatorních chorob dnes čítá dle zprávy International Union of Immunological Societies celkem 37 monogenních autoinflamatorních chorob. Dále pak obsahuje autoinflamatorní choroby, u kterých zatím nebyla odhalena genetická podstata, ke kterým patří syndrom Schnitzlerové a Stillova choroba (8). Obě mají některé podobné projevy. A protože ke Stillově nemoci a syndromu Schnitzlerové nebyly přiřazeny genetické determinanty, jsou obě jmenované nemoci diagnostikovány na základě klinických kritérií. Kritéria syndromu Schnitzlerové uvádí tabulka 1. Možné příznaky této nemoci nejpodrobněji analyzovala Heleen de Koning, která shromáždila záznamy celkem 281 případů publikovaných ve světě. Její analýzu příznaků považujeme za velmi edukativní a proto ji uvádíme v tabulce 4 (1). V literatuře lze nalézt výjimečně i další projevy nemoci, jako je poškození ledvin (9).
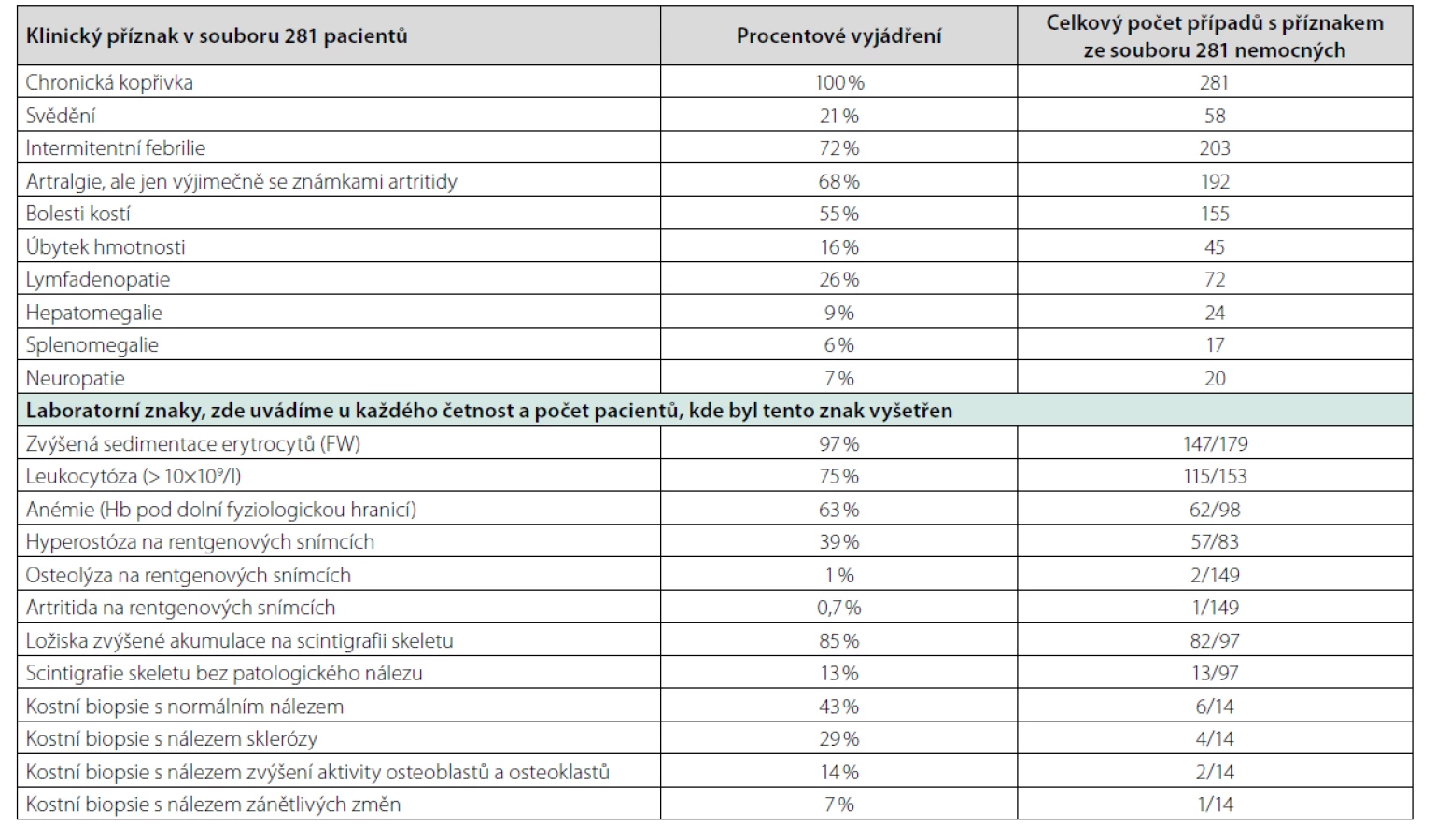
Incidence či prevalence syndromu Schnitzlerové není zmapována.
Heleen de Koning byla poslední, kdo vyhledal ve světovém písemnictví všechny publikované případy, 281 nemocných, a analyzovala je. Novější údaje o počtu publikovaných případů jsme nenašli.
Je zřejmé, že tato nemoc je stále poddiagnostikována, neboli často zůstává u pacientů trvale nerozpoznána. Již před lety na to upozornila práce z Mayo Clinic, kde evidovali v letech 1972–2010 pouze 16 pacientů se syndromem Schnitzlerové. Ve sledování však měli 4 103 pacientů s IgM-MGUS. V tomto velkém souboru provedli skrínink syndromu Schnitzlerové a odhalili dalších 46 nerozpoznaných případů, ačkoliv tito nemocní si často stěžovali svým lékařům na febrilie či subfebrilie (54 %) a/nebo na revmatické bolesti kostí a kloubů (78 %). Takže počet nerozpoznaných nemocných 2x převyšoval počet rozpoznaných (10).
Z Evropských center nejvíce případů popsali v univerzitní nemocnici Nantes (Francie). Za posledních 20 let má jejich soubor 25 nemocných s mediánem věku při stanovení diagnózy 68 let (medián stanovení diagnózy v našem souboru je 58 let). V jejich souboru nemocných měli všichni kožní příznaky typu chronických kopřivkových výsevů, které mnoho let předcházely nástupu projevů systémové zánětlivé reakce a nástupu bolestí kostí, svalů případně kloubů, skupinově označovaných jako „revmatické příznaky“ (7). A tak příznaky nemoci napřed nasměrovaly kroky nemocných k dermatologům, a teprve později kroky nemocných přesměrovaly k revmatologům. Z uvedeného souboru z Nantes si 72 % stěžovalo na bolesti kostí, které často byly interpretovány jako „revmatické bolesti“. Nejčastěji byly bolesti popisovány v okolí kolenního kloubu, v proximální tibii a distálním femuru (7). I další autoři uvádějí, že v 70–80 % případů si nemocní stěžují na špatně lokalizovatelné bolesti dolních končetin. Často byly popisovány i exacerbace bolesti v noci. Na rozdíl od revmatických chorob zde není popisována ranní ztuhlost a známky artritidy (7).
Z našich pacientů si na bolesti kostí, kloubů a případně svalů stěžovali všichni. Při popisování těchto bolestí pacienti často špatně odlišují, zda je bolí kloub, nebo kost či sval. V diferenciální diagnóze a řešení otázky, zda jsou to revmatoidní bolesti kloubů, nebo bolesti kostí typické pro tuto autoinflamatorní chorobu, pomohou zobrazovací vyšetření skeletu s cílem detekovat ložiska hyperostózy. Průkaz hyperostotických či osteosklerotických změn na skeletu je důležitý, protože tvoří malé kritérium této diagnózy, viz tabulka 1. Bolesti kostí souvisejí se změnami ve skeletu, pro které je charakteristická ložiskově zvýšená novotvorba kosti a ložiskově zvýšená denzita, jak zdokumentoval Terpos (11). Změny na kostech jsou popisovány jako fokální osteoskleróza, hyperostóza, periostální reakce, což vede ke zvýšené akumulaci radiofarmaka na kostním skenu (11).
Niederhauser nalezl při klasickém radiografickém vyšetření abnormality hlavně kolem kolenou u 14 z 22 pacientů (12). Ostatní autoři s použitím klasické radiografie zachytili kostní změny méně často, jen 10–30 %, jak uvádí velká přehledová práce (1). Proto klasické rentgenové snímky nejsou vhodné pro vyhledávání těchto kostních změn.
Mnohem větší senzitivu pro průkaz ložiskových hyperostotických kostních změn má klasická kostní scintigrafie (1). Abnormálně zvýšené vychytávání radiofarmaka prokázala kostní scintigrafie u 15 (85 %) z 18 pacientů, ponejvíce ve femurech a v tibii, případně v pánvi, jen ojediněle ve skeletu horních končetin. Míra vychytávání radiofarmaka (techneciumpyrofosfátu) korelovala s aktivitou nemoci. V případě úspěšné léčby anakinrou docházelo k poklesu vychytávání radiofarmaka v kostních ložiscích (13).
Dnes se do popředí zájmu dostává PET/CT vyšetření. Ve studii 10 pacientů bylo nejčastější anomálií difuzně zvýšené vychytávání 18FDG v kostní dřeni a/nebo zvýšené vychytávání 18FDG ve femurech, ale nepozorovali korelaci s léčebnou odpovědí, zatímco výsledky kostní scintigrafie korelovaly aktivitou nemoci (14). Je to vysvětlitelné faktem, že 18FDG-PET/CT, které bylo použito v této práci, odráží aktivitu buněčného metabolismu, zatímco scintigrafie skeletu odráží míru vychytávání kalcia ve skeletu.
U našich 5 pacientů bylo také provedeno 18FDG-PET/CT a hyperostotické či osteosklerotické kostní změny, odpovídající kritériím této nemoci, byly popsány na low-dose CT zobrazení celého skeletu. V těchto případech je prováděno zobrazení nazvané „total body“ neboli od „hlavy po paty“, které je schopné ve skeletu detekovat tyto hyperostotické změny při zobrazení kostí metodou low-dose CT. Akumulace 18FDG byla mírně zvýšená ve femurech případně v pánvi, podobně popisům v citované studii (14).
U jednoho pacienta byl místo 18FDG použit jako radiofarmakum Na18F. Toto radiofarmakum dobře znázorňuje ložiska novotvorby kosti, například osteosklerotické metastázy karcinomu prostaty. Radiofarmakum Na18F je možné použít k detekci hyperostotických změn, které jsou malým kritériem této nemoci, jak ilustruje Obr. 2. Tento typ vyšetření detekuje osteosklerotická či hyperostotická ložiska zřejmě s vyšší senzitivitou než klasická scintigrafie skeletu. Použít 18FDG-PET/ CT nebo Na18F PET/CT? To je otázka. Klasické 18FDG-PET/CT je schopno detekovat další patologie, jako například aortitidu (15), jak uvádí citovaná práce, zatímco Na18F-PET/CT má zatím nejvyšší senzitivu pro průkaz hyperostózy a zřejmě by mohlo zachytit dobře i dynamiku těchto změn. Lze tedy zobecnit, že sledování aktivity nemoci je možné i metodami, které detekují kostní ložiska se zvýšenou depozicí kalcia.
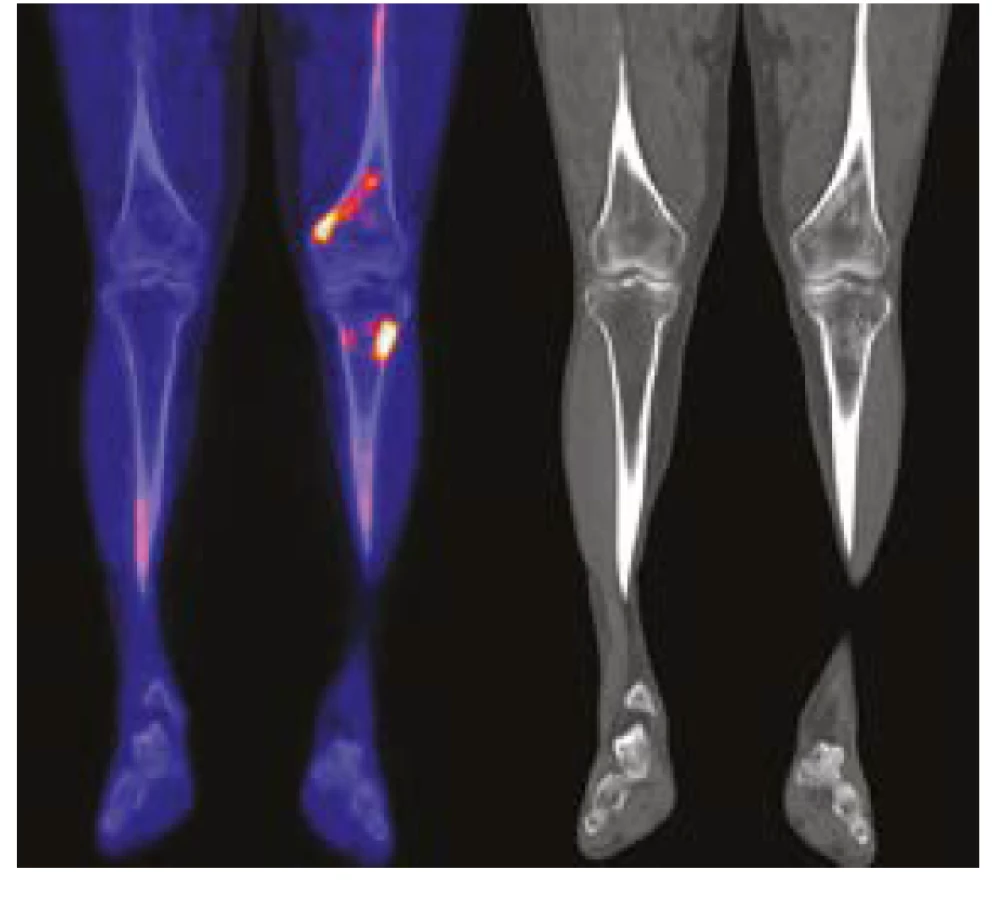
Diagnóza syndromu Schnitzlerové bývá obvykle rozpoznána v době, kdy M-IgM je jen mírně zvýšený a monoklonální gamapatie má charakter IgM-MGUS, který má však tendenci k vývoji, jak prokazuje vývoj koncentrací M-IgM i celkového imunoglobulinu typu IgM v našem souboru pacientů (tabulka 3, graf 3). V práci dvou center z Velké Británie popisují celkem 21 pacientů, kteří splnili kritéria syndromu Schnitzlerové (urtika, teploty, artralgie a kostní bolesti), navíc uvádějí v 47 % hmotnostní úbytek, ve 40 % patologickou únavu a ve 21 % lymfadenopatie. M-IgM detekovali u 86 %. Při mediánu sledování 13 let dekovali transformaci do Waldenströmovy makroglobulinemie nebo IgM myelomu u 20 % osob se syndromem Schnitzlerové (7). Tento počet odpovídá známému údaji z Mayo Clinic o počtu transformací IgM-MGUS do morbus Waldenström, případně IgM myelomu: 18 % po 10 letech sledování (16).
Takže náš počet dvou transformací při mediánu 8 let sledování s tím koresponduje. Proces transformace je kontinuální, nicméně je přijato arbitrární dělení na IgM-MGUS, asymptomatickou a symptomatickou Waldenströmovu makroglobulinémii.
Základem stanovení diagnózy je vždy histologické vyšetření válečku kostní dřeně, získaného trapanobiopticky z lopaty kosti pánevní. Stále platí histologická kritéria, jak je popsal Owen v roce 2003 (17). Mezinárodní klasifikace nemocí definuje diagnózu Waldenströmova makroglobulinemie (MKN-10 kód C88.0) následovně: Přítomnost M-IgM, infiltrace kostní dřeně lymfoplazmocytárním lymfomem – tedy klonálními lymfocyty, lymfoplazmocytoidními buňkami a plazmocyty. Typický imunofenotyp je CD19, CD20, and negativní CD5, CD10, and CD23.
Tento typ lymfomu není tvořen jen jedním morfologickým typem buněk, proto stanovit diagnózu z aspirátu a myelogramu se obvykle nedaří. Histologická diagnóza dle Owena (2003) na rozdíl od mnohočetného myelomu neobsahuje procentové vyjádření patologických buněk v kostní dřeni, i když autoři z Mayo Clinic jednou publikovali jejich verzi vyžadující 10% podíl patologických buněk, což však nebylo akceptováno. Definice dle Owena také neobsahuje hraniční koncentraci M-IgM, protože nekoreluje přesně s mírou infiltrace kostní dřeně. Koncentrace M-IgM odvisí také poměru klonálních lymfocytů, lymfoplazmocytů a plazmocytů. Pokud má lymfoplazmocytární infiltrace kostní dřeně více patologických lymfocytů a méně patologických lymfoplazmocytů a plazmocytů, tak je tvorba M-IgM nižší, než při vyšším zastoupení patologických plazmocytů, lymfoplazmocytů a při nižším zastoupení patologických lymfocytů (17). To je rozdíl od sekrečního mnohočetného myelomu, u něhož výše koncentrace M-Ig v séru je v přímém vztahu k počtem myelomových buněk v kostní dřeni.
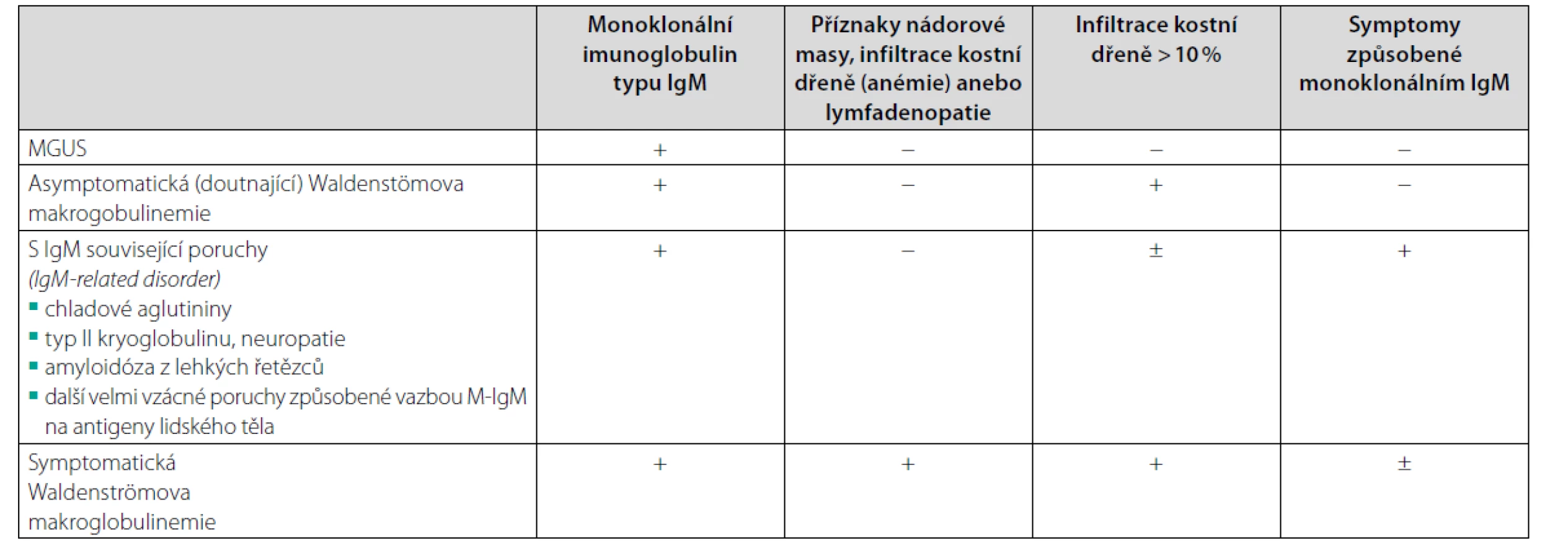
Alternativou pro případ nemožnosti provést trepanobiopsii, třeba pro závažnou koagulopatii, je flowcytometrické vyšetření kostní dřeně. Uvádí se i možnost transformace do mnohočetného myelomu typu IgM, ale tyto případy jsou podstatně vzácnější a odlišit tyto dvě jednotky není zcela jednoduché, jak popisuje Elba (18). Při průkazu M-IgM připadají v úvahu diagnózy uvedené v tabulce 5 (19).
Termín asymptomatická (doutnající) Waldenströmova makroglobulinemie se používá pro případy, kdy v kostní dřeni je jednoznačná infiltrace odpovídající lymfoplazmocytárnímu lymfomu a nejsou přítomny příznaky nemoci, které by zhoršovaly kvalitu života člověka. Termín symptomatická Waldenströmova makroglobulinemie znamená, že tato nemoc již snižuje kvalitu života člověka a je třeba ji léčit. V tabulce 6 uvádíme indikace pro léčbu z roku 2018 (20). Iniciální léčba je v roce 2021 založena stále na kombinaci monoklonální protilátky antiCD20 v kombinaci s alkylačním cytostatikem (cyklofosfamid) nebo s bendamustinem a dexametazonem (21). A tuto léčbu jsme použili u našeho pacienta a dosáhli téměř kompletní remise, viz graf 1. Tento pacient zjistil, že po ukončení léčby si již nepotřebuje aplikovat Kineret denně, ale stačí interval 48 hodin.

Jedna práce popisuje použití chemoterapie obsahující antiCD20 protilátku rituximab u pacienta se syndromem Schnitzlerové, jehož IgM gamapatie již měla charakter Waldenströmovy makroglobulinemie. Léčba, která dosáhla kompletní hematologické remise, vedla i k remisi syndromu Schnitzlerové (22). Nicméně ne ve všech případech vedl rituximab k léčebnému efektu, k vymizení příznaků syndromu Schnitzlerové, ale také nevedl k dosažení kompletní nebo téměř kompletní remise Waldenströmovy makroglobulinemie (23–26). Naše zkušenost však potvrzují publikovanou zkušenost (22), že účinná léčba Waldenströmovy makroglobulinemie, která dosáhne kompletní hematologické remise, nebo téměř kompletní remise, výrazně sníží intenzitu příznaků syndromu Schnitzlerové. Po ukončení této léčby již více než rok se příznaky syndromu Schnitzlerové nevrací při aplikace anakinry v intervalu 48 hodin, zatímco dříve se vracely již následující den, tedy za 24 hodin, pokud si ji pacient zapomněl píchnout. Z této zkušenosti plyne, že pokud původní MGUS, přítomný při stanovení diagnózy syndromu Schnitzlerové, se postupně transformuje do symptomatické Waldenströmovy makroglobulinemie, je vhodné použít kombinaci obsahující antiCD20 monoklonální protilátku (například rituximab) s nadějí, že při dosažení remise Waldenströmovy makroglobulinemie se zmírní příznaky syndromu Schnitzlerové (22, 24–26).
Role monoklonálního imunoglobulinu typu IgM zůstává nejasná. Zdá se, že jeho kvantita není v přímém vztahu se závažností nemoci (1, 2, 27, 28). V jednom případě se monoklonální imunoglobulin stal prokazatelným až 4 roky po prvních symptomech, takže samotný monoklonální imunoglobulin zřejmě není příčinou této nemoci (2, 27, 28). Z druhé strany jak naše, tak i popsané zkušenosti potvrzují, že léčba kombinací obsahující rituximab redukuje intenzitu choroby. Nevíme však, zda je to díky redukci patologické lymfoplazmocytární populace, nebo redukce fyziologických C20+ lymfocytů.
Je zde další publikace, potvrzující, že potlačení monoklonální gamapatie vedlo k ústupu symptomů nemoci, tentokráte k tomu byl použit ibrutinib (29).
Přehled léčebných možností syndromu Schnitzlerové
Anakinra
Anakinra je do roku 2007 lékem číslo jedna pro tyto pacienty Přípravek anakinra (Kineret) má ověřenou účinnost u revmatoidní artritidy. Dalšími indikacemi jsou: periodické horečky u dospělých, dospívajících, dětí a kojenců, kryopyrin-asociované periodické syndromy (CAPS). Anakinra je indikována k léčbě CAPS včetně multisystémového zánětlivého onemocnění se začátkem v novorozeneckém věku (NOMID), chronického infantilního neurologického kožního a kloubního syndromu (CINCA), Muckle–Wellsova syndromu (MWS), familiárního chladového autoinflamatorního syndromu (FCAS), familiární středozemní horečky (FMF). Další indikací je Stillova nemoc včetně systémové juvenilní idiopatické artritidy (SJIA) a Stillovy nemoci dospělých (Adult-Onset Still’s Disease, AOSD). Syndrom Schnitzlerové je natolik vzácný, že by se firmě nevyplatilo organizovat registrační studii. Proto pro léčbu syndromu Schnitzlerové preparátem anakinra je nutno si vždy vyžádat schválení úhrady plátcem zdravotní péče a opakovaně o ně žádat každé 3 nebo 6 měsíců, dle domluvy se zdravotní pojišťovnou. Anakinra je léčbou volby syndromu Schnitzlerové. Pozitivní efekt v této indikaci byl potvrzen velmi četnými publikacemi, citujeme jen vybrané (1, 2, 30, 31, 32). První větší klinická studie je opět z Francie. Léčili celkem 29 pacientů, u všech pozorovali rychlý nástup účinku. Při mediánu sledování 36 (2–79) měsíců se účinnost neměnila. Výsledek léčby hodnotili u 24 (83 %) jako kompletní remisi a u 5 (17 %) jako parciální remisi. V průběh sledování se u dvou pacientů byla prokázána transformace do Waldenströmovy makroglobulinemie a v jednom se zjistila AA-amyloidóza (32). Další větší studie obsahovala 21 pacientů, anakinra dosáhla u 95 % kompletní dlouhodobé remise (33). Velmi dobrou účinnost potvrdila i další evropská studie popisující léčbu 11členného souboru pacientů s anakinrou (34). A není bez zajímavosti, že francouzští specialisté na autoinflamatorní choroby zveřejnili popis nové klinické jednotky, blízké syndromu Schnitzlerové: kombinaci monoklonál - ní gamapatie, artralgií a rekurentních horeček také léčitelné anakinrou(35). V jednom případně byla anakinra použita v případě souběhu syndromu Schnitzlerové a Castlemanovy nemoci s léčebným úspěchem (36) a tato publikace otevírá cestu k použití anakinry také u Castlemanovy nemoci.
Canakinumab
Canakinumab (Ilaris) je monoklonální protilátka proti IL-1β. Canakinumab má dlouhý poločas (23–26 dní), a to umožňuje další intervaly mezi injekcemi. V roce 2020 je dle SÚKLu canakinumab indikován pro léčbu kryopyrin asociovaného periodického syndromu (CAPS) u dospělých, dospívajících a dětí ve věku od 2 let, pro Muckle-Wellsův syndrom (MWS), pro multisystémové zánětlivé onemocnění se začátkem v novorozeneckém věku (NOMID), nebo pro léčbu chronického infantilního neurologického kožního a kloubního syndromu (CINCA), pro léčbu těžkých forem familiárního chladového autoinflamatorního syndromu (FCAS), pro léčbu familiární chladové kopřivky (FCU). Podobně jako u anakinry, syndrom Schnitzlerové sice není ve výčtu oficiálních indikací, ale jeho účinnost u syndromu Schnitzlerové byla doložena četnými studiemi (37–40). V letech 2011–2020 bylo identifikováno 7 publikací popisujících léčbu celkem 34 pacientů se syndromem Schnitzlerové. Kumulativní doba sledování byla 253 měsíců a pět studií mělo medián sledování delší než 12 měsíců. Léčba canakinumabem dovedla 58,6 % pacientů do kompletní remise, u všech ostatních pacientů byla dosažena parciální remise. Nejčastějším nežádoucím účinkem byly infekce. Jeden pacient zemřel na sepsi způsobenou atypickou mykobakteriální infekcí. Tato analýza tedy potvrzuje velmi dobrou účinnost. Srovnávací studie anakinry a canakinumabu zatím nebyla provedena. Canakinumab je dle této analýzy nutno považovat za lék podobně účinný jako anakinra (41). Měsíční léčba tímto preparátem je však podstatně nákladnější, což limituje jeho použití v praxi.
Tocilizumab
Léčba pomocí protilátky proti interleukinu-6 zvaná tocilizumab byla popsána taktéž jako účinná u syndromu Schnitzlerové, nicméně v roce 2021 je průběh léčby tocilizumabem popsán v nevelkém počtu popisů případů. V jedné publikaci je sice popsána kompletní léčebná odpověď, další publikace hodnotí léčebný efekt jako variabilní (42–46). V roce 2021 byla publikována berlínskými autory klinická studie, v níž bylo 9 pacientů léčeno tocilizumabem. Autoři došli k závěru, že tocilizumab snižuje intenzitu klinických příznaků a snižuje hodnotu zánětlivých markerů. V průběhu léčby však docházelo ke ztrátě účinnosti, a tak autoři dospěli k závěru, že tocilizumab je vhodné otestovat u pacientů, kteří nereagují na jiné anticytokinové terapie. Takže tocilizumab se zatím nedostal do rutinní praxe v této indikaci (47).
Závěr
Domníváme se, že i u nás je hodně pacientů s nediagnostikovaným syndrom Schnitzlerové, podobně jak to bylo odhaleno při skrínink této choroby u všech osob s IgM-MGUS na Mayo Clinic (10). K diferenciální diagnóze zde může pomoci detekce osteosklerotických kostních změn u pacientů stěžujících si na bolesti kostí, které u revmatických chorob nebývají. Je však nutno použít citlivé detekční metody, scintigrafii skeletu, CT skeletu. Nejcitlivější je PET/CT s fluoridovým radiofarmakem jak ilustruje Obr. 2. V našem případě osteosklerotická ložiska odhalilo často low-dose CT zobrazení skeletu při provedení 18FDG-PET/CT (2, 13). Osteosklerotická ložiska mohou vzbudit podezření na syndrom Schnitzlerové, i když bývají i u dalších autoinflamatorních chorob. A pak již srovnání příznaků a nálezů s kritérii v tabulce 1 umožní stanovit diagnózu.
Anakinra přestavuje v roce 2021 lék volby pro syndrom Schnitzlerové (7, 32, 33, 34). Jeho účinnost s délkou podávání se nesnižuje. Dle počtu publikací je vidět, že v zahraničí již canakinumab (Ilaris), začíná anakinře konkurovat. Pří účinné léčba anakinrou je zřetelná normalizace CRP, amylopeptidu-A, případně dalších zánětlivých markerů, ale i míry vychytávání radiofarmaka při scintigrafii skeletu nebo při Na18F-PET/CT (1, 7).
V průběhu léčby je nutno monitorovat M-IgM, ale i celkový imunoglobulin typu IgM a volné lehké řetězce s cílem včas podchytit transformaci IgM-MGUS do symptomatické formy Waldenströmovy makroglobulinemie a podat odpovídající léčbu. Úspěšná léčba symptomatické formy Waldenströmovy makroglobulinemie může snížit aktivitu syndromu Schnitzlerové. Ojediněle jsou popsány i případy kompletního vymizení syndromu Schnitzlerové při dosažení kompletní remise Waldenströmovy makroglobulinemie (22). A proto tato nemoc patří současně do autoinflamatorních chorob, ale i do monoklonálních gamapatií, budou ji i nadále léčit hematologové, kteří mají praxi v diagnostice a léčbě monoklonálních gamapatií.
KORESPONDENČNÍ ADRESA AUTORA:
prof. MUDr. Zdeněk Adam, CSc.
Interní hematologická a onkologická klinika LF MU a FN BRNO
Jihlavská 20, 625 00 Brno
Článek přijat redakcí: 27. 11. 2020
Článek přijat po recenzích k publikaci: 15. 4. 2021
Sources
1. de Koning HD. Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy 2014; 4 : 41–45.
2. Mulla E, Neame R. Delayed development of the IgM paraprotein in Schnitzler’s syndrome. Scand J Rheumatol 2015; 44 : 521–522.
3. Willekens I, Walgraeve N, Goethals L et al. Correlative bone imaging in a case of Schnitzler’s syndrome and brief review of the literature. Hell J Nucl Med 2015; 18 : 71–73.
4. Hrodek O. Schnitzler syndrome: an under-diagnosed clinical entity Transfuze a hematologie dnes. 2013; 19(4): 214–215.
5. Janíková-Obořilová, Adam Z. Schnitzlerův syndrom Vnitřní lék 1998; 44 (7): 423–4274.
6. Lipsker D, Veran Y, Grunenberger F et al. The Schnitzler syndrome. Four new cases and review of the literature. Medicine (Baltimore) 2001; 80 : 37–44.
7. Simon A, Asli B, Braun-Falco M et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy 2013; 68 : 562–568.
8. Kacar M, Pathak S, Savic S. Hereditary systemic autoinflammatory diseases and Schnitzler’s syndrome Rheumatology (Oxford) 2019; 58(Suppl 6): 31–43.
9. Basile C, Rossi L, Casucci F et al. Kidney involvement in the Schnitzler syndrome, a rare disease. Clin Kidney J. 2017; 10(6): 723–727. doi: 10.1093/ckj/sfx077.
10. Jain T, Offord CP, Kyle RA, Dingli D. Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica. 2013; 98(10): 1581–1585. doi: 10.3324/haematol.2013.084830.
11. Terpos E, Asli B, Christoulas D, Brouet J-C, Kastritis E, Rybojad M et al. Increased angiogenesis and enhanced bone formation in patients with IgM monoclonal gammopathy and urticarial skin rash: new insight into the biology of Schnitzler syndrome. Haematologica. 2012; 97(11): 1699–1703.
12. Niederhauser BD, Dingli D, Kyle RA, Ringler MD. Imaging findings in 22 cases of Schnitzler syndrome: characteristic Para-articular osteosclerosis, and the “hot knees” sign differential diagnosis. Skeletal Radiol. 2014; 43(7): 905–915.
13. Darrieutort-Laffite C, Ansquer C, Aubert H et al.Rheumatic involvement and bone scan features in Schnitzler syndrome: initial and follow-up data from a single-center cohort of 25 patients. Arthritis Res Ther. 2020; 22(1): 272. doi: 10.1186/s13075-020-02318-5.
14. Alix L, Néel A, Cador B et al. Diagnostic value of 18-F fluorodeoxyglucose PET/CT and bone scan in Schnitzler syndrome. Autoimmunity. 2019; 52(7–8): 264–271.doi: 10.1080/08916934.2019.1680649.
15. Bursztejn AC, Imperiale A, Lipsker D. Aortitis: A new feature of Schnitzler syndrome. JAAD Case Rep. 2017; 3(5): 454–456. doi: 10.1016/j.jdcr.2017.06.016. PMID: 28971133; PMCID: PMC5602823.
16. Kyle RA, Therneau TM, Rajkumar SV et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood. 2003; 102(10): 3759–3764.
17. Owen RG, Treon SP, Al-Katib A, Fonseca R, Greipp PR, McMaster ML et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003; 30(2): 110–115.
18. Elba S, Castellino A, Soriasio R et al. Immunoglobulin M (IgM) multiple myeloma versus Waldenström macroglobulinemia: diagnostic challenges and therapeutic options: two case reports. J Med Case Rep. 2020; 14(1): 75. doi: 10.1186/s13256-020-02380-2. PMID: 32564775; PMCID: PMC7310052.
19. Gerz MA: Waldenström macroglobulinemia: 2021 update on diagnosis, risk stratification, and management. Amer J Hematol. 2021; 96(2): 258–269.
20. Gavriatopoulou M, Musto P, Caers J et al. European myeloma network recommendations on diagnosis and management of patients with rare plasma cell dyscrasias. Leukemia. 2018 Sep;32(9):1883-1898. doi: 10.1038/s41375-018-0209-7. Epub 2018 Jul 23. PMID: 30038381.
21. Kastritis E, Leblond V, Dimopoulos MA, ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018; 29(Suppl 4): iv41–iv50. doi: 10.1093/annonc/mdy146.
22. Aouba A, Pressiat C, Pricopi M et al. Complete remission of Schnitzler syndrome and Waldenström macroglobulinemia under rituximab-cyclophosphamide-dexamethasone. Dermatology 2015; 230(1): 18–22.
23. Cascavilla N, Bisceglia M, D’Arena G. Successful treatment of Schnitzler’s syndrome with anakinra after failure of rituximab trial. Int J Immunopathol Pharmacol 2010; 23(2): 633–636.
24. Murota H, Shoda Y, Ishibashi T et al. Improvement of recurrent urticaria in a patient with Schnitzler syndrome associated with B-cell lymphoma with combination rituximab and radiotherapy. J Am Acad Dermatol 2009; 61(6): 1070–1075.
25. Eiling E, Möller M, Kreiselmaier I et al. Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol 2007; 57(2): 361–364.
26. Ramadan KM, Eswedi HA, El-Agnaf MR. Schnitzler syndrome: a case report of successful treatment using the anti-CD20 monoclonal antibody rituximab. Br J Dermatol 2007; 156(5): 1072–1074.
27. Husak R, Nestoris S, Goerdt S et al. Severe course of chronic urticaria, arthralgia, fever and elevation of erythrocyte sedimentation rate: Schnitzler’s syndrome without monoclonal gammopathy? Br J Dermatol 2000; 142 : 581–582.
28. Cristina T, Varella N, Nishimura MY et al. Schnitzler’s syndrome without monoclonal gammopathy. Acta Derm Venereol 2005; 85 : 272–273.
29. Jani P, Vissing MB, Ahmed S et al. Ibrutinib for the Management of Schnitzler Syndrome: A Novel Therapy for a Rare Condition. J Oncol Pract. 2018; 14(6): 387–388. doi: 10.1200/ JOP.18.00050. Epub 2018 Mar 20.
30. Doležalová P. Nové indikace léčby anakinrou Farmakoterapie (Praha, Print) 2019; 15(3): 333–337.
31. Šedivá, A, Slíva J, Doležalová P et al. Anakinra Farmakoterapie (Praha, Print) 2011; 7(6): 621–629.
32. Néel A, Henry B, Barbarot S et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler’s syndrome: A French multicenter study. Autoimmun Rev 2014; 13 : 1035–1041.
33. Rowczenio DM, Pathak S, Arostegui JI et al. Molecular genetic investigation, clinical features, and response to treatment in 21 patients with Schnitzler syndrome. Blood. 2018; 131(9): 974–981. doi:10.1182/blood-2017-10-810366.
34. Vanderschueren S, van der Veen A. The Schnitzler syndrome: chronic urticaria in disguise: a single-centre report of 11 cases and a critical reappraisal of the literature. Clin Exp Rheumatol. 2017; 35(1): 69–73.
35. Giurgea I, Grateau G, Georgin-Lavialle S; French Network of Dysimmune Disorders Associated with Hemopathies. Monoclonal Gammopathy, Arthralgias, and Recurrent Fever Syndrome: A New Autoinflammatory Syndrome? J Rheumatol. 2019; 46(11): 1535–1539. doi: 10.3899/jrheum.181204.
36. Soudet S, Fajgenbaum D, Delattre C et al. Schnitzler syndrome co-occurring with idiopathic multicentric Castleman disease that responds to anti-IL-1 therapy: A case report and clue to pathophysiology. Curr Res Transl Med. 2018; 66(3): 83–86. doi: 10.1016/j. retram.2018.06.001.
37. Gorodetskiy VR, Salugina SO, Fedorov ES. Increasing the Interval of Canakinumab Administration Effectively Supports the Remission of Schnitzler’s Syndrome. Case Rep Rheumatol. 2018;2018 : 5416907. doi: 10.1155/2018/5416907. PMID: 29850358; PMCID: PMC5925130.
38. Fujita Y, Asano T, Sakai A et al. A case of Schnitzler’s syndrome without monoclonal gammopathy successfully treated with canakinumab. BMC Musculoskelet Disord. 2021;22(1):257. doi: 10.1186/s12891-021-04120-z. Betrains A, Staels F, Vanderschueren S. Efficacy and safety of canakinumab treatment in schnitzler syndrome: A systematic literature review. Semin Arthritis Rheum. 2020; 50(4): 636–642. doi: 10.1016/j.semarthrit.2020.05.002.
39. Krause K, Tsianakas A, Wagner N et al. Efficacy and safety of canakinumab in Schnitzler syndrome: A multicenter randomized placebo-controlled study. J Allergy Clin Immunol. 2017; 139(4): 1311–1320.
40. Krause K, Bonnekoh H, Ellrich A et al. Long-term efficacy of canakinumab in the treatment of Schnitzler syndrome. J Allergy Clin Immunol. 2020; 145(6): 1681–1686.e5. doi:10.1016/j.jaci.2019.12.909.
41. Betrains A, Staels F, Vanderschueren S. Efficacy and safety of canakinumab treatment in schnitzler syndrome: A systematic literature review. Semin Arthritis Rheum. 2020; 50(4): 636–642. doi: 10.1016/j.semarthrit.2020.05.002.
42. Krause K, Feist E, Fiene M et al. Complete remission in 3 of 3 anti-IL-6-treated patients with Schnitzler syndrome. J Allergy Clin Immunol 2012; 129 : 848–850.
43. Claus J, Vanderschueren S. Variable Responses to Tocilizumab in Four Patients with Schnitzler Syndrome. J Clin Immunol 2019; 39(4): 370–372.
44. Yan R, Cao W, Liu X, Li F, Shen M. A Chinese case series of Schnitzler syndrome and complete remission in one tocilizumab-treated patient. Clin Rheumatol. 2020; 39(12): 3847–3852.
45. Kluger N, Bessis D, Guillot B. Tocilizumab as a potential treatment in Schnitzler syndrome. Med Hypotheses 2009; 72(4): 479–480.
46. Więsik-Szewczyk E, Felis-Giemza A et al. Schnitzler Syndrome in a 27-Year-Old Man: Diagnostic and Therapeutic Dilemma in Adult Auto-Inflammatory Syndromes A Case Report and Literature Review. Int J Gen Med. 2020; 13 : 713–719. doi: 10.2147/IJGM.S265482
47. Bonnekoh H, Frischbutter S, Roll S et al. Tocilizumab treatment in patients with Schnitzler syndrome: An open-label study. J Allergy Clin Immunol Pract. 2021; S2213–2198(21)00152 - 5. doi: 10.1016/j.jaip.2021.01.024.
48. Castillo JJ, Advani RH, Branagan AR et al. Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol. 2020; 7(11): e827–e837. doi:10.1016/S2352-3026(20)30224-6.
49. Castillo JJ, Garcia-Sanz R, Hatjiharissi E, Kyle RA et al. Recommendations for the diagnosis and initial evaluation of patients with Waldenström Macroglobulinaemia: A Task Force from the 8th International Workshop on Waldenström Macroglobulinaemia. Br J Haematol. 2016; 175(1): 77–86. doi: 10.1111/bjh.14196.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2021 Issue E-3
Most read in this issue
- Jaterní cirhóza a těhotenství: kazuistika a přehled literatury
- Biomarker GDF-15 v kardiologii
- Recidivující asfyktická zástava oběhu – neobvyklá manifestace SLE
- Rituximab v liečbe primárnych glomerulopatií – naše skúsenosti