
Aterogenní dyslipidémie a metabolický syndrom: patofyziologické mechanismy
Atherogenic Dyslipidemia and the Metabolic Syndrome: Pathophysiological Mechanisms
Atherogenic dyslipidemia (ADL), a frequent metabolic derangement found in patients with manifest atherosclerosis, is characterized by hypertriglyceridemia, low plasma HDL cholesterol and prevalence of small dense LDL particles. The key pathogenetic mechanisms of ADL are closely linked to insulin resistance, the lack of appropriate responses to insulin in peripheral cells, especially in adipose tissue, skeletal muscles and liver. Impaired insulin signalling leads to a decreased suppression of lipolysis, defective fat storage in adipocytes, and increased flux of free fatty acids to the liver, which together with posttranslational stabilization of apolipoprotein B enhances the assembly and secretion of VLDL particles. Decreased activity of lipoprotein lipase contributes to slow clearance of triglyceride-rich particles, with negative consequences in LDL metabolism. Impaired HDL synthesis, intravascular remodelling, and catabolism decreases reverse cholesterol transport from peripheral tissues, hepatocytes and macrophages. Small dense LDL particles are considered to be highly atherogenic, due to increased penetration of arterial intima, and decreased antioxidant capacity. Better understanding of pathophysiological mechanisms involved in ADL could promote new therapeutic methods, as well as increase the compliance with essential lifestyle interventions.
Key words:
atherogenic dyslipidemia, insulin resistance, hypertriglyceridemia, reverse cholesterol transport, low-density lipoprotein size.
:
A. Žák; A. Slabý
:
IV. interní klinika 1. LF UK a VFN, Praha
:
Čas. Lék. čes. 2008; 147: 459-470
:
Review Article
Aterogenní dyslipidémie (ADL), metabolická porucha častá u pacientů s manifestní aterosklerózou, se vyznačuje souborem biochemických příznaků, k nimž patří hypertriglyceridémie, nízká plazmatická koncentrace HDL-C a převaha malých denzních částic v třídě LDL. Základní patogenetické mechanismy ADL mají těsnou kauzální spojitost s inzulínovou rezistencí, nedostatečnou odpovědí na působení inzulínu, která se projevuje zejména v tukové tkáni, kosterním svalstvu a játrech. V důsledku porušené inzulínové signalizace nedochází v tukové tkáni k supresi lipolýzy, snižuje se schopnost adipocytů skladovat triglyceridy a stoupá přítok volných mastných kyselin do jater, který se spolu s posttranslační stabilizací molekul apolipoproteinu B podílí na zvýšené tvorbě a sekreci částic VLDL. Snížená aktivita lipoproteinové lipázy přispívá ke zpomalenému štěpení lipoproteinů bohatých na triglyceridy, což má též negativní vliv na metabolismus LDL. Porucha syntézy, intravaskulární remodelace a katabolismu HDL vede k omezení zpětného transportu cholesterolu z periferních tkání, z hepatocytů a makrofágů. Malým denzním částicím LDL se připisuje vysoký aterogenní potenciál, vzhledem ke snadné penetraci intimou a snížené antioxidační kapacitě. Lze očekávat, že lepší porozumění patofyziologickým mechanismům ADL přinese nové terapeutické metody, ale současně též zvýší pozornost k nezbytným změnám životního způsobu.
Klíčová slova:
aterogenní dyslipidémie, inzulínová rezistence, hypertriglyceridémie, zpětný transport cholesterolu, velikost částic LDL.
Porucha lipidového metabolismu, která se u pacientů s manifestní aterosklerózou vyskytuje nejčastěji, úzce souvisí s inzulínovou rezistencí a řadí se dnes jako aterogenní dyslipidémie (ADL, syn. aterogenní lipoproteinový fenotyp) do jednotícího rámce metabolického syndromu (MS). U pacientů s ischemickou chorobou srdeční lze příznaky ADL zjistit v 50–70 % případech a epidemiologické studie prokázaly, že zdraví nositelé aterogenního lipoproteinového fenotypu mají tři - až čtyřnásobně zvýšené kardiovaskulární riziko (1).
V současné době existuje všeobecná shoda, že ADL je plně charakterizována triádou biochemických příznaků, kterou tvoří hypertriglyceridémie (HTG), snížená plazmatická koncentrace HDL-C a zvýšené zastoupení malých denzních částic LDL (sd-LDL), tzv. fenotyp B velikosti LDL.
Podle diagnostických kritérií MS (IDF 2005), která uvádí tabulka 1, se HTG definuje jako plazmatická koncentrace TG nalačno ≥ 1,7 mmol/l. Vzhledem k značné inter - a intraindividuální variabilitě hodnot nalačno se předpokládá, že osoby s MS od osob bez tohoto syndromu nejlépe odlišují koncentrace TG v pozdní postprandiální fázi (> 6 hodin po tukové zátěži). K potvrzení HTG u ADL lze užít také dvou ukazatelů, a to poměru TG/HDL-C a TG/LDL-C. Za hraniční hodnotu normy pro HDL-C byla arbitrárně stanovena plazmatická koncentrace 1,3 mmol/l pro muže a 1,0 mmol/l pro ženy. Fenotyp B velikosti LDL charakterizuje převaha subfrakcí III a IV, s velikostí částic < 25,5 nm a hustotou 1,038 až 1,065 g/ml. Vzhledem k náročnosti přímého laboratorního průkazu se v klinické praxi usuzuje na fenotyp B nepřímo, a to podle zvýšené plazmatické koncentrace apolipoproteinu B (apo B-100 ≥ 1,3 g/l) při normální nebo hraničně zvýšené koncentraci LDL-C (3,4–4,1 mmol/l), nebo podle poměru LDL–C/apo B v LDL (< 0,85; w/w). Nepřímá diagnostická kritéria pro určení fenotypu B velikosti LDL (resp. převahy sd-LDL) ukazuje tabulka 2.


K dalším nálezům, které konstantně provázejí ADL, patří zvýšená průměrná 24hodinová plazmatická koncentrace volných mastných kyselin (FFA – free fatty acids), která je patrná nalačno a zejména postprandiálně. Dochází k typickým změnám ve spektru mastných kyselin (FA – fatty acids), což lze prokázat v hlavních lipidových třídách analýzou triglyceridů (TG), fosfolipidů (PL – phospholipids) a esterů cholesterolu (CE – cholesteryl esters). Jako nová komponenta MS byl v poslední době doplněn vzestup syntézy cholesterolu v játrech, kompenzovaný jeho sníženou střevní resorpcí.
Na změny v metabolismu lipoproteinů u MS lze usuzovat mimo jiné ze zvýšené aktivity jaterní lipázy (HL), respektive z vysokého poměru aktivity tohoto enzymu k aktivitě lipoproteinové lipázy (LPL). Rovněž transferový protein esterů cholesterolu (CETP) a mikrozomální triglyceridový transferový protein (MTTP) vykazují vyšší aktivitu.
Inzulínová rezistence
Patofyziologické mechanismy, které vyvolávají ADL, mají kauzální souvislost s inzulínovou rezistencí (IR), nedostatečnou odpovědí cílových tkání na působení inzulínu. Tato častá metabolická porucha se v rozvinuté formě projevuje ve všech tkáních (včetně centrálního nervového systému, cévního endotelu a imunokompetentních buněk) a je spojena s poklesem oxidativní fosforylace. V buňkách kosterního svalstva byla popsána zvýšená koncentrace TG a snížená denzita mitochondrií (2). Intracelulární akumulace FFA působí závažné změny glukózového a lipidového metabolismu (blokuje transport a oxidaci jak glukózy, tak i FFA).
Předpokládá se, že v etiopatogenezi IR má klíčovou úlohu interakce genetických a zevních faktorů (3). Genetický výzkum se soustřeďuje na variabilitu genů, které řídí glycidový a lipidový metabolismus. Inzulínovou senzitivitu ovlivňují zejména geny pro transmembránový glykoprotein PC-1 a pro substrát inzulínového receptoru IRS-1. Přímý vztah k ADL mají polymorfismy genů pro některé apolipoproteiny (apo) (A-I až A-V, B, C-III, E), LPL, HL, střevní izoformu proteinu vázajícího mastné kyseliny (FABP-2), CETP, MTTP i transkripční faktory PPARα a PPARγ. Genetické faktory se podílejí na vzniku IR nejméně v rozsahu 30–40 %. Ze zevních faktorů je nejvýznamnější nadměrný příjem živin v potravě, který spolu s nízkou fyzickou aktivitou vede k pozitivní energetické bilanci. Byl prokázán též vliv kvalitativního složení stravy, například vysokého podílu fruktózy, změn v příjmu základních skupin FA i spojitost s různými neurohumorálními a imunitními mechanismy (4).
Výsledky experimentálních prací naznačují, že IR vzniká jako převážně postreceptorová porucha inzulínové signalizační kaskády (5). Jde o složitý komplex na sebe navazujících nitrobuněčných mechanismů, který se spouští po navázání inzulínu na povrchový inzulínový receptor. Autofosforylace receptoru a aktivace tyrozinkinázy má za následek fosforylaci jednotlivých substrátů inzulínového receptoru (IRS1-4) a dalších proteinů. Ze základních signalizačních drah, které ovlivňují intracelulární metabolické děje, je při IR postižena hlavně dráha fosfatidylinozitol-3-kinázová (PI3K). Této dráze se připisují změny transmembránového transportu glukózy, syntézy glykogenu a dalších biologicky významných reakcí, které souvisejí s translokací glukózového transportéru GLUT4 k buněčné membráně. Funkční poruchu dráhy PI3K lze vyvolat u zdravých osob zvýšením plazmatické koncentrace FFA, například infuzí lipidové emulze. Naproti tomu signalizační dráha označovaná jako MAP–kinázová, s mitogenními a prozánětlivými účinky, zůstává při IR intaktní a podílí se na rozvoji endoteliální dysfunkce (6).
Až donedávna byla IR studována hlavně v kosterním svalstvu a v játrech, teprve v poslední době se pozornost obrací též k tukové tkáni, kde se porucha manifestuje dříve než v jiných orgánových systémech. Pravidelně lze v tukové tkáni prokázat nedostatečnou supresi lipolýzy, zejména v postprandiálním období, která je důsledkem chybějícího inhibičního účinku inzulínu na hormon-senzitivní lipázu a na nově objevenou specifickou lipázu tukové tkáně ATGL (adipose triglyceride lipase) (7).
Při IR je podstatně omezena hlavní adaptační funkce tukové tkáně, která spočívá v uskladňování nadbytečné energie ve formě intracelulárních zásob TG (8). Za fyziologických podmínek inzulín stimuluje v cévách tukové tkáně LPL, která hydrolyzuje VLDL a CM. Uvolněné mastné kyseliny vstupují do adipocytů za pomoci specifických transportérů. Syntéza TG v adipocytech vyžaduje dostupnost glycerol-3-fosfátu, která závisí na stimulaci membránového transportu glukózy inzulínem; významný je též účinek inzulínu na izoformu 1 diacylglycerolacyltransferázy (DGAT-1). U stavů spojených s IR dochází v adipocytech k dysfunkci endoplazmatického retikula (9). Navíc je IR spojena s poruchou diferenciace preadipocytů ve zralé adipocyty, na níž se podílí také zvýšená aktivita lokálního systému renin–angiotensin (10). Jak ve viscerálním, tak v podkožním tuku se hromadí velké dysfunkční adipocyty, s vystupňovanou IR, přeplněné tukovými kapkami a neschopné přijímat další FA.
Snížená schopnost uskladňovat TG a nedostatečná suprese lipolýzy jsou příčinou trvale zvýšeného toku FFA z tukové tkáně do jiných orgánových systémů. V důsledku této poruchy dochází k ektopické akumulaci tuku v játrech (rozvoj nealkoholické steatózy), v kosterních svalech (vzestup intramyocelulárních TG prokazatelný magnetickou rezonancí), v pankreatu (porucha funkce i regenerační schopnosti β–buněk) a v dalších tkáních, se všemi negativními metabolickými důsledky lipotoxicity a oxidačního stresu. Zvláště závažné patogenní účinky mají FA s dlouhým řetězcem a jejich metabolické produkty (2). Obezita i intracelulární ektopická akumulace lipidů (v játrech a kosterním svalstvu) souvisí se změnami aktivit odpřahujících proteinů (UCP – uncoupling proteins), které jsou lokalizovány na vnitřní straně mitochondriální membrány buněk řady tkání; UCP1 je exprimován především v bílé tukové tkáni, UCP2 navíc v pankreatu, izoforma UCP3 pak v buňkách kosterního svalstva a myokardu. Odpřahující proteiny 1. ovlivňují termogenezi (UCP2, -3) a snižují riziko obezity; 2. chrání před oxidačním stresem (UCP1, -2, -3), který propojuje vzájemně jednotlivé komponenty MS; 3. negativně regulují sekreci inzulínu (UCP2) a 4. vykazují neuroprotektivní účinky (UCP2). Aktivace UCP má vztah k ochraně mitochondrií před toxickým účinkem aniontů FA a lipoperoxidů v důsledku stimulace transportu aniontů FA z mitochondrií do cytoplazmy a antioxidačních mechanismů. Jako nejvýznamnější ve vztahu k MS se jeví aktivace UCP2, který chrání mitochondrie před poškozením reaktivními kyslíkovými sloučeninami a podílí se též na řízení sekrece inzulínu v ß-buňkách pankreatu, a aktivace UCP3, která ovlivní inzulínovou rezistenci kosterního svalstva (11).
V tukové tkáni dochází při IR ke kvantitativním změnám v produkci fyziologicky aktivních peptidů – adipocytokinů. V popředí zájmu je zejména pokles sekrece peptidového hormonu adiponektinu, který má příznivé metabolické účinky. Prostřednictvím AMP dependentní proteinové kinázy (AMPK) stimuluje adiponektin glukózový metabolismus a snižuje IR, dále inhibuje aktivační kaskádu nukleárního faktoru kappa B (NFκB) a chrání β-buňky pankreatu před apoptózou indukovanou akumulací lipidů. Na metabolickém působení adiponektinu má hlavní podíl jeho vysokomolekulární multimér (hmw-adiponectin), jehož plazmatická koncentrace významně koreluje s inzulínovou senzitivitou při vyšetření euglykemickým hyperinzulínovým clampem. U pacientů s HTG se pravidelně zjišťují nízké hodnoty plazmatické koncentrace adiponektinu. Bylo zjištěno, že hmw-adiponectin je významným prediktorem rizika DM 2. typu i MS. Jeho koncentrace, resp. jeho poměr k celkovému adiponektinu pozitivně koreluje s množstvím intraabdominálního tuku a negativně koreluje s koncentracemi velkých VLDL, sd-LDL a velkých HDL (12).
Paralelně se sníženou tvorbou adiponektinu a leptinu stoupá v tukové tkáni sekrece prozánětlivých adipocytokinů, na níž se kromě adipocytů podílejí též zmnožené makrofágy (13). Zvýšená genová exprese byla prokázána pro tumor nekrotizující faktor α (TNFα), interleukiny IL-6, IL-1 a IL 9, rezistin a angiotensinogen. K nepříznivým účinkům prozánětlivých adipocytokinů patří inhibice inzulínové signalizační kaskády, zpomalení katabolismu VLDL a CM v důsledku snížené aktivity LPL, jakož i aktivace některých lipogenních enzymů, např. acyl–koenzym A–karboxylázy. Sekrece prozánětlivých cytokinů vykazuje kauzální souvislost s oxidačním stresem, zvýšenou produkcí reaktivních kyslíkových a dusíkových sloučenin (RONS) a sníženou expresí antioxidačních enzymů, zejména superoxiddismutázy. Omezená dostupnost oxidu dusnatého (NO), toxické působení oxidativně modifikovaných částic LDL (ox-LDL) a další důsledky oxidačního stresu zhoršují endotelovou dysfunkci a urychlují rozvoj aterosklerotických plátů (14).
Hypertriglyceridémie
Na podkladě výsledků experimentálního a klinického výzkumu lze považovat za prokázané, že v patogenezi ADL hraje klíčovou úlohu komplexní metabolická porucha, která se projevuje jako HTG. Druhé dvě složky lipidové triády, tj. nízká plazmatická koncentrace HDL-C a převaha sd-LDL, vznikají druhotně, v kauzální souvislosti s HTG (8).
V současné době se na rozdíl od starších epidemiologických studií, které uznávaly pouze vliv cholesterolu, HTG považuje za samostatný rizikový faktor pro ischemickou chorobu srdeční (ICHS). Vzestup plazmatické koncentrace TG o 1 mmol/l zvyšuje riziko ICHS u mužů o 13 %, u žen dokonce o 37 %, po adjustaci na hmotnostní index, kouření cigaret a jiné významné kovarianty (15).
Za primární příčinu HTG se považuje zvýšená produkce VLDL v játrech, a to zejména velkých částic VLDL-1 (Sf 100–400), při neměnícím se celkovém množství VLDL-2 (Sf 60–100). Intenzita jaterní sekrece VLDL-1 závisí především na přítoku FFA do jater, za předpokladu dysfunkce inzulínové signalizační kaskády (16). K dalším faktorům, které zvyšují jaterní produkci VLDL-1, patří chronická stimulace lipogeneze de novo a vychytávání remnantních částic lipoproteinů bohatých na TG z cirkulace.
Nadměrný přívod FFA do jater je především důsledkem IR tukové tkáně, jak bylo vysvětleno v předchozím oddíle, ale též sníženého využití FFA v kosterním svalstvu a zvýšené střevní resorpce TG. Frakční extrakce FFA v játrech činí 20–30 %. V závislosti na aktuální energetické potřebě je část FFA v játrech oxidována, kdežto zbývající, zpravidla větší část esterifikována a ukládána jako cytosolová zásoba (pool) TG v hepatocytech. Předpokládá se, že intracelulární ukládání TG slouží jako ochrana před toxickým působením nadměrně vysokých koncentrací FFA. Proporcionální podíl cytosolových TG je po částečné nebo kompletní hydrolýze a následné reesterifikaci inkorporován do nově vznikajících částic VLDL. Kromě velikosti cytosolové zásoby TG ovlivňuje sekreci VLDL dostupnost jaterních PL, aktivita MTTP a stabilita hlavní proteinové komponenty, apo B-100. Byl prokázán též vliv variability genů pro apo C-III, apo E a pro transportéry ABCA1, ABCG5/8 (17).
Kinetické studie potvrdily vzájemnou podmíněnost tvorby apo B-100 a TG v hepatocytech (18). Zatímco exprese genu pro apo B zůstává stabilní, v endoplazmatickém retikulu dochází k neustálému, různě intenzivnímu odbourávání nově syntézovaného proteinu. Zvýšená sekrece TG spolu se zvýšenou aktivitou MTTP stabilizuje molekuly apo B in status nascendi, které jsou spojovány s lipidy v procesu sestavování a sekrece zralých částic VLDL (19).
Chronická hyperinzulinémie spolu se zvýšeným obsahem sacharidů ve stravě vede k vzestupu lipogeneze de novo, která u pacientů s ADL zvyšuje jaterní produkci VLDL až o 30 %. Ke stimulaci hlavních lipogenních enzymů dochází v hepatocytech díky zvýšené transkripci proteinu vázajícího sterolový regulační element (SREBP-1c). Významné funkce v intermediárním metabolismu se přičítají malonyl-koenzymu A (Mal-CoA). Jeho molekula má signalizační funkcí a současně vlastnosti energetického senzoru, regulátoru příjmu potravy; Mal-Co A je substrátem pro syntézu FA a působí současně jako inhibitor karnitin-acyltransferázy (CAT-1), jejíž aktivita je nezbytná pro β-oxidaci FA v mitochondriích. V přímé závislosti na množství Mal-CoA stoupá v játrech podíl esterifikace FFA v neprospěch jejich oxidace. Akumulace Mal-CoA inhibuje syntézu neuropeptidu Y (NPY) v hypothalamu s potlačením příjmu potravy (20).
Částice VLDL a ostatních lipoproteinů (LP) obsahujících apo B (remnantní CM, IDL a LDL) jsou v plazmě předmětem neustálých delipidačních a výměnných metabolických dějů. Lipoproteinová lipáza, jejíž aktivita je u stavů spojených s IR snížena, uvolňuje z lipoproteinů bohatých na TG (TGR) mastné kyseliny. Volný cholesterol a PL jsou z povrchové vrstvy částic TGR přenášeny do HDL. Další důležitý mechanismus spočívá v tom, že působením CETP dochází ke směně TG obsažených ve VLDL za CE, které jsou uloženy v jádru částic HDL a LDL. Čím déle zůstávají LP bohaté na TG v cirkulaci, tím více jsou vystaveny účinku CETP a tím větší množství TG odevzdávají do HDL a LDL. Tímto způsobem vznikají jednak velké částice ß-VLDL ochuzené o TG a obohacené o CE, jednak malé částice HDL a LDL obohacené o TG a ochuzené o CE. Pouze nevelký podíl částic TGR předává TG do hepatocytů, prostřednictvím receptorů LDL R (apo B/E).
V patogenezi HTG se dále uplatňuje snížený frakční katabolismus LP obsahujících apo B. Kinetické studie v postprandiálním období prokázaly především zpomalení katabolismu VLDL-1 a remnantních CM, které spolu kompetují o LPL (8). Rovněž frakční katabolismus IDL je snížen zhruba o jednu třetinu. Denní obrat TG je ekvivalentní 2–6 % z celkového poolu v organismu (21).
Jak již bylo uvedeno, jedním z faktorů zodpovědných za prodloužení postprandiální lipémie je pokles aktivity LPL ve svalstvu, tukové tkáni a dalších orgánech, respektive snížený poměr aktivit LPL a HL. Za hlavní příčinu snížené aktivity LPL se považuje zvýšená plazmatická koncentrace FFA. U jedinců, kteří mají v důsledku nedostatečné tělesné aktivity nízký průtok krve kosterním svalstvem, dochází navíc k omezení kontaktu LP částic s LPL, navázanou k cévnímu endotelu. Na zpomalení katabolismu VLDL a CM u stavů s IR se podílí též snížená exprese nukleárního transkripčního faktoru PPARα v hepatocytech, spolu se sníženou plazmatickou koncentrací apo A-V a výrazným vzestupem (až o 50 %) sekrece apo C-III. Apo C-III snižuje frakční katabolismus VLDL a současně též zvyšuje sekreci VLDL v játrech (22).
Lipolýza remnantních částic (β-VLDL a IDL) je funkcí HL, která se podílí též na dalším katabolismu těchto částic (23). Ke vzestupu aktivity HL dochází u ADL především v důsledku hyperinzulinémie. Do popředí se dostává proaterogenní působení HL při remodelaci VLDL-1 a CM na sd LDL, o čemž bude pojednáno níže.
Patologické změny v metabolismu HDL
V souvislosti s HTG vznikají poruchy v metabolismu LP třídy HDL, které se týkají jejich sekrece, remodelace v cirkulaci a intenzity odbourávání (24). Mezi hodnotami plazmatické koncentrace TG (nalačno i postprandiálně) a koncentracemi HDL-C byly prokázány významné negativní korelace. Nízké hodnoty HDL-C v plazmě (hypoalfacholesterolémie) slouží v klinice jako výsledný ukazatel poruchy, nemohou však informovat o jejím typu ani o negativních důsledcích pro zpětný transport cholesterolu a další specifické funkce jednotlivých subfrakcí HDL.
Epidemiologické studie, včetně Framingham Heart Study, jednoznačně prokázaly, že nízké koncentrace HDL-C jsou spojeny se zvýšeným rizikem ICHS a patří k nejsilnějším prediktorům kardiovaskulárních příhod (25). Pokles HDL-C o 0,3 mmol/l zvýší riziko ICHS u mužů dvojnásobně, u žen trojnásobně (26).
Částice HDL vznikají v játrech a z menší části též ve stěně tenkého střeva z nově secernovaných molekul apo A-I, které se spojují s PL a s malým množstvím volného (neesterifikovaného) cholesterolu. Tyto částice označované jako nascentní, stejně jako samotné molekuly apo A-I, mají funkci silného akceptoru cholesterolu v rámci tzv. iniciální lipidace. Sekrece apo A-I je regulována na transkripční i posttranskripční úrovni; byl prokázán vliv celé řady faktorů genetických, nutričních (množství a složení tuku ve stravě, zastoupení mastných kyselin, retinoidy, alkohol) a hormonálních (glukokortikoidy, estrogeny, androgeny, hormony štítné žlázy) (27). Změny sekrece apo A-I však většinou nemají významný vliv na jeho plazmatickou koncentraci; daleko více záleží na intenzitě katabolismu a výši renální clearance (28).
V procesu iniciální lipidace vzniká několik subpopulací HDL, které se podle elektroforetických vlastností označují jako prae-β LP. Obsahují v každé částici dvě molekuly apo A-I a méně než 10 % hmotnosti tvoří lipidy. Během postupného zrání přibývá v částicích HDL dalších funkčně relevantních složek. Z apolipoproteinů jsou zastoupeny zejména apo A-II, A-IV, C-III a E, z enzymů lecitin-cholesterol-acyltransferáza (LCAT) a paraoxonáza 1 (PON1), konstantně se vyskytuje též ceruloplasmin, transferrin aj. Současně stoupá množství volného cholesterolu. Při vzniku zralých částic HDL2 hraje významnou úlohu enzym LCAT, jehož působením vznikají z volného cholesterolu hydrofobní CE přesunované do jádra, čímž se původní diskoidní tvar mění na sférický. V subfrakci HDL2 se vyskytují též sférické částice, které neobsahují apo A-I, nýbrž pouze samotný apo E (29). Zralé částice HDL2 odevzdávají cholesterol v játrech díky interakci s receptory SR-B1a HL a znovu přecházejí do cirkulace, ochuzené o CE a PL.
Remodelace HDL probíhá v plazmě, v rámci neustálé směny lipidů mezi různými LP třídami. Pro ADL je charakteristická nadměrně intenzivní výměna CE v subfrakci HDL2 za TG z VLDL-1 a jiných LP částic bohatých na TG (remnantní CM, IDL, LDL), které jsou zvláště v postprandiálním období k dispozici ve velkém množství. Jak již bylo uvedeno, k jejich akumulaci přispívá snížená aktivita LPL. Směnu neutrálních lipidů (CE za TG) zprostředkuje CETP, jehož aktivita je u stavů spojených s IR rovněž zvýšená. Vznikají tak částice HDL bohaté na TG a ochuzené o CE, což se projeví poklesem plazmatické koncentrace HDL-C a zpomalením reverzního transportu cholesterolu (RCT – reverse cholesterol transport).
Částice HDL bohaté na TG jsou preferenčním substrátem HL, specifického enzymu, který vzniká v hepatocytech a váže se k proteoglykanu heparansulfátu na vnitřním povrchu sinusoidálních kapilár. Aktivita HL kolísá v širokém rozmezí, přičemž 40–60 % rozptylu je podmíněno geneticky a k významnému vzestupu dochází v důsledku IR jaterní tkáně, příjmu SFA a intraabdominální akumulace tukové tkáně. V experimentu na zvířeti byl zjištěn stimulační vliv chronické hyperinzulinémie (23). Částice HDL bohaté na TG se působením HL mění na nestabilní remnantní částice, prae-β HDL a částice apo A-I chudé na lipidy. Apolipoproteiny uvolněné z povrchové vrstvy HDL jsou urychleně katabolizovány.
Kromě HL se na přeměně HDL bohatých na TG podílí též transferový protein fosfolipidů (PLTP), který přenáší PL mezi HDL a VLDL, ale též mezi částicemi HDL různých velikostí, přičemž dochází k fúzi částic a k disociaci apo A I; PLTP působí v interakci s endoteliální lipázou (30) a se skupinou sekrečních fosfolipáz, zejména sPLA2-IIA, jejichž aktivita stoupá u zánětlivých stavů (31).
Katabolismus HDL probíhá hlavně v ledvinách, játrech a steroidogenních tkáních (nadledviny, ovaria). V důsledku HTG je relativně omezena převládající cesta katabolické přeměny HDL2, která spočívá v selektivním předání cholesterolu a jiných lipidů do buněk shora uvedených tkání (selective cholesterol uptake). Jde o dvojstupňový proces zahájený vazbou HDL k „zametačovým“ receptorům SR-BI, které umožní difuzi lipidů buněčnou membránou. Částice HDL zbavené lipidů mohou být opět remodelovány na HDL2, ale vždy dochází ke ztrátě apolipoproteinů. U stavů s IR pravděpodobně nabývá na významu minoritní katabolická cesta – endocytóza celých částic HDL zbavených lipidů a jejich desintegrace v lyzozymech (holoparticle HDL uptake). Studie metabolického obratu prokázaly, že jedinci s ADL mají ve srovnání se zdravými osobami významně zvýšený katabolismus apo A-I. V plazmě přítomné molekuly apo A-I, volné nebo spojené s malým množstvím lipidů, přestupují v ledvinách do glomerulárního filtrátu, odkud jsou absorbovány tubulárními buňkami a rozloženy za účasti systému cubilin/megalin (32).
Poruchy remodelace a urychlení katabolismu HDL mají za následek nedostatek funkčních částic, které jsou nezbytné pro RCT. Vyplavení nadbytečného cholesterolu z buněk periferních tkání (včetně tepen) probíhá jako řízený proces zprostředkovaný transportérem ABCA1, jen výjimečně prostou difuzí, přičemž příjemcem jsou převážně zralé částice HDL2. Akumulace FFA, která je typická pro MS, snižuje v buňkách expresi ABCA1. Jako alternativní mechanismus RCT z periferních buněk byl prokázán přenos cholesterolu do HDL2 pomocí receptorů SR-BI (33). Kvantitativně významnou součástí RCT je přenos cholesterolu do nascentních HDL a prae-β HDL v játrech. Mutace ABCA1 jsou spojeny s poruchou iniciální lipidace HDL, což se v extrémní formě projevuje u vzácné homozygotní formy, Tangierské nemoci. Z hlediska aterogeneze má zvláštní význam RCT z makrofágů, na němž se podílejí transportéry ABCG1 a ABCG4, přičemž příjemcem cholesterolu jsou pouze zralé částice HDL2. Nadbytek cholesterolu signalizují nukleární receptory LXR (liver X receptors), které expresi transportérů zvyšují.
Jak bylo potvrzeno izotopovými studiemi, většinu zpětně transportovaného volného cholesterolu předávají v játrech částice HDL2, naproti tomu významný podíl CE se z HDL přemístí do LP obsahujících apo B (34). V játrech jsou CE vychytávány specifickými receptory hepatocytů, a to SR-B1 pro HDL, LRP (LDL-receptor related protein) pro IDL a β VLDL, LDL-receptory pro IDL a LDL.
Také další ochranné funkce HDL mohou být v důsledku IR v různém rozsahu porušeny. Předmětem výzkumu je stimulační účinek HDL na syntézu NO a prostacyklinu (PGI2) v endotelových buňkách, jakož i potlačení oxidačního stresu (pomocí aktivity PON1) a podíl HDL při odstraňování oxidovaných LP a amyloidogenních proteinů z cirkulace. Fosfolipidy na povrchu částic HDL neutralizují aktivitu C-reaktivního proteinu (35). V experimentech na zvířatech bylo prokázáno, že nitrožilní podání rekonstituovaných diskoidních HDL tlumí akutní i chronický zánět cévní stěny (36).
Převaha malých denzních LDL
Za třetí kardinální příznak ADL se dnes všeobecně považuje fenotyp B velikosti LDL, tj. převaha malých denzních částic (sd-LDL), které jsou obsaženy ve III. a IV. subfrakci. Zastoupení subfrakcí LDL u fenotypu A, intermediárního fenotypu (fenotyp I) a fenotypu B je znázorněno na obrázku 1.
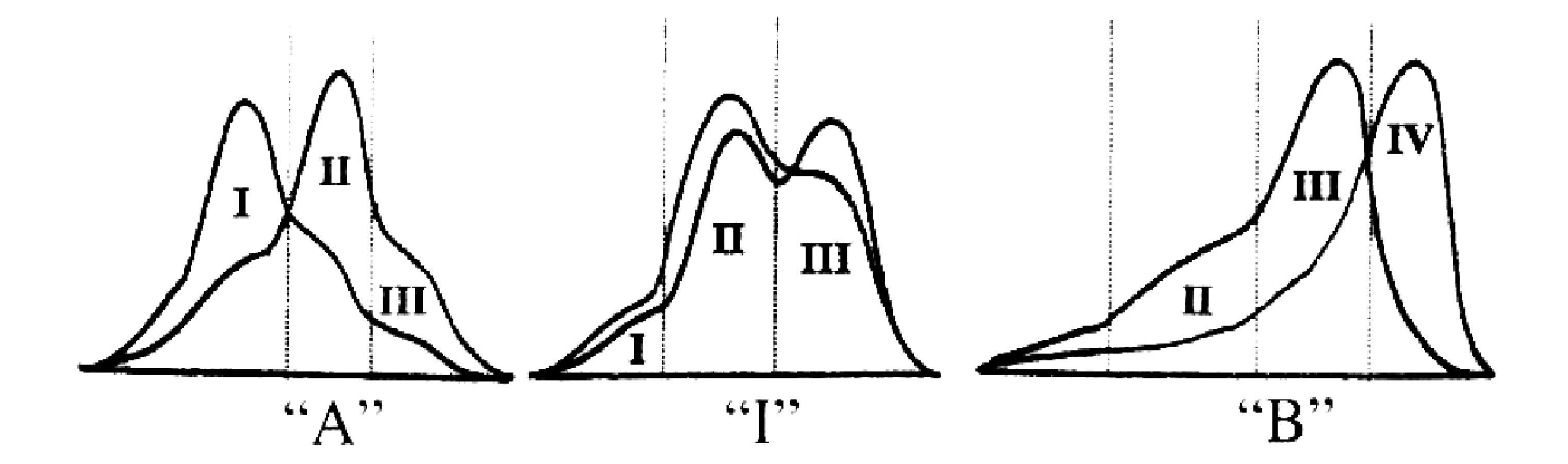
Z epidemiologických studií vyplynul závěr, že fenotyp B představuje samostatný rizikový faktor pro ICHS, který je zčásti nezávislý na hodnotách plazmatické koncentrace TG, HDL-C a apo B, ale v kombinaci s těmito faktory významně potencuje jejich účinky (37).
Ve srovnání s částicemi LDL převažujícími u zdravých jedinců (subfrakce I a II) mají sd-LDL odlišné fyzikálně-chemické a biologické vlastnosti. V experimentech in vitro bylo zjištěno, že sd–LDL jsou méně odolné vůči oxidaci, ať už vyvolané buňkami, nebo kovovými katalyzátory, a ochranný vliv různých antioxidačních látek trvá kratší dobu (38). Malé denzní LDL obsahují méně glycidů a kyseliny sialové. In vivo se sd-LDL vyznačují tím, že díky malému průměru a příznivým elektrostatickým vlastnostem snáze prostupují cévní stěnou. Oxidativně modifikované sd-LDL působí toxicky na endotel (inhibují syntázu NO a zvyšují expresi cytoadhezních molekul), v subintimálním prostoru atrahují monocyty a makrofágy, čímž udržují subklinický zánět. Částice sd-LDL přítomné v cirkulaci mají v důsledku odlišné konformace apo B-100 nižší afinitu k LDL receptorům (apo B/E receptory) v hepatocytech, což se projevuje významně nižším frakčním katabolismem, zejména v postprandiálním období (22). Díky zvýšené afinitě k receptorům SR-B1 mohou sd-LDL přednostně pronikat do makrofágů v aterosklerotických plátech (26). U osob s MS se částice sd-LDL vyznačují vyšším obsahem apo C-III a vyšší afinitou k proteoglykanům cévní stěny (39).
Převaha sd-LDL má, obdobně jako pokles HDL-C, přímou kauzální spojitost s HTG. K hlavním metabolickým prediktorům fenotypu B patří kromě HTG zvýšená plazmatická koncentrace apo B, zvýšená aktivita HL a CETP. Jak již bylo uvedeno, na vzniku HTG se významně podílí vzestup produkce VLDL-1 v játrech. Velké částice VLDL-1, které jsou bohaté na TG a obsahují kromě apo B též apo C-III a apo E, dosahují maximální koncentrace v plazmě v pozdním postprandiálním období a delší dobu setrvávají v cirkulaci. Bylo prokázáno, že existují různé metabolické dráhy, které rozhodují o vlastnostech TGR secernovaných v játrech a o způsobu jejich postupné delipidace, která vede přes IDL 1 nebo IDL-2 k jednotlivým subfrakcím LDL. Nepřímo úměrně k plazmatické koncentraci HDL-C se uplatňuje výměna TG z velkých VLDL-1 nebo z jejich remnantních částic za CE v LDL, kterou zprostředkuje CETP. Tento faktor však není pro diferenciaci subfrakcí LDL nezbytný, jak vyplynulo ze studie rodin s jeho deficitem (40). Částice LDL bohaté na TG a ochuzené o CE jsou preferovaným substrátem HL, která vykazuje zvýšenou aktivitu a působí současně jako lipáza i fosfolipáza. Po hydrolýze TG a PL vznikají sd-LDL, s vysokým poměrem apo B-100 k cholesterolu. Na obrázku 2 je schematicky znázorněn mechanismus vzniku sd-LDL.

Metabolické faktory, které ovlivňují převahu sd-LDL, nejsou dosud v plném rozsahu objasněny; jejich další výzkum směřuje k novým možnostem dietní a farmakologické intervence.
Přeměna mastných kyselin
S patofyziologickými pochody vedoucími k ADL úzce souvisejí změny v profilu FA, které lze prokázat kapilární plynovou chromatografií v základních lipidových třídách (TG, PL a CE) separovaných z plazmy. Kvantitativní zastoupení jednotlivých FA v TG, CE a PL reflektuje příjem FA potravou, jejich metabolickou přeměnu (desaturaci, elongaci) a degradaci (β-oxidaci a lipoperoxidaci).
Z faktorů, které ovlivňují profil FA u jedinců s MS, přicházejí v úvahu především zvýšený přísun nasycených tuků v potravě, urychlení lipogeneze de novo, změny rychlosti metabolické přeměny a oxidační stres (41).
Typický profil FA asociovaný s MS je charakterizován zvýšenou koncentrací nasycených (SFA) a hlavních mononenasycených (MUFA) kyselin, naproti tomu se pravidelně zjišťuje pokles vícenenasycených kyselin (PUFA) řady n-6, zejména kyseliny linolové (LA, C18 : 2n-6). Podle švédských autorů je prediktorem vzniku MS také pokles PUFA řady n 3 (42).
Ze skupiny SFA je zvýšena především kyselina palmitová (PA, C16 : 0), a to úměrně k množství přijímanému v potravě. Vzestup koncentrace dvou MUFA, kyseliny palmitolejové (PAO, C16 : 1n-7) a olejové (OA, C18 : 1n-9), svědčí o zvýšené aktivitě delta-9-desaturázy (Δ9D), enzymu, který se podílí na jejich přeměně desaturací příslušných SFA (kyselina palmitolejová vzniká z palmitové, olejová ze stearové kyseliny). Bylo prokázáno, že vzestup aktivity ∆9D má přímou kauzální souvislost s hyperinzulinémií při IR, stejně jako změny v aktivitě dalších desaturáz a některých elongáz. Aktivita Δ6D je u MS zvýšena, jak vyplývá z poměru kyseliny γ linolenové (GLA, C18 : 3n-6) k LA (43). Dále dochází k poklesu aktivity Δ5D, který je indikován sníženým poměrem kyseliny arachidonové (AA, C20 : 4n-6) ke kyselině dihomo-γ-linolenové (DHGLA, C20 : 3n-6). Protože shora uvedené FA (PAO, GLA a DHGLA) nejsou obsaženy v běžných tucích přijímaných v potravě, vzestup jejich relativních koncentrací může být jednoznačně interpretován jako důsledek enzymatických změn v rámci IR. Pokles LA, téměř konstantní nález u MS, zpravidla není způsoben nutriční karencí, nýbrž zvýšenou enzymatickou degradací v důsledku oxidačního stresu (44).
Recentní klinické a epidemiologické studie potvrzují souvislost popsaných změn FA s rozvojem MS, porušené glukózové tolerance a DM 2. typu (45). Typický profil FA, zejména výrazný vzestup SFA, se pravidelně vyskytuje u mužů s MS po infarktu myokardu (46). Pokles LA spolu se vzestupem GLA a DHGLA byl zjištěn u obézních dětí, které měly ještě další příznaky MS, nikoli u prosté obezity (47). Při sledování souboru adolescentů s MS bylo prokázáno, že profil FA významně koreluje s indikátory IR a s markery systémového zánětu (48).
Metabolické, endokrinní a nutriční faktory mohou u MS indukovat změny ve složení PUFA s dlouhým řetězcem, které působí jako modulátory specifických nukleárních receptorů (PPAR, SREBP, LXR), a tím dalekosáhle ovlivňují lipidový metabolismus.
Z uvedených poznatků o LA vyplývá důležité upozornění pro praxi. Zjistí-li se u jedinců s MS nízká plazmatická koncentrace této kyseliny, většinou není vhodné se pokoušet o korekci potravinovými doplňky. Nejde-li o karenci esenciálních FA řady n-6, suplementace LA pouze zvyšuje oxidační stres a vytváří podmínky pro udržování chronického zánětu a prokoagulačního stavu. Dosud zůstává otevřená otázka, mají-li se u pacientů s DM 2. typu suplementovat některé FA (např. DHGLA), či PUFA n-3, jejichž snížené koncentrace jsou dávány do souvislosti s přechodem stavů s IR k MS a se vznikem mikrovaskulárních komplikací, zejména diabetické polyneuropatie.
Homeostáza cholesterolu
Plazmatické koncentrace celkového cholesterolu (TC), stejně jako LDL-C a non–HDL-C, jsou u MS zpravidla normální nebo jen mírně zvýšené. Před několika lety popsali finští autoři jako novou komponentu MS sníženou střevní resorpci cholesterolu, která kompenzuje jeho zvýšenou syntézu de novo (49). Syntéza cholesterolu hodnocená podle plazmatické koncentrace jeho prekurzorů (skvalen, desmosterol, lathosterol, lanosterol), respektive podle poměru těchto prekurzorů k TC, může být u MS zvýšena až o 50 %. Současně dochází k vzestupu metabolického obratu cholesterolu a katabolismu apo B-100 obsaženého v LDL. Zvýšení syntézy cholesterolu v játrech se vysvětluje stimulací proteinu SREBP-1c. Bylo zjištěno, že markery syntézy cholesterolu korelují pozitivně s tělesnou hmotností, inzulinémií a glykémií (50).
Resorpce cholesterolu ve střevě se stanoví izotopovými metodami nebo na podkladě plazmatické koncentrace doprovodných fytosterolů (campesterol, β-sitosterol, brassicasterol). U MS klesá resorpce cholesterolu zhruba o 20 %, v závislosti na celkové náloži neutrálních a kyselých sterolů ve střevním lumen. Množství exogenního cholesterolu přijímaného v potravě kolísá v širokém rozmezí od 50 do 1000 mg za den; k tomu je třeba přičíst 500–2500 mg cholesterolu obsaženého ve žluči.
Resorpce cholesterolu je aktivní proces, na němž participuje specifický přenašeč NPN 1L1 (Niemann-Pick C1-like protein), lokalizovaný v apikální membráně enterocytů. Opačný mechanismus představují transportní proteiny ABCG8 a ABCG5, které naopak vylučují cholesterol z enterocytu do lumen střeva (obr. 3). Tytéž transportní proteiny jsou přítomny v kanalikulárním pólu hepatocytů, kde urychlují transport cholesterolu do žlučovodů. Změny střevní resorpce a žlučové exkrece modifikují bazální nastavení homeostázy cholesterolu. U MS a DM 2. typu byla prokázána zvýšená aktivita ABCG8/5. Naproti tomu DM 1. typu je spojen s nízkou expresí genů pro ABCG8/5, což se projevuje významně vyšší střevní resorpci a nižší syntézou cholesterolu (51).

V praxi je třeba mít na paměti, že pokles tělesné hmotnosti vede u obézních jedinců ke zpomalení syntézy cholesterolu a vzestupu jeho resorpce ve střevě. Proto by se měl klást zvýšený důraz na doporučení, aby redukční dieta obsahovala méně než 200 mg cholesterolu za den, popřípadě aby byla obohacena potravinami speciálního určení (rozpustná vláknina, fytosteroly), které potlačují resorpci cholesterolu ve střevě. V úvahu přicházejí též léky snižující resorpci exogenního cholesterolu (ezetimib).
Metabolické účinky některých preventivních a terapeutických metod
Patofyziologické mechanizmy ADL mohou být příznivě ovlivněny metodami užívanými v prevenci a léčbě MS a DM 2. typu. Existují jednoznačné důkazy o účinnosti dvou základních nefarmakologických přístupů, nutriční intervence a pohybové aktivity (52). Cílevědomou změnou životního způsobu lze v krátké době dosáhnout zlepšení všech parametrů IR včetně endotelové dysfunkce (53).
Dietní opatření
Obecným principem dietní prevence a léčby MS je redukce celkového energetického příjmu, spolu se selektivním omezením nasycených tuků, cholesterolu, alkoholu a jednoduchých cukrů, zejména fruktózy, která je v nadměrném množství obsažena ve slazených nealkoholických nápojích (54).
V dietologických studiích se věnuje velká pozornost složení stravy, která byla dříve typická pro oblasti kolem Středozemního moře (mediterranean diet), s vysokým obsahem olivového oleje. U osob s MS bylo prokázáno, že i částečná náhrada nasycených tuků (SFA) mononenasycenými (MUFA) v množství okolo 7 % energetického příjmu významně sníží plazmatické koncentrace LDL-C, zvýší koncentrace HDL-C a potlačí inzulínovou rezistenci za předpokladu, že příjem lipidů nepřesáhne 30–35 % celkového energetického příjmu (53, 55). Redukce tělesné hmotnosti, jíž lze dietními opatřeními poměrně snadno dosáhnout, vede u jedinců s viscerální obezitou k poklesu jaterní produkce VLDL a k vzestupu HDL-C (17). Speciální dietní doporučení zohledňující genetické faktory a stravovací zvyklosti konkrétního jedince jsou předmětem nutrigenetického výzkumu.
Pohybová aktivita
Samotná nízkoenergetická dieta by vyvolala nežádoucí úbytek svalové tkáně, a proto je nezbytné, aby byla vždy doplněna pravidelnou pohybovou aktivitou. K příznivým účinkům pohybové aktivity patří pokles IR kosterního svalstva a redukce viscerální tukové tkáně. Bylo zjištěno, že ve svalových buňkách přibývá mitochondrií a oxidačních enzymů, stoupá příjem a oxidace FFA. Zvýšený průtok krve kosterním svalstvem vede k vzestupu aktivity LPL (17). Doporučení týkající se druhu, trvání a intenzity pohybové aktivity by měla být individuálně přizpůsobena.
Vícenenasycené mastné kyseliny řady n-3
V prevenci a léčbě MS se stále více používají PUFA n-3, esenciální mastné kyseliny s dlouhým řetězcem (převážně kyselina eikosapentaenová a dokosahexaenová). Tyto kyseliny jsou důležitou součástí buněčných membrán. Jejich účinky na LP metabolismus se vysvětlují tím, že jako přirozené ligandy stimulují nukleární receptory PPARα, zčásti též PPARγ, a snižují expresi genu pro protein SREBP-1. Dochází tak k významnému zvýšení inzulínové senzitivity ve svalstvu a v tukové tkáni, k poklesu jaterní syntézy VLDL a k vzestupu jejich frakčního katabolismu (56). Podáváním PUFA n-3 lze dosáhnout značného poklesu plazmatické koncentrace TG, nalačno i postprandiálně, současně se mírně zvýší HDL-C; i když se po podávání PUFA n-3 zvyšuje koncentrace LDL-C, redukuje se významně podíl částic sd-LDL. Byly popsány též příznivé změny ve složení žluči. Se zřetelem na prevenci ICHS jsou významné protizánětlivé a antiagregační účinky (57).
Fibráty
Hypolipidemika ze skupiny fibrátů působí jako exogenní ligandy nukleárních receptorů PPARα, takže modulují expresi četných genů, které řídí lipidový a glycidový metabolismus. Se zřetelem na ADL je významné zvýšení syntézy apo A-I, apo A-II, ABCA1 a LDL, při současném poklesu apo C-III. Podávání fibrátů příznivě ovlivňuje HTG i HDL C, zvyšuje zastoupení subfrakcí I a II v třídě LDL. Bylo prokázáno, že fibráty u MS zvyšují oxidaci FFA, urychlují katabolismus LP obsahujících apo B a snižují syntézu cholesterolu. Dosud se však nepodařilo potvrdit předpokládaný pokles sekrece VLDL v játrech (58). Aktivací receptorů PPARα v cévní stěně se vysvětluje mimo jiné zlepšení endotelové dysfunkce a snížení exprese mediátorů zánětu.
Kyselina nikotinová
Obnovený zájem o kyselinu nikotinovou v terapii MS lze vysvětlit tím, že ze všech běžných hypolipidemik nejvýrazněji zvyšuje plazmatickou koncentraci HDL-C (59). Má rovněž konzistentní příznivé účinky na HTG a velikost částic LDL. Mechanismus účinku spočívá ve snížení toku FFA z tukové tkáně do jater, svalstva a pankreatu, díky inhibici hormon-senzitivní lipázy. Dochází ke značnému poklesu sekrece VLDL1 a glukoneogeneze v játrech. Přípravky kyseliny nikotinové s prodlouženým uvolňováním mají minimalizované nežádoucí účinky na glukózovou homeostázu a na metabolismus purinů. Totéž platí o dlouhodobě působících derivátech kyseliny nikotinové (např. acipimox).
Statiny
Kromě známého účinku na syntézu cholesterolu působí statiny u MS ještě v dalším směru. Primárně zvyšují katabolismus všech LP obsahujících apo B-100 i remnantních CM, současně však dochází ke vzestupu střevní resorpce sterolů. Produkci apo B-100 mohou statiny snížit, je-li nadměrně vysoká. Hypotriglyceridemický efekt se vysvětluje zvýšenou aktivitou receptorů LDL-R, které váží VLDL2. S poklesem TG je asociován mírný vzestup HDL-C (17).
Léky a látky omezující resorpci cholesterolu
Podávání ezetimibu a potravinových doplňků (vláknina, fytosteroly), které omezují resorpci cholesterolu ze střevního lumen, je zdůvodněno u MS v těch situacích, kdy klesne syntéza cholesterolu de novo (např. v důsledku redukce tělesné hmotnosti, nebo po léčbě statiny) a pokles syntézy cholesterolu je doprovázen jeho zvýšenou resorpcí, bez příznivého dopadu na koncentrace TC a LDL-C (22).
Glitazony (thiazolidindiony)
Nová antidiabetika označovaná též jako inzulínové senzitizéry jsou exogenními ligandy nukleárních receptorů PPARγ. Aktivace těchto receptorů exprimovaných převážně v tukové tkáni ovlivňuje diferenciaci adipocytů a ukládání TG, snižuje odtok FFA a zlepšuje IR. Mění se též produkce adipocytokinů (vzestup adiponektinu, pokles TNFα, rezistinu a angiotensinogenu). Druhotně dochází ke zvýšení oxidace FA a ke zlepšení utilizace glukózy v kosterním svalstvu a snižuje se IR v játrech. Vzestup frakční clearance cirkulujících VLDL se vysvětluje zvýšenou aktivitou LPL při poklesu syntézy apo C-III (60). Klinicky se účinek glitazonů na lipidový metabolismus projevuje poklesem plazmatické koncentrace TG, vzestupem HDL-C a snížením převahy sd LDL, naproti tomu mírně stoupá koncentrace TC a LDL C (61). Mezi hlavní nežádoucí účinky glitazonů patří retence tekutin, která může vést k srdečnímu selhání. Na přírůstku hmotnosti se podílí též zvětšení objemu subkutánní tukové tkáně.
Glitazary
Snaha o současné ovlivnění nukleárních receptorů PPARα a PPARγ vedla k vývoji duálních agonistů, o nichž se předpokládalo, že budou mít všechny příznivé účinky fibrátů i glitazonů (62). Jednotlivé preparáty z této skupiny se liší stupněm afinity k oběma podtypům PPAR; snižují plazmatickou koncentraci TG o 30–60 % a koncentraci TC o 15 až 20 %, HDL-C zvyšují o 15–30 %. Ve fázi klinického zkoušení byly zjištěny závažné nežádoucí účinky. Ze souhrnné analýzy studií o muraglitazaru vyplynulo, že podávání tohoto léčiva bylo spojeno s významným vzestupem kardiovaskulárních příhod a s pozitivním trendem ve výskytu srdečního selhání (63). Hodnocení glitazarů není dosud uzavřeno, na základě dosavadních zkušeností je však oprávněná zdrženlivost nejen ke glitazarům, ale též ke kombinaci fibrátů s glitazony.
Inhibitory transferového proteinu esterů cholesterolu (CETP)
Poznatek, že inhibice CETP vede k výraznému vzestupu plazmatické koncentrace HDL-C, inicioval vývoj několika syntetických preparátů, jejichž očekávaný účinek se plně potvrdil. V klinickém zkoušení postoupil nejdále torcetrapib, který u dobrovolníků zvyšoval HDL-C o více než 50 %, v ojedinělých případech až o 100 %, a současně snižoval LDL-C. Mechanismus účinku spočívá v potlačení frakčního katabolismu HDL a apo A-I (17). Torcetrapib však nebyl vzhledem k nežádoucím účinkům (zvýšený výskyt koronárních příhod) uveden do klinické praxe. Navíc existuje zásadní pochybnost týkající se inhibice CETP, protože přenos esterů cholesterolu z HDL do LP obsahujících apo B představuje kvantitativně nejvýznamnější cestu reverzního transportu cholesterolu. Zvýšení plazmatické koncentrace HDL C samo o sobě nevypovídá o funkčních vlastnostech částic HDL, které přibyly. Dosud není známo, zda velké částice HDL s vysokým obsahem CE, které se akumulují během inhibice CETP, jsou dostatečně účinné z hlediska prevence aterosklerózy (24).
Blokátory kanabinoidních receptorů
Receptory endokanabinoidního systému CB1 se vyskytují v mozku i v některých periferních tkáních (tuková tkáň, gastrointestinální trakt). Chronická stimulace těchto receptorů zvyšuje příjem potravy a ukládání tuků, udržuje závislost na nikotinu a některých drogách. Rimonabant, nově zaváděný blokátor receptorů CB1, příznivě ovlivňuje IR, mimo jiné zvyšuje tvorbu adiponektinu (64). Ve studii 4. fáze RIO Lipids došlo u obézních osob po jednom roce podávání rimonabantu k významnému snížení hmotnosti a obvodu pasu, HDL-C se zvýšil o 23 %, TG poklesly o 15 %, zlepšila se glukózová tolerance. Procento osob, které splňovaly kritéria MS, kleslo na polovinu. Dlouhodobé abstinence kouření bylo dosaženo u motivovaných jedinců dvakrát častěji než v placebové skupině. Jako nežádoucí účinek se popisuje zvýšený výskyt depresivních stavů.
Kombinovaná léčba
Klinická zkušenost ukazuje, že u značného počtu pacientů s MS nelze dosáhnout cílových hodnot plazmatických lipidů monoterapií. Z poznatků o patofyziologických mechanismech ADL vyplývá, že zvláště vhodné jsou kombinace statinů s fibráty, s kyselinou nikotinovou nebo s n-3 PUFA. I když dosud nejsou k dispozici důkazy z intervenčních studií, lze předpokládat, že výsledný účinek smysluplných kombinací bude více než aditivní.
Závěr
Aterogenní dyslipidémie vzniká jako integrální komponenta metabolického syndromu v kauzální souvislosti s inzulínovou rezistencí. Za hlavní patogenetický faktor se považuje pozitivní energetická bilance, která spolu se sníženou oxidativní kapacitou tkání vede k akumulaci mastných kyselin a triglyceridů. Chronický zánětlivý stav a oxidační stres ohrožují buněčnou homeostázu. Logickou, ale obtížně realizovatelnou cestou k prevenci a léčbě metabolického syndromu je změna životního způsobu, zejména nutričních zvyklostí a pohybové aktivity. Farmakoterapií lze dyslipidémii příznivě ovlivnit, ale normalizace komplexní poruchy zůstává vzdáleným cílem klinického výzkumu.
Zkratky
ADL – aterogenní dyslipidémie
apo – apolipoprotein
CETP – transferový protein esterů cholesterolu
CE – cholesterylestery
CM – chylomikrony
CRP – C-reaktivní protein
DM – diabetes mellitus
FA – mastné kyseliny
FABP-2 – střevní izoforma proteinu vázajícího mastné kyseliny
FFA – volné mastné kyseliny
HDL – lipoprotein o vysoké hustotě
HDL-C – cholesterol lipoproteinu o vysoké hustotě
HL – jaterní lipáza
HTG – hypertriglyceridémie
ICHS – ischemická choroba srdeční
IR – inzulínová rezistence
LCAT – lecitin-cholesterol-acyltransferáza
LDL – lipoprotein o nízké hustotě
LDL-C – cholesterol lipoproteinu o nízké hustotě
LDLR – receptor LDL (apo B/E)
LPL – lipoproteinová lipáza
MS – metabolický syndrom
MTTP – mikrozomální triglyceridový transferový protein
MUFA – mononenasycené mastné kyseliny
ox-LDL – oxidativně modifikované LDL
PGI2 –prostacyklin
PL – fosfolipidy (phospholipids)
PLTP – transferový protein pro fosfolipidy (phospholipid transfer protein)
PPAR – receptor aktivovaný peroxisomálními proliferátory
PUFA – vícenenasycené mastné kyseliny
RCT – zpětný transport cholesterolu (reverse cholesterol transport)
sd-LDL – malé denzní LDL
SFA – nasycené mastné kyseliny
SR-B1 – zametací receptor B1
SREBP – protein vázající sterolový regulační element
TC – celkový cholesterol
TG – triglyceridy
TRL – lipoproteiny bohaté na triglyceridy
UCP – odpřahující proteiny (uncoupling proteins)
VLDL – lipoprotein o velmi nízké hustotě
Podporováno výzkumným záměrem MŠMT ČR MSM 0021620820.
prof. MUDr. Aleš Žák, DrSc.
IV. interní klinika 1. LF UK a VFN
U Nemocnice 2, 128 08 Praha 2
fax: +420 224 923 524, e-mail: azak@vfn.cz
Sources
1. Superko, H. R.: Beyond LDL cholesterol reduction. Circulation, 1996, 94, s. 2351–2354.
2. Petersen, K. F., Shulman, G. I.: Etiology of insulin resistance. Am. J. Med, 2006, 119 (Suppl. 5A), s. 10S–16S.
3. Laakso, M.: Gene variants, insulin resistance, and dyslipidaemia. Curr. Opin. Lipidol., 2004, 15, s. 115–120.
4. Miranda, P. J., DeFronzo, R. A., Califf, R. M. et al.: Metabolic syndrome: Definition, pathophysiology, and mechanisms. Am. Heart J., 2005, 149, s. 33–45.
5. Le Roith, D., Zick, Y.: Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care, 2001, 24, s. 588–597.
6. Cusi, K., Maezono, K., Osman, A. et al.: Insulin resistance differentially affects the PI 3-kinase - and MAP kinase-mediated signaling in human muscle. J. Clin. Invest., 2000, 105, s. 311–320.
7. Avramoglu, R. K., Basciano, H., Adeli, K.: Lipid and lipoprotein dysregulation in insulin resistant states. Clin. Chim. Acta, 2006, 368, s. 1–19.
8. Lewis, G. F., Carpentier, A., Adeli, K. et al.: Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev, 2002, 23, s. 201–229.
9. Gregor, M. F., Hotamisligil, G. S.: Thematic review series: Adipocyte biology: The endoplasmic reticulum and metabolic disease. J. Lipid Res., 2007, 48, s. 1905–1914.
10. Sharma, A. M.: The obese patient with diabetes mellitus: From research targets to treatment options. Am. J. Med., 2006, 119 (Suppl. 5A), s. 17S–23S.
11. Fisler, J. S., Warden, C. H.: Uncoupling proteins, dietary fat and the metabolic syndrome. Nutr. Metab., 2006, 3, 38, doi:10.1186/1743-7075-3-38.
12. Lara-Castro, C., Luo, N., Wallace, P. et al.: Adiponectin multimeric complexes and the metabolic syndrome trait cluster. Diabetes, 2006, 55, s. 249–259.
13. Bouloumié, A., Curat, C. A., Sengenés, C. et al.: Role of macrophage tissue infiltration in metabolic diseases. Curr. Opin. Clin. Nutr. Metab. Care, 2005, 8, s. 347–354.
14. Sonnenberg, G. E., Krakower, G. R., Kissebah, A. H.: A novel pathway to the manifestations of metabolic syndome. Obes. Res., 2004, 12, s. 180–186.
15. Hokanson, J. E., Austin, M. A.: Plasma triglyceride level is a risk factor for cardiovascular disease independent of high density lipoprotein cholesterol level: A meta-analysis of population-based prospective studies. J. Cardiovasc. Risk, 1996, 3, s. 213–219.
16. Adeli, K., Taghibiglou, C., Van Iderstine, S. C. et al.: Mechanisms of hepatic very low-density lipoprotein overproduction. Trends Cardiovasc. Med., 2001, 11, s. 170–176.
17. Chan, D. C., Barrett, P. H., Watts, G. F.: Recent studies of lipoprotein kinetics in the metabolic syndrome and related disorders. Curr. Opin. Lipidol., 2006, 17, s. 28–36.
18. Krauss, R. M., Siri, P. W.: Metabolic abnormalities: Triglyceride and low-density lipoprotein. Endocrinol. Metab. Clin. North Am., 2004, 33, s. 405–415.
19. Ginsberg, H. N., Zhang, Y.-L., Hernandez-Ono, A.: Metabolic syndrome: Focus on dyslipidemia. Obesity, 2006, 14 (Suppl.), s. 41S–49S.
20. Rangan, V. S., Smith, S.: Fatty acid synthesis in eukaryotes. In: Biochemistry of lipids, lipoproteins and membrane. Vance, D. E. Vance, J. E. (eds.), 4th ed. Amsterdam, Elsevier Science B. V., 2002, s. 151–179.
21. Olivecrona, T., Bergo, M., Hultin, M. et al.: Nutritional regulation of lipoprotein lipase. Can. J. Cardiol., 1995, 11 (Suppl.), s. 73G–78G.
22. Chan, D. C., Barrett, P. H., Watts, G. F.: Lipoprotein transport in the metabolic syndrome: Pathophysiological and interventional studies employing stable isotopy and modelling metods. Clin. Sci., 2004, 107, s. 233–249.
23. Deeb, S. S., Zambon, A., Carr, M. C. et al.: Hepatic lipase and dyslipidemia: Interactions among genetic variants, obesity, gender, and diet. J. Lipid Res., 2003, 44, s. 1279–1286.
24. Lewis, G. F., Rader, D. J.: New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res., 2005, 96, s. 1221–1232.
25. Després, J. P., Lemieux, I., Dagenais, G. R. et al.: HDL-cholesterol as a marker of coronary heart disease risk: The Quebec Cardiovascular Study. Atherosclerosis, 2000, 153, s. 263–272.
26. Gotto, A. M., Pownall, H. J.: Manual of Lipid Disorders. 3rd ed. Philadelphia, Lippincott, Williams & Wilkins, 2003.
27. Malik, S.: Transcriptional regulation of the apolipoprotein AI gene. Front. Biosci., 2003, 8, s. d360–d368.
28. Horowitz, B. S., Goldberg, I. J., Merab, J. et al.: Increased plasma and renal clearance of an exchangeable pool of apolipoprotein A-I in subjects with low levels of high density lipoprotein cholesterol. J. Clin. Invest., 1993, 91, s. 1743–1752.
29. Rye, K. A., Bright, R., Psaltis, M. et al.: Regulation of reconstituted high density lipoprotein structure and remodeling by apolipoprotein E. J. Lipid Res., 2006, 47, s. 1025–1036.
30. Jahangiri, A., Rader, D. J., Marchadier, D. et al.: Evidence that endothelial lipase remodels high density lipoproteins without mediating the dissociation of apolipoprotein A-I. J. Lipid Res., 2005, 46, s. 896–903.
31. Murakami, M., Kudo, I.: New phospholipase A(2) isozymes with a potential role in atherosclerosis. Curr. Opin. Lipidol., 2003, 14, s. 431–436.
32. Hammad, S.M., Barth, J. L., Knaak, C. et al.: Megalin acts in concert with cubilin to mediate endocytosis of high density lipoproteins. J. Biol. Chem., 2000, 275, s. 12003–12008.
33. Rothblat, G. H., Llera-Moya, M., Atger, V. et al.: Cell cholesterol efflux: Integration of old and new observations provides new insights. J. Lipid. Res., 1999, 40, s. 781–796.
34. Schwartz, C. C., VandenBroek, J. M., Cooper, P. S.: Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J. Lipid. Res., 2004, 45, s. 1594–1607.
35. Wadham, C., Albanese, N., Roberts, J. et al.: High-density lipoproteins neutralize C-reactive protein proinflammatory activity. Circulation, 2004, 109, s. 2116–2122.
36. Barter, P. J., Puranik, R., Rye, K. A.: New insights into the role of HDL as an anti-inflammatory agent in the prevention of cardiovascular disease. Curr. Cardiol. Rep., 2007, 9, s. 493–498.
37. Lamarche, B. A., St-Pierre, A. C., Ruel, I. L. et al.: A prospective, population-based study of low density lipoprotein particle size as a risk factor for ischemic heart disease in men. Can. J. Cardiol., 2001, 17, s. 859–865.
38. Sevanian, A., Hwang, J., Hodis, H. et al.: Contribution of an in vivo oxidized LDL to LDL oxidation and its association with dense LDL subpopulations. Arteioscler. Thromb. Vasc. Biol., 1996, 16, s. 784–793.
39. Davidsson, P., Hulthe, J., Fagerberg, B. et al.: A proteomic study of the apolipoproteins in LDL subclasses in patients with the metabolic syndrome and type 2 diabetes. J. Lipid Res., 2005, 46, s. 1999–2006.
40. Berneis, K. K., Krauss, R. M.: Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res., 2002, 43, s. 1363–1379.
41. Vessby, B.: Dietary fat, fatty acid composition in plasma and the metabolic syndrome. Curr. Opin. Lipidol., 2003, 14, s. 15–19.
42. Warensjö, E., Sundström, J., Lind, L. et al.: Factor analysis of fatty acids in serum lipids as a measure of dietary fat quality in relation to the metabolic syndrome in men. Am. J. Clin. Nutr., 2006, 84, s. 442–448.
43. Žák, A., Vecka, M., Tvrzická, E. et al.: Složení esterifikovaných mastných kyselin a lipoperoxidace u metabolického syndromu. Čas. Lék. čes., 2007, 146, s. 484–491.
44. Evans, J. L., Goldfine, I. D., Maddux, B. A. et al.: Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocrine Rev., 2002, 23, s. 599–622.
45. Laaksonen, D. E., Lakka, T. A., Lakka, H.-M. et al.: Serum fatty acid composition predicts development of impaired fasting glycaemia and diabetes in middle-aged men. Diabet. Med., 2002, 19, s. 456–464.
46. Leskinen, M. H., Solakivi, T., Kunnas, T. et al.: Serum fatty acids in postinfarction middle-aged men. Scand. J. Clin. Lab. Med., 2005, 65, s. 485–490.
47. Decsi, T., Csábi, G., Török, K. et al.: Polyunsaturated fatty acids in plasma lipids of obese children with and without metabolic cardiovascular syndrome. Lipids, 2000, 35, s. 1179–1184.
48. Klein-Platat, C., Drai, J., Oujaa, M. et al.: Plasma fatty acid composition is associated with the metabolic syndrome and low grade inflammation in overweight adolescents. Am. J. Clin. Nutr., 2005, 82, s. 1178–1184.
49. Simonen, P. P., Gylling, H., Miettinen, T. A.: Introducing a new component of the metabolic syndrome: low cholesterol absorption. Am. J. Clin. Nutr., 2000, 72, s. 82–88.
50. Simonen, P. P., Gylling, H., Miettinen, T. A.: Body weight modulates cholesterol metabolism in non-insulin dependent type 2 diabetics. Obesity Res., 2002, 5, s. 328–335.
51. Gylling, H., Tuominen, J. A., Koivisto, V. A. et al.: Cholesterol metabolism in type 1 diabetes. Diabetes, 2004, 53, s. 2217–2222.
52. Haffner, S. M.: Risk constellations in patients with the metabolic syndrome: Epidemiology, diagnosis, and treatment patterns. Am. J. Med., 2006, 119 (Suppl. 5A), s. 3S–9S.
53. Richardi, G., Giacco, R., Rivellese, A. A.: Dietary fat, insulin sensitivity and the metabolic syndrome. Clin. Nutr., 2004, 23, s. 447–456.
54. Chong, M. F., Fielding, B. A., Frayn, K. N.: Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr., 2007, 85, s. 1511–1520.
55. Berglund, L., Lefevre, M., Ginsberg, H. N. et al.: Comparison of monounsaturated fat with carbohydrates as a replacement for saturated fat in subjects with a high metabolic risk profile: studies in the fasting and postprandial states. Am. J. Clin. Nutr., 2007, 86, s. 1611–1620.
56. Lombardo, Y. B., Chicco, A. G.: Effects of dietary polyunsaturated n-3 fatty acids on dyslipidemia and insulin resistance in rodents and humans. A review. J. Nutr. Biochem., 2006, 17, s. 1–13.
57. Žák, A., Tvrzická, E., Zeman, M. et al.: Patofyziologie a klinický význam vícenenasycených mastných kyselin řady n‑3. Čas. Lék. čes., 2005, 144 (Suppl. 1), s. 6–18.
58. Watts, G. F., Barrett, P. H., Ji, J. et al.: Differential regulation of lipoprotein kinetics by atorvastatin and fenofibrate in subjects with the metabolic syndrome. Diabetes, 2003, 52, s. 803–811.
59. Shepherd, J., Betteridge, J., Van Gaal, L. et al.: Nicotinic acid in the management of dyslipidaemia associated with diabetes and metabolic syndrome: A position paper developed by a European Consensus Panel. Curr. Med. Res. Opin., 2005, 21, s. 665–682.
60. Nagashima, K., Lopez, C., Donovan, D. et al.: Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J. Clin. Invest., 2005, 115, s. 1323–1332.
61. Lefebvre, F., Chinetti, C., Fruchart, J.-C. et al.: Sorting out the role of PPARalpha in energy metabolism and vascular homeostasis. J. Clin. Incest., 2006, 116, s. 571–580.
62. Štulc, T., Češka, R.: Duální blokáda receptorů PPAR. Farmakoterapie, 2006, 2, s. 13–16.
63. Nissen, S. E., Wolski, K., Topol, E. J.: Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA, 2005, 294, s. 2581–2586.
64. Bensaid, M., Gary-Bobo, M., Esclangon, A. et al.: The cannabinoid CB1 receptor antagonist SR 141716 increases Acrp30 mRNA expression in adipose tissue of obese fa/fa rats and in cultured adipocyte cells. Mol. Pharmacol., 2003, 63, s. 908–914.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Hydration in Wound Healing
Most read in this issue
- Atherogenic Dyslipidemia and the Metabolic Syndrome: Pathophysiological Mechanisms
- Acute Abdomen in Elderly Patients
- Laparoscopic Surgery in Senior Age
- Acyclic Nucleoside Phosphonates as Potential Antineoplastic Agents