
Vrozený těžký deficit FVII a získaná idiopatická trombocytopenická purpura – vzácná kombinace dvou krvácivých stavů
Congenital Deficiency of FVII and Acquired Idiopathic Thrombocytopenic Purpura – the Rare Combination of Two Bleeding Disorders
The congenital deficiency of FVII is an autosomal recessive rare disorder. We have observed 15 patients with this deficiency in our haematology department including the only one patient with the FVII level < 1%. The deficiency with the FVII level < 2% is associated with serious bleeding complications including the joint bleeding, bleeding in the retroperitoneum, gastrointestinal system and in central nervous system (CNS). Serious acquired thrombocytopenia can originate from the impairment of platelets production, from their higher sequestration in the spleen but even from the elevated thrombocytes disintegration. The raised thrombocytes disintegration caused by the autoantibodies is the most frequent source of the acute life threatening thrombocytopenia with the massive bleeding in tissues, gastrointestinal system or CNS. We list the case report of a young patient with congenital FVII deficiency (FVII level < 1%) and acute idiopathic thrombocytopenic purpura with yet unknown aetiology of which has not been fully clarified although the very favourable effect of prednisone therapy indicates the autoimmune mechanism.
Key words:
congenital deficiency of FVII, idiopathic thrombocytopenic purpura, haptoglobin.
Authors:
J. Šlechtová
Authors‘ workplace:
Ústav klinické biochemie a hematologie LF UK a FN, Plzeň
Published in:
Čas. Lék. čes. 2008; 147: 431-433
Category:
Case Report
Overview
Vrozený deficit FVII je autozomálně recesivní vzácné onemocnění. V hematologické ambulanci sledujeme 15 pacientů s tímto deficitem, z toho pouze jednoho pacienta s hladinou FVII < 1 %. Deficit s hladinou F VII < 2 % bývá spojen se závažnými krvácivými komplikacemi včetně krvácení do kloubů, retroperitonea, GIT i CNS. Těžká získaná trombocytopenie může mít původ v porušené tvorbě destiček, ve zvýšené sekvestraci ve slezině, ale i ve zvýšeném rozpadu destiček. Zvýšený rozpad destiček, způsobený autoprotilátkami, je nejčastější příčinou akutní těžké, život ohrožující trombocytopenie s masivním krvácením do tkání, GIT či CNS. V naší kazuistice uvádíme případ mladého pacienta s vrozeným těžkým deficitem FVII (hladina FVII < 1 %) a akutní těžkou idiopatickou trombocytopenickou purpurou, jejíž etiologie není zatím plně objasněna, byť velmi příznivý účinek terapie prednisonem by nasvědčoval autoimunitnímu mechanizmu.
Klíčová slova:
vrozený deficit FVII, idiopatická trombocytopenická purpura, haptoglobin.
Dnes 32letý muž je od dětství léčen pro těžký vrozený deficit FVII s hladinou FVII < 1 %. První krvácivou epizodu (epistaxi) prodělal v necelém roce věku. Později se epistaxe objevila vždy při běžném infektu horních cest dýchacích nebo při neopatrných dětských hrách, protrahované krvácení se pravidelně objevovalo i v lůžku po spontánní extrakci zubů mléčného chrupu. Pacient musel být opakovaně hospitalizován na Dětské klinice nebo ORL klinice FN v Plzni nebo na dětském či ORL oddělení nemocnice v Klatovech či na dětském oddělení nemocnice v Domažlicích. Zpočátku byl léčen pouze skupinovou plazmou. Transfuze erytrocytů mu dosud nikdy podány nebyly. Těžký deficit FVII byl diagnostikován ve 3 letech. Později byl lehký deficit FVII prokázán i u matky (hladina FVII 34 %) a u dědečka z matčiny strany (hladina FVII 37 %). Ani u matky ani u dědečka se žádné spontánní krvácivé epizody nikdy nevyskytly. Matka měla fyziologicky probíhající menarche a rodila 2× bez komplikací s přiměřenými krevními ztrátami. Bratr a ostatní příbuzní z matčiny strany byli zdrávi.
Asi v 9 letech se u našeho nemocného objevilo první intraartikulární krvácení do levého kolenního kloubu. Později se přidružilo občasné krvácení do loketních kloubů a v dalších letech opakované krvácení do obou kloubů hlezenních. Pacient byl postupně během let léčen skupinovou plazmou, vitaminem K1 (Kanavit), koncentrátem FVII (Factor VII Concentrate TIM 4 Immuno), faktory protrombinového komplexu (Prothromplex Total TIM 4) a pro hypochromní mikrocytární anémii vznikající na podkladě opakovaných krvácivých epizod preparáty železa. Asi ve 20 letech mu byla už prokázána těžká artróza pravého hlezenního kloubu s cystami až nekrotickými změnami v oblasti chrupavky kosti hlezenní a doporučena intenzivní léčba chondroprotektivy, šetřící fyzioterapie a výhledové provedení radiační synovektomie. Radiační synovektomii později pacient odmítl. V prosinci 2001 byla u nemocného zahájena domácí léčba rekombinantním FVIIa (preparátem NovoSeven) s myšlenkou, že k největším krvácivým komplikacím dochází při transportu mezi místem bydliště a hematologickým pracovištěm, na němž je nemocnému podávána substituční léčba. Pacient má proto v posledních letech pro akutní potřebu stále k dispozici 2 balení rFVIIa (NovoSeven) o síle 1,2 mg. Pro zástavu krvácení u deficitu FVII stačí oproti deficitům koagulačních faktorů VIII nebo IX nebo jiným závažným krvácivým příhodám podstatně nižší hladina deficitního faktoru VII – i pouhých 20 % aktivity FVII (norma 50–150 % aktivity FVII).
Pacientovi bylo provedeno v časné dospělosti v období plánovaného rodičovství genetické vyšetření, které potvrdilo autozomální dědičnost onemocnění.
Největšími problémy jsou v posledních letech opakované krevní výrony do obou hlezenních kloubů (více vpravo). Na doporučení ortopeda je však pacient i nadále léčen pouze konzervativně chondroprotektivy a při krevních výronech si aplikuje rFVIIa (self service).
Dne 2. 7. 2007 se pacient akutně dostavil do hematologické ambulance s výraznou purpurou. Krvácel z ústních sliznic, z dásní, nosu a po celém těle měl rozsáhlé hematomy, mnohočetné petechie a sufuze (obr. 1 a 2).
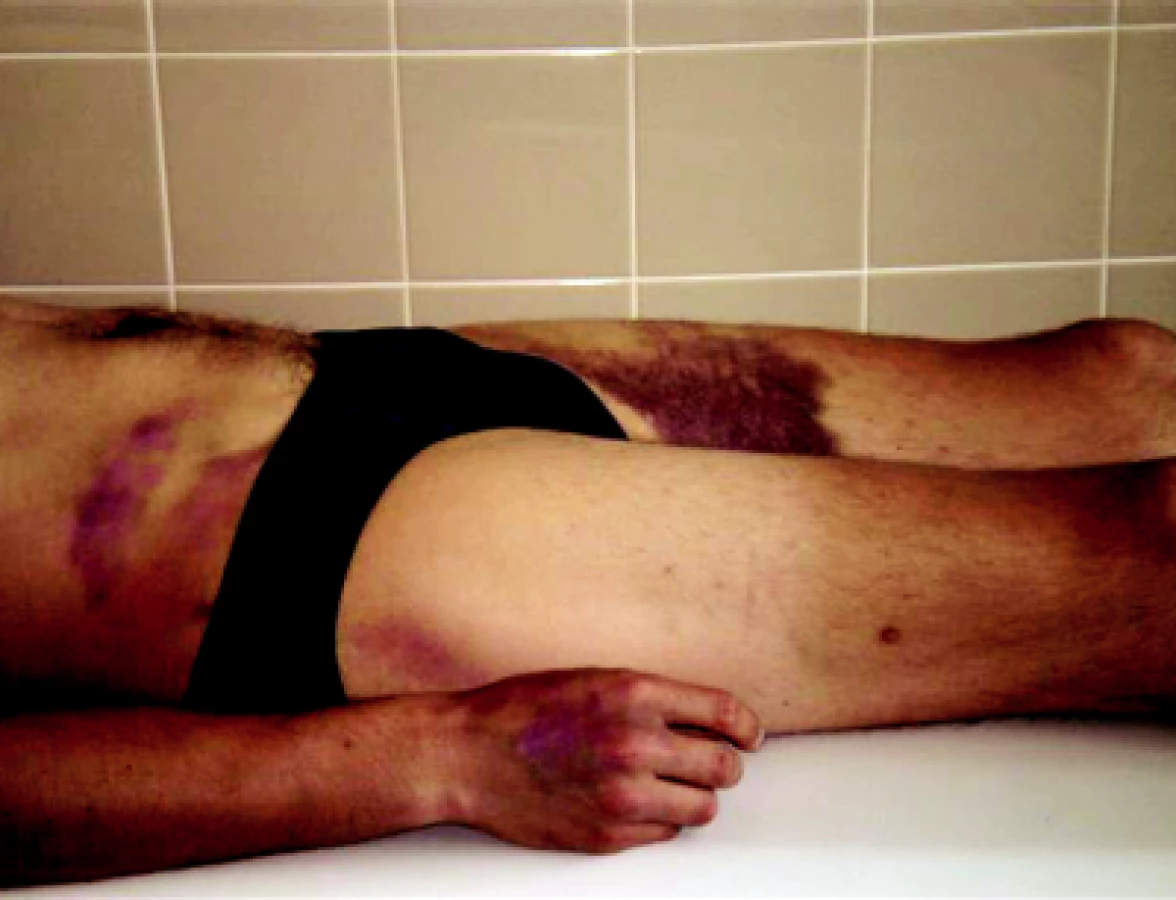
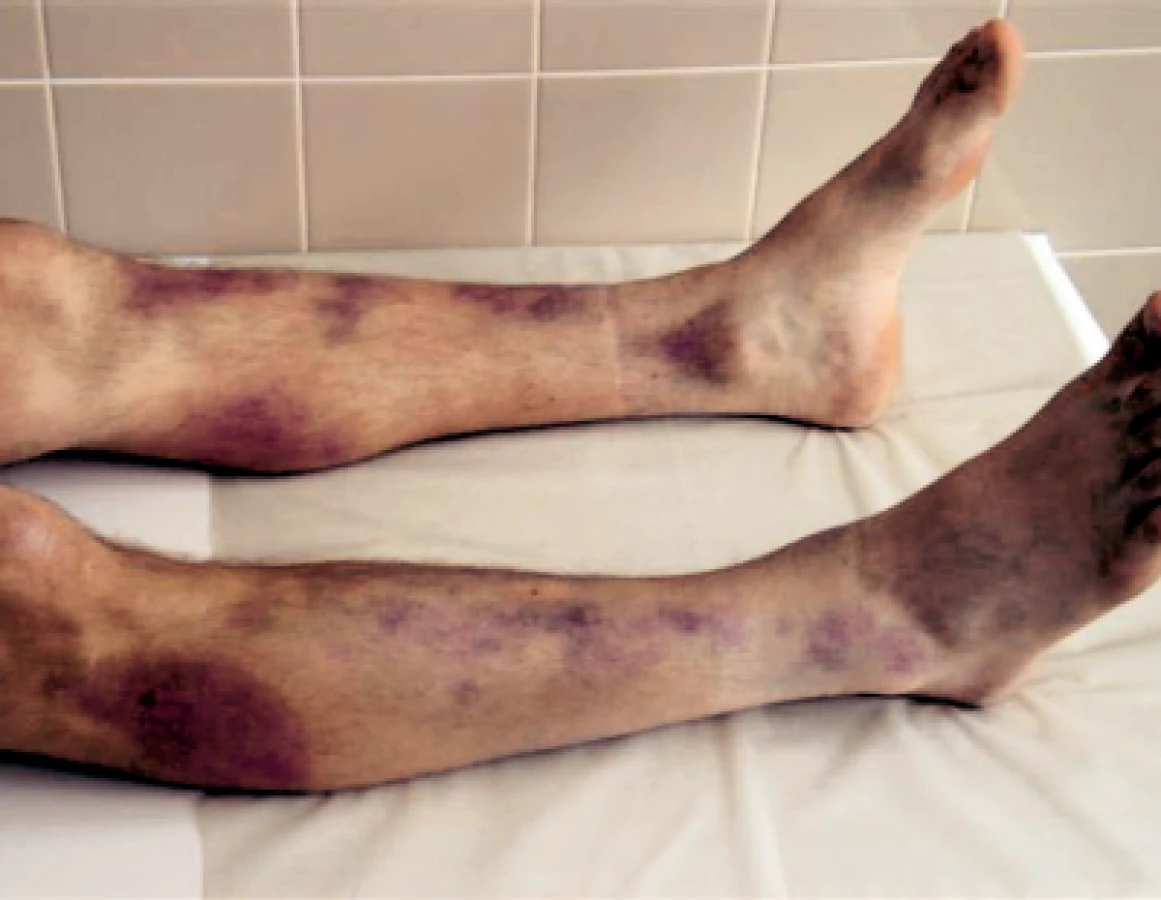
Anamnéza předchozího i běžného infektu byla negativní, mladý muž přiznával pouze zvýšenou fyzickou zátěž (přípravu dřeva na zimu). Laboratorní vyšetření prokázalo těžkou trombocytopenii s hodnotou trombocytů pouhé 3 × 109/l! Další parametry KO (krevního obrazu): leukocyty 4,5 × 109/l, hemoglobin 124 g/l, hematokrit 0,374, střední objem ery 72 fl, Hb ery (barvivo erytrocytu) 24,5 pg, Hb konc (střední barevná koncentrace hemoglobinu) 342 g/l, retikulocyty 0,048. V diferenciálním rozpočtu byla výrazná neutrofilie (0,819) a lymfopenie (0,093) s absolutní lymfopenií 0,60 × 109/l. Hodnota APTT-R 1,29, PT-R 3,35, ostatní koagulační parametry byly v normě. Hodnota protrombinového času (PT) odpovídala těžkému deficitu FVII. Z dalších výsledků: bilirubin 29 μmol/l, LD 9,96 μkat/l, Fe 12,9 μmol/l, saturace transferinu 16 %, haptoglobin < 0,3 g/l. Okamžitě byly nabrány protilátky proti trombocytům a proti erytrocytům a zahájena kortikoterapie prednisonem v dávce 1 mg/1 kg hmotnosti. Byly podány 2 trombocytární koncentráty ze separátoru – i s vědomím toho, že v případě imunologicky podmíněné trombocytopenie bude podání trombo nálevů potencovat další tvorbu protilátek. Po trombocytárních nálevech trombocyty stouply pouze na 8 × 109/l, ale pacient přestal spontánně krvácet z dutiny ústní a z nosu. První 4 dny léčby trvala těžká trombocytopenie s hodnotou trombo mezi 3–8 × 109/l, ale pacient již spontánně nekrvácel ze sliznic ani se netvořily čerstvé petechie či hematomy. Pátý den léčby stoupla hodnota trombocytů na 55 × 109/l a 7. den léčby došlo k normalizaci trombocytů. Třebaže anémie a mírná hyperbilirubinémie byly prokazovány pouze v prvních dnech hospitalizace a zvýšené retikulocyty přetrvávaly necelý týden, pro přetrvávající vyšší hodnotu LD (3 týdny od začátku ataky) a trvající výrazné snížení haptoglobinu jsme pomýšleli na možný Evansův syndrom (imunologicky podmíněná hemolytická anémie a trombocytopenie). Pacientovi jsme postupně snižovali dávku prednisonu tak, že do 3 měsíců od počátku ataky těžké idiopatické trombocytopenické purpury byl prednison vysazen. Po zlepšení stavu (zhruba od 2. týdne onemocnění) bylo pacientovi provedeno celkové přešetření za účelem zjištění eventuální prvotní příčiny těžké idiopatické trombocytopenie. Všechna provedená vyšetření včetně pátrání po možné malignitě byla negativní, prvotní příčinu onemocnění se nepodařilo prokázat. V rámci celkového vyšetřovacího programu bylo provedeno v Ústavu hematologie a krevní transfuze v Praze i stanovení metaloproteinázy ADAMTS 13 k vyloučení či potvrzení možné mikroangiopatické hemolytické anémie typu trombotické trombocytopenické purpury (TTP) i přesto, že pacient neměl alterované renální funkce ani žádnou neurologickou symptomatologii a možná hemolýza byla velmi slabá. Ani toto onemocnění se nepodařilo prokázat. Protilátky proti trombocytům byly opakovaně – poprvé před podáním kortikoidů, podruhé 3. den léčby – negativní a také přímý a nepřímý antiglobulinový (Coombsův) test byly negativní. Po celou dobu hospitalizace i při dalších ambulantních kontrolách trvala extrémně nízká hodnota haptoglobinu. Charakter anémie posléze svědčil spíše pro anémii posthemoragickou než hemolytickou. Hemolytickou anémii jsme proto později vyloučili, ale otázkou zůstává stále trvající výrazně snížená hladina haptoglobinu. Dnes proto nevylučujeme získaný či hereditární deficit haptoglobinu. Získaný deficit je spojován buď s chronickou hepatopatií, kterou nemocný nemá, nebo s malnutricí, kterou také nemá. Možný vrozený deficit je poměrně dobře dokumentován např. u japonské populace (5, 6). Jde o mutaci v oblasti haptoglobinového genu Hpdel , která bývá spojována s protilátkami třídy IgE a IgG proti haptoglobinu i s možnými těžkými anafylaktickými reakcemi po podání krevních transfuzí.
I když náš pacient zareagoval příznivě na kortikoterapii, není jednoznačně prokázáno, že šlo o imunologicky podmíněnou trombocytopenii charakteru idiopatické trombocytopenické purpury na podkladě autoprotilátek. Pacienta nadále pečlivě sledujeme. Je nyní bez pravidelné léčby, při „běžných“ krvácivých příhodách aplikuje rFVIIa a hodnoty KO včetně trombocytů má opakovaně v normě. V souvislosti s těžkým deficitem FVII přetrvává jen patologická hodnota protrombinového času a zatím neobjasněné snížení hodnoty haptoglobinu.
Vrozený autozomálně recesivně podmíněný deficit FVII je vzácné onemocnění. Získaná akutní idiopatická trombocytopenická purpura není onemocněním vzácným, ale výskyt obou těchto chorobných jednotek u jednoho nemocného je onemocněním raritním.
Zkratky
KO – krevní obraz
LD – laktikodehydrogenáza
ITP – idiopatická trombocytopenická purpura
PT – protrombinový čas
TTP – trombotická trombocytopenická purpura
MUDr. Jitka Šlechtová
ÚKBH FN a LF UK, Plzeň
Alej Svobody 80, 323 00 Plzeň
fax: +420 377 104 234, e-mail: slechtova@fnplzen.cz
Sources
1. Friedman, K. D., Rodgers, G. M.: Inherited Coagulation Disorders,. Wintrobe’s Clinical Hematology (11th edition) Lippincott Wiliams & Wilkins, 2004, s. 1620-1667
2. Penka, M., Buliková, A., Matýšková, M., Zavřelová, J.: Hematologie I, neonkologická hematologie. Praha, Grada Publishing, 2001, s. 101–117, s. 137–138.
3. Berkow, R., Fletcher, A. J. et al.: Kompendium klinické medicíny. Krvácivé poruchy. Praha, X-Egem, 1996, s. 1057–1068
4. Cetkovský, P. et al.: Intenzivní péče v hematologii. Praha, Galén, 2004, s. 268–269
5. Koda, Y. et al.: Simple PCR detection of haptoglobin gene deletion in anhaptoglobinemic patiens with antihaproglobin antipody that cause anaphylactic transfusion reactions. Blood, 2000,95, s. 1138–1143.
6. Shimada, E. et al.: Anaphylactic transfusion reactions in haptoglobin-deficient patiens with IgE and IgG haptoglobin antibodies. Transfusion, 2002, 42, s. 766–773.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Spasmolytic Effect of Metamizole
Most read in this issue
- Vrozený těžký deficit FVII a získaná idiopatická trombocytopenická purpura – vzácná kombinace dvou krvácivých stavů
- Vyšetřovací metody na pomezí biochemie a imunologie
- Lékaři a sestry – dialog o profesních rolích a kompetencích
- Změna bazálního inzulínu z NPH na inzulínový analog detemir vede k dlouhodobému zlepšení ranní glykémie nalačno u dětí s diabetes mellitus 1. typu: výsledky roční multicentrické studie