
Preleukemické fúzne gény typické pre akútnu myeloidnú leukémiu
Preleukemic fusion genes typical for acute myeloid leukemia
Acute myeloid leukemia (AML) is a highly heterogeneous subtype of leukemia, accounting for 25 % of childhood leukemias. By the presence of genetic mutations in hematopoietic/ progenitor stem cells, the bone marrow produces a large number of abnormal undifferentiated leukocytes (blasts), which significantly impairs the proper differentiation of cells. AML is induced by two interventions. Chromosomal translocation during hematopoiesis of intrauterine development is the first intervention. This creates preleukemic fusion genes (PFG), which can later be transformed by a second intervention (point genetic mutation – deletion, insertion …) into a functional malignant clone. Characteristic AML fusion genes include AML1-ETO, PML-RARA or MLL-AF9, which in turn produce hybrid proteins with altered function. Several studies suggest that these PFGs are considered an important prognostic tool in disease assessment. While the incidence of PFG characteristic of acute lymphoblastic leukemia (ALL) has been relatively well studied by several research groups and has been estimated at 1 to 5% in the umbilical cord blood of healthy neonates, PFG relevant to AML are still not sufficiently clarified.
Keywords:
Acute myeloid leukemia – AML1-ETO – MLL-AF9 – PML-RARA – preleukemic fusion genes
Authors:
Daniela Klimová 1; Jakub Styk 1; Michal Svoboda 1; Simona Humplíková 1,2; Vanda Repiská 1
Authors‘ workplace:
Univerzita Komenského v Bratislave, Lekárska fakulta, Ústav lekárskej biológie, genetiky a klinickej genetiky, Bratislava
1; Klinika anestéziológie a intenzívnej medicíny, Nemocnica Ružinov, Bratislava
2
Published in:
Vnitř Lék 2021; 67(E-5): 9-12
Category:
Review Articles
Overview
Akútna myeloidná leukémia (AML) je značne heterogénny podtyp leukémie, ktorý predstavuje 25 % všetkých detských leukémií. Prítomnosťou genetických mutácií v kmeňových krvotvorných/progenitorových bunkách produkuje kostná dreň veľké množstvo abnormálnych nediferencovaných leukocytov (blastov), čo výrazne narúša správnu diferenciáciu buniek. AML je indukovaná dvomi zásahmi. Chromozomálna translokácia počas hematopoézy vnútromaternicového vývoja predstavuje prvý zásah. Tak vzniknú preleukemické fúzne gény (PFG), ktoré sa môžu neskôr transformovať druhým zásahom (bodová genetická mutácia – delécia, inzercia…) na funkčný malígny klon. Medzi charakteristické fúzne gény AML patria napríklad AML1-ETO, PML‑RARA či MLL‑AF9, ktoré následne produkujú hybridné proteíny so zmenenou funkciou. Viaceré štúdie poukazujú na to, že tieto PFG sú považované za dôležitý prognostický nástroj pri hodnotení ochorení. Zatiaľ čo výskyt PFG charakteristických pre akútnu lymfoblastickú leukémiu (ALL) je relatívne dobre preskúmaný a odhadoval sa na 1 až 5 % v pupočníkovej krvi zdravých novorodencov, PFG relevantné pre AML stále nie sú dostatočne objasnené.
Klíčová slova:
akútna myeloidná leukémia – AML1-ETO – MLL‑AF9 – PML‑RARA – preleukemické fúzne gény
Úvod
Detská leukémia má svoj pôvod v vnútromaternicovom vývoji. Na základe klinických prejavov alebo molekulárnych zmien sa delí na akútnu myeloidnú leukémiu (AML) a akútnu lymfoblastoidnú leukémiu (ALL). ALL je najbežnejšia forma detskej leukémie. AML sa vyskytuje prevažne u dospelých jedincov, je zriedkavejšia u detí, s prevalenciou u starších detí. Úspešnosť liečby AML u detí je v porovnaní s ALL nižšia, vylieči sa 40–50 % všetkých pacientov (70–90 % u ALL). Pre najefektívnejšiu liečbu AML je rozhodujúca správna a presná diagnóza. To si vyžaduje spoľahlivé morfologické, imunologické a cytogenetické vyšetrenia kostnej drene a periférnej krvi pacienta v čase stanovenia diagnózy.
Vznik a výskyt PFG
Viacero detských leukémií je pravdepodobne spôsobených tvorbou preleukemických klonov u novorodencov, ktoré vznikajú transformáciou hematopoetických kmeňových/progenitorových buniek (HSPC) (1). Vývoj akútnej detskej leukémie je viacstupňový proces, ktorý je riadený akumuláciou genetických abnormalít. Počiatočná udalosť, ako je chromozomálna translokácia, často generuje preleukemický fúzny gén, ktorý nadobúda niektoré nové aktivity (často ovplyvnené transkripčné faktory alebo tyrozínkinázy) a blokuje diferenciáciu HSPC. Takáto udalosť sa nazýva „prvý zásah“ a primárne vzniká nesprávnym opravením dvojvláknových zlomov DNA (DSB) počas fetálnej hematopoézy (2,3). Tento preleukemický fúzny klon môže prejsť konverziou na úplný malígny klon „druhým zásahom“, čo môže byť spôsobené bodovými mutáciami, akými sú napríklad delécie, inzercie alebo duplikácie (Obr. 1). Väčšina PFG je zvyčajne neskôr eliminovaná v postnatálnom vývoji (4).
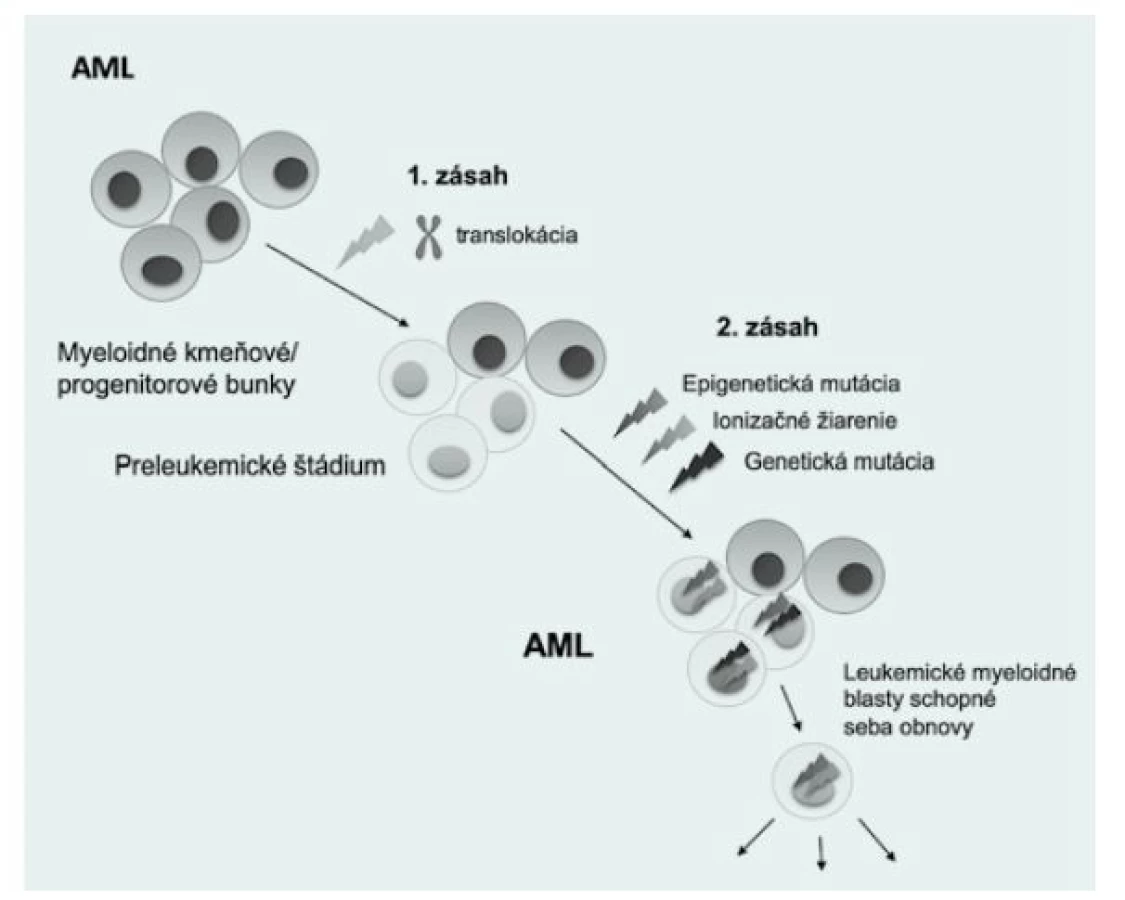
Prítomnosť PFG u zdravých novorodencov sa uvádza vo viacerých štúdiách, a predpokladá sa, že majú pôvod už v maternici (5). Údaje o výskyte najčastejších AML PFG u novorodencov zatiaľ nie sú súhrnne opísané. Konkrétne sa odhadoval výskyt AML1-ETO vo veľmi širokom rozmedzí medzi 0,2 % a 40 % (3, 6). Táto variabilita bola spôsobená najmä rozdielmi vo veku, etnickej príslušnosti a metodickými problémami. Výskyt PML‑RARA bol analyzovaný iba v jednej štúdii, ktorá detegovala incidenciu tohto génu až na 70 % (7). Podľa dostupných informácií ešte neprebehla žiadna štúdia na stanovenie frekvencie génu MLL‑AF9. Včasná detekcia PFG u zdravých novorodencov, rovnako aj špecifické zacielenie konkrétnej subpopulácie hematopoetických kmeňových buniek, môže významne pomôcť v prevencii a liečbe AML (8). Charakterizácia PFG u tehotných žien by bola výrazne nápomocná pri identifikácii a diferenciálnej diagnostike ostatných ochorení, akým je pri preeklampsii HELLP syndróm, nakoľko AML u tehotných žien s preeklampsiou simuluje parametre krvi HELLP syndrómu (9).
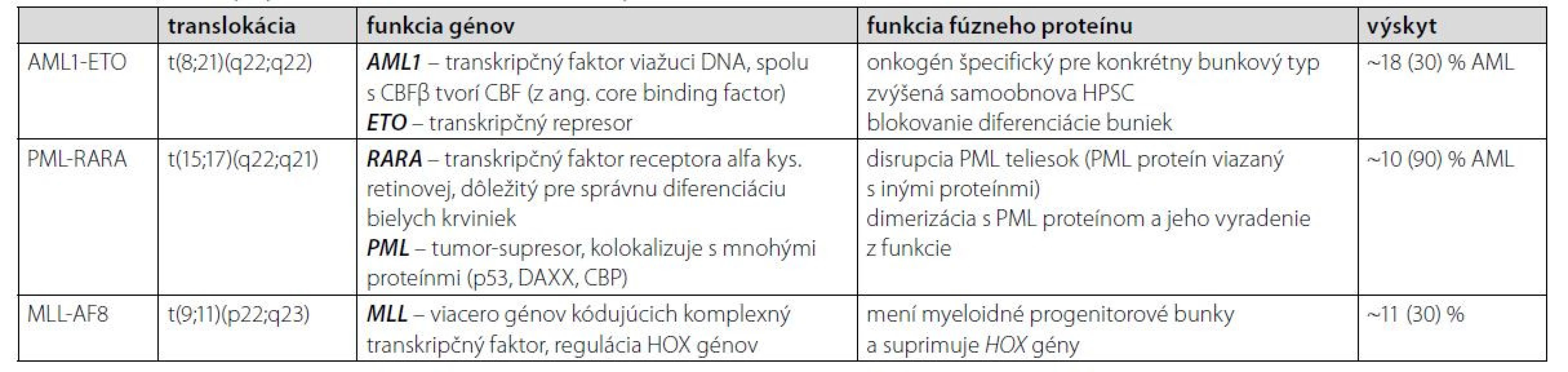
AML1-ETO
Fúzny gén AML1-ETO vzniká chromozomálnou translokáciou t (8;21) (q22;q22) a predstavuje jednu z najbežnejších cytogenetických abnormalít v AML, najmä v podtype M2. AML1 funguje ako transkripčný faktor, ktorý aktivuje hematopoetické kontrolné gény a ETO sa naopak považuje za transkripčný korepresor (10). Tento typ translokácie nesie približne 10 % všetkých prípadov AML, zatiaľ čo v detstve je diagnostikovaných 10–15 %. Aj napriek priaznivej prognóze pacientov je päťročné prežitie iba 50 %. V rámci diagnostiky je táto translokácia vhodným markerom na detekciu leukémie. Terapia AML s translokáciou t (8;21) je v počiatočnom štádiu štandardne liečená antracyklínom a cytarabínom. V porovnaní s inými subtypmi je chemosenzitivita vyššia, čo môže byť zapríčinené aktiváciou kaskády p53, vedúcej v konečnom dôsledku k apoptóze bunky (11). Liečba môže byť pre zlepšenie výsledku doplnená o anti‑CD33 protilátku gemtuzumab ozogamicín. Niektoré štúdie sa zameriavajú na degradáciu fúzneho génu na molekulárno‑genetickej úrovni, a to vývojom modifikovaných molekúl schopných blokovať aktivitu abnormálneho génu (12).
PML ‑ RARA
Translokácia t (15;17) (q22; q21) je diagnostickým znakom akútnej promyelocytovej leukémie (APL) a vedie k fúzii génov PML a RARA. APL je podtriedou AML a podľa klasifikačného systému WHO je chromozomálna aberácia v 95 % prípadoch APL t (15;17) (q22;q21), ktorá zabraňuje správnej diferenciácii buniek (13). PML proteín dokáže vďaka svojmu zloženiu prechodne interagovať s viac ako 170 proteínmi, čím je zapojený v rôznych dráhach (regulujúcich genómovú instabilitu, apoptózu a senescenciu, samoobnovu kmeňových buniek či epigenetickú reguláciu a transkripciu hematopoetických kmeňových buniek). Väčšina týchto interakcií je sprostredkovaná jeho RBCC doménou, ktorou dokáže multimerizovať a vytvárať tzv. PML jadrové telieska, alebo inými, izoformovo špecifickými doménami (14–16). Izoformy proteínu RARA regulujú transkripciu v závislosti od prítomnosti ligandu. Bez potrebného ligandu interagujú s korepresorom, čím reprimujú transkripciu génov. Pomocou tohto mechanizmu proteínu možno rozlišovať medzi zdravými a abnormálnymi myeloidnými hematopoetickými bunkami (17). Prognóza pacientov je pri použití liečby ATRA (kyselina all‑trans retinová, aktívny metabolit vitamínu A) a pri náležitej starostlivosti pomerne priaznivá. Liečba pomocou ATRA v kombinácii s ATO (oxid arzenitý) je vhodná pri počiatočných štádiách APL (18). Keďže gén PML má za normálnych okolností funkciu tumor supresora, vo fúzii s RARA je jeho funkcia blokovaná a bunka nemá kontrolovanú bunkovú smrť. ATRA je schopná degradovať fúzny gén PML‑RARA a ATO podporuje nematurované leukocyty k samo deštrukcii. Liečba pomocou ATRA+ATO má oproti kombinácii ATRA+chemoterapia výrazne silnejšie a trvalejšie antileukemické účinky, pri vysoko rizikových pacientoch sa stále štandardne využíva kombinácia ATRA+chemoterapia (20, 21). Celkové prežitie pacientov dosahuje až 89 % (19).
Fúzie génu MLL
Gén MLL je lokalizovaný na 11q23 a fúzuje s viac ako 60 rôznymi partnerskými chromozómami. Najbežnejšie translokácie v AML sú MLL‑AF9 t (9;11) a MLL‑AF6 t (6;11). Mutácie MLL vznikajú de novo a vedú u ľudí k vzniku AML (5–6 %) a ALL (5–10 %) (22). Prítomnosť prešmykov 11q23 je podstatne vyššia u pediatrickej ALL a dojčenskej AML. Asociácia medzi MLL translokáciami a jej dopadom na AML je oveľa komplikovanejšia ako u ALL. Uvádza sa, že najbežnejšia fúzia MLL v AML, MLL‑AF9, je spojená s prechodnou až dobrou prognózou (23). Sľubnou cestou na liečbu AML indukovanej MLL génom je inhibícia DOT1L (DOT1-podobná histón H3K79 metyltransferáza (24). Zhrnutie AML PFG a ich charakteristík je v Tabuľke 1.
Výhody pupočníkovej krvi pre transplantáciu PFG+ detí
Pupočníková krv (UCB) predstavuje alternatívny zdroj, ktorý umožňuje spätnú analýzu prenatálneho potenciálneho PFG. Jednou z možností sú aj Guthrieho karty, ktoré ale zriedka obsahujú viac ako 105 buniek (31), preto je pri identifikácii detí s rizikom vzniku PFG+ leukémie vhodnejší skríning UCB. Všeobecne má UCB aj oproti kostnej dreni (BM) niekoľko výhod, medzi ktoré patrí bohatá a okamžitá dostupnosť, ľahký odber, darovanie bez rizika, znížené riziko infekcie prenášanej krvou a znížené riziko „graft‑versus‑host“ kompilácií. V porovnaní s dospelou BM sú bunky UCB menej zrelé, majú dlhšie teloméry a väčší proliferačný potenciál. U pediatrickej ALL je transplantácia krvotvorných kmeňových buniek (HSCT) bežná u detí s mimoriadne vysokými rizikovými vlastnosťami, napr. hypodiploidia, zlyhanie indukcie (32). Vďaka relatívne nízkej účinnosti chemoterapeutickej liečby detskej AML sa u detí v remisii bežne používa alogénna HSCT (33). Alogénna HSCT je navyše jedinou liečebnou metódou na terapiu zriedkavých detských leukémií, ako je chronická myeloidná leukémia, juvenilná myelomonocytová leukémia a myelodysplastické syndrómy (32). Autológna transplantácia UCB sa používa predovšetkým pri poraneniach mozgu (82 %) (34). Okrem toho sa jej prínosné terapeutické účinky prejavili najviac u detí s hemoglobinopatiami a dedičnými metabolickými chorobami, ako sú Hurlerov syndróm, Krabbeov syndróm a metachromatická leukodystrofia (35).
Záver
Akútna myeloidná leukémia je charakterizovaná nekontrolovanou proliferáciou nediferencovaných blastov v periférnej krvi a kostnej dreni. Ak nie je včas diagnostikovaná, pacient umiera v priebehu niekoľkých týždňov až mesiacov. Inhibícia správnej tvorby krvných elementov je spôsobená génovými mutáciami, ktoré nastávajú v génoch zodpovedných za reguláciu bunkového cyklu, rastu a delenia. Leukemické bunky majú v svojej DNA špecifické miesta zlomu, vďaka ktorým sú leukemické klony spoľahlivo detekovateľné. Protoonkogény často fúzujú so silnými promótormi, čo spôsobuje kaskádu udalostí vedúcich k tumorigenéze. Celosvetová incidencia tohto ochorenia je 2,5–4/100 000 ročne, a zvyšuje sa s pribúdajúcim vekom. Incidencia AML na Slovensku je približne 6–8 prípadov za rok. Pacienti pod 60 rokov dosahujú kompletnú remisiu v 60–80 %. Po dosiahnutí kompletnej remisie prežíva dlhodobo 25–30 % pacientov. Prognóza AML je variabilná a závislá hlavne od molekulárno‑genetických zmien. K nepriaznivým prognostickým faktorom patrí typ M0, M5 M6 a M6 AML, vyšší vek a celkový horší zdravotný stav, zlá reakcia na prvú indukciu, pridružená leukémia, či vysoká koncentrácia LDH. Terapia prebieha na špecializovaných pracoviskách, s cieľom maximálnej redukcie leukocytov, eradikácie leukemických klonov a obnovy hematopoézy. Pacienti mladší ako 60 rokov sú liečení chemoterapiou podľa guidelinov, prípadne alogénnou transplantáciou kostnej drene. V porozumení patobiológie AML sa postupne dosahujú veľké pokroky, najmä v oblasti molekulárnych technológií. Špecifikovali sa mutačné oblasti a ich úloha v leukemogenéze.
Preleukemické fúzne gény predstavujú spoľahlivý marker včasnej detekcie AML.
Poďakovanie: Tento článok vznikol za podpory grantu SYNCYTÍN-1 ako nový marker preeklampsie, druhej najčastejšej príčiny úmrtia gravidných žien. MZSR (MZSR 2018/40-LKUK-14). Štúdium etiológie preeklampsie – druhej najčastejšej príčiny úmrtí gravidných žien. VEGA (1/0168/18).
KORESPONDENČNÍ ADRESA AUTORA:
Mgr. Daniela Klimová
Univerzita Komenského v Bratislave, Lekárska fakulta, Ústav lekárskej biológie, genetiky a klinickej genetiky, Sasinkova 4, 811 08 Bratislava
Cit. zkr: Vnitř Lék 2021; 67(e5): e9–e12
Článek přijat redakcí: 18. 2. 2021
Článek přijat po recenzích k publikaci: 14. 6. 2021
Sources
1. Škorvaga M, Nikitina E, Kubes M et al. Incidence of Common Preleukemic Gene Fusions in Umbilical Cord Blood in Slovak Population. PLoS ONE 2014; 9(3).
2. Greaves M F, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer 2003; 3 : 639–649.
3. Mori H, Colman S M, Xiao Z et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci 2002; 99 : 8242–8247.
4. Košík P, Škorvaga M, Belyaev I. Incidence of preleukemic fusion genes in healthy subjects. Neoplasma 2016; 63(5): 659–672.
5. Košík P, Škorvaga M, Durdík M et al. Low numbers of preleukemic fusion genes are frequently present in umbilical cord blood without affecting DNA damage response. Oncotarget 2017; 8 : 35824–35834.
6. Basecke J, Cepek L, Mannhalter C et al. Transcription of AML1/ETO in bone marrow and cord blood of individuals without acute myelogenous leukemia. Blood 2002; 100(6): 2267–2268.
7. Song J, Mercer D, Hu X et al. Common leukemia - and lymphoma‑associated genetic aberrations in healthy individuals. J Mol Diagn 2011; 13(2): 213–219.
8. Kosik P, Durdik M, Skorvaga M, Klimova D. Induction of AML Preleukemic Fusion Genes in HSPCs and DNA Damage Response in Preleukemic Fusion Gene Positive Samples. Antioxidants 2021; 10(3), 481.
9. Laguna‑Olmos M et al. Leucemia mieloide aguda y pre‑eclampsia coexistente. Algunas dificultades diagnósticas. A propósito de un caso. Revista chilena de obstetricia y ginecologia 2020; 85(2): 155–161.
10. https://ghr.nlm.nih.gov/gene/AML1 [cit. 2021-02-18]
11. Krejci O, Wunderlich M, Geiger H. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress ‑ induced death. Am. J. Hematol 2008; 111(4), 2190–2199.
12. Oo Z M, Illendula A, Grembecka J. A tool compound targeting the core binding factor Runt domain to disrupt binding to CBFβ in leukemic cells. Leukemia & lymphoma 2018; 59(9), 2188–2200.
13. Zahedipour F, Ranjbaran R, Behzad A et al. Development of Flow Cytometry‑Fluorescent In Situ Hybridization (Flow‑FISH) Method for Detection of PML/RARa Chromosomal Translocation in Acute Promyelocytic Leukemia Cell Line. Avicenna J. Med. Biotechnol 2017; 9(2): 104–108.
14. Lallemand‑Breitenbach, V. PML nuclear bodies. Cold Spring Harb Perspect Biol 2010; 2(5).
15. Matt S, Hofmann T. G. Crosstalk between p53 modifiers at PML bodies. Molecular & cellular oncology 2018; 5(3).
16. Wang, G, Tian Y, Hu Q. PML/RARa blocks the differentiation and promotes the proliferation of acute promyelocytic leukemia through activating MYB expression by transcriptional and epigenetic regulation mechanisms. J. Cell. Biochem 2019; 120(2): 1210–1220.
17. Zhu H H, Yang M C, Wang F. Identification of a novel NUP98–RARA fusion transcript as the 14th variant of acute promyelocytic leukemia. Am. J. Hematol 2020; 95(7): E184–E186.
18. Testi A M, Biondi A, Coco F L et al. Protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 2005; 106(2), 447–453.
19. Soignet S L, Maslak P, Wang Z et al. Complete Remission After Treatment of Acute Promyelocytic. NEJM 1998; 339 : 1341–1348.
20. Burnett A K, Russell N H, Hills R K. Myeloid Leukaemia Working Group. Arsenic trioxide and all‑trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): results of a randomised, controlled, phase 3 trial. The Lancet Oncology 2015; 16(13): 1295–1305.
21. Platzbecker U, Avvisati G, Cicconi L. Improved outcomes with retinoic acid and arsenic trioxide compared with retinoic acid and chemotherapy in non‑high‑risk acute promyelocytic leukemia: final results of the randomized Italian‑German APL0406 trial. Am. J. Clin. Oncol 2017.
22. Bernard O, Berger R. Molecular basis of 11q23 rearrangements in hematopoietic malignant proliferations. Genes Chromosomes Cancer 1995;13 : 75–85.
23. Balgobind B V, Raimondi S C, Harbott J et al Novel prognostic subgroups in childhood 11q23/MLL‑rearranged acute myeloid leukemia: results of an international retrospective study. Blood 2009; 114(12), 2489–2496.
24. Kuntimaddi A, Achille N J, Thorpe J. Degree of recruitment of DOT1L to MLL‑AF9 defines level of H3K79 di - and tri‑methylation on target genes and transformation potential. Cell Rep 2015; 11 : 808–820.
25. Rowley JD. The critical role of chromosome translocations in human leukemias. Annu. Rev. Genet 1998; 32(1), 495–519.
26. Tonks A, Tonks A J, Pearn L et al. Expression of AML1-ETO in human myelomonocytic cells selectively inhibits granulocytic differentiation and promotes their self‑renewal. Leukemia 2004; 18(7): 1238–1245.
27. https://ghr.nlm.nih.gov/gene/RARA [cit. 2021-02-18]
28. Nunes V S, Moretti N S. Nuclear subcompartments: an overview. Cell biology international 2016; 41 : 2–7.
29. Wood A M, Garza‑Gongora AG, Kosak A. A crowdsourced nucleus : understanding nuclear organization in terms of dynamically networked protein function, BBA 2014; ISSN 1874-9399.
30. Lang M, Jegou T, Chung I, Three‑dimensional organization of promyelocytic leukemia nuclear bodies. J. Cell. Sci 2010; 123(3): 392–400.
31. Lausten‑Thomsen U, Madsen HO, Vestergaard TR et al. Prevalence of t(12;21)[ETV6-RUNX1] - positive cells in healthy neonates. Blood 2011; 117 : 186–189.
32. Wayne AS, Baird K, Egeler RM. Hematopoietic stem cell transplantation for leukemia. Pediatr Clin North Am 2010; 57 : 1–25.
33. Oliansky DM, Rizzo JD, Aplan PD. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute myeloid leukemia in children: an evidence‑based review. Biol Blood Marrow Transplant 2007; 13 : 1–25.
34. Rosenthal J, Woolfrey AE, Pawlowska A et al. Hematopoietic cell transplantation with autologous cord blood in patients with severe aplastic anemia: an opportunity to revisit the controversy regarding cord blood banking for private use. Pediatr Blood Cancer 2011.
35. Ballen K K, Verter F, Kurtzberg J. Umbilical cord blood donation: public or private? Bone Marrow Transplant 2015; 50 : 1271–1278.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2021 Issue E-5
Most read in this issue
- D-laktátová acidóza – zriedkavá komplikácia syndrómu krátkeho čreva
- Věnujeme medikaci seniorů dostatečnou péči? (Případ Domova Vlčí mák ÚVN Praha)
- Whippleova nemoc – systémová choroba s gastrointestinálními projevy
- Mortalita pacientů s covidem-19 na JIP – naše zkušenosti