
Sekvenování lidského genomu – technologie nové generace aneb budeme rutinně sekvenovat lidské genomy?
Human genome sequencing – next generation technology or will the routine sequencing of human genome be possible?
DNA sequencing has become a standard method widely used in molecular genetic analysis of biological materials. Its use in medicine is widespread, especially in diagnostics of inherited disorders and cancer related diseases. Development of DNA diagnostics has been strongly accelerated by publication of the human genome sequence in 2001. During the last few years one can observe rapid development of novel sequencing technologies, which have led to the introduction of so called „New Generation Sequencing“. These new technologies based on principles of massive parallel sequencing (e.g. Roche/454, Illumina Genome Analyzer IIx, Life Technologies SOLiD 3 and others) enable a massive increase of sequencing capacity and in parallel also a fundamental decrease of costs. This major technological breakthrough allowed development of the whole-genome sequencing including analyses of individual human genomes. It also started the era of personal genomics. The first sequenced individual human genomes belonged to famous geneticists J. C. Venter (2007) and J. D. Watson (2008), but they were rapidly followed by sequencing analyses of other individuals from various ethnic groups. These studies brought substantial information about interpersonal differences in genome structure (through characterization of nucleotide polymorphisms, DNA deletions and amplifications etc.). Sequencing of cancer cell genomes, e.g. acute myeloid leukemia has already brought first important clinically relevant results. Although currently we are still unable to interpret the relevance of all detected genome variants, it is obvious, that the possibility to sequence individual human genomes represents a fundamental breakthrough not only in DNA diagnostics but also in clinical medicine.
Key words:
genome, DNA, sequencing, next-generation sequencing.
Authors:
Š. Pospíšilová; B. Tichý; J. Mayer
Authors‘ workplace:
Fakultní nemocnice Brno a LF MU, Interní hematoonkologická klinika, Centrum molekulární biologie a genové terapie
Published in:
Čas. Lék. čes. 2009; 148: 296-302
Category:
Topic
Overview
Sekvenování DNA patří již řadu let ke standardním postupům při molekulárně-genetických analýzách biologického materiálu. V medicíně nachází široké uplatnění, zejména v oblasti diagnostiky dědičných chorob a nádorových onemocnění, přičemž rozvoj DNA diagnostiky byl významně podpořen zveřejněním sekvence lidského genomu v roce 2001. V posledních několika letech dochází k rychlému technologickému rozvoji nových sekvenačních technologií, který umožnil vznik sekvenátorů nové generace („tzv. New Generation Sequencing“). Nové technologie založené na principu masivního paralelního sekvenování (např. Roche/454, Illumina Genome Analyzer IIx, Life Technologies SOLiD 3 a další) umožňují zásadní navýšení kapacity sekvenátorů a výrazné snížení ceny. Tento významný technologický pokrok umožnil rozvoj celogenomového sekvenování včetně analýz individuálních lidských genomů a nastartoval rozvoj personální genomiky. První osekvenované individuální lidské genomy patřily významným genetikům J. C. Venterovi (2007) a J. D. Watsonovi (2008), avšak rychle následovaly sekvenační analýzy dalších jedinců z různých etnik, které přinesly podstatné informace o interpersonálních rozdílech ve struktuře genomů (byly např. charakterizovány nukleotidové polymorfismy, delece a amplifikace úseků DNA). První významné aplikovatelné výsledky již přineslo sekvenování genomů nádorových buněk, např. akutní myeloidní leukémie. Ačkoli v současné době ještě nejsme schopni interpretovat význam všech detekovaných variant genomu, znamená možnost sekvenování individuálních lidských genomů zásadní zlom v DNA diagnostice i celé medicíně.
Klíčová slova:
genom, DNA, sekvenování, sekvenování nové generace.
Vývoj metodických přístupů k sekvenování genomu
Lidský genom reprezentovaný molekulami DNA rozdělenými do 46 chromozomů (22 párů autozomů a jednoho páru gonozomů) má velikost 3,2 miliardy nukleotidů (tj. 3200 Mb = megabází) a celkově obsahuje přibližně 25 000 genů. Kódující oblasti, dominantně tvořené exony strukturních genů, zaujímají zhruba 1,5 % genomu. Zbytek genomu tvoří introny, pseudogeny, mobilní elementy, repetitivní sekvence a další úseky, jejichž význam dosud nebyl zcela objasněn. Pro lepší představu o velikosti lidského genomu lze uvést srovnání s jinými organismy. Například velikost genomu baktérií se pohybuje v rozmezí 0,5–10 Mb, což odpovídá počtu cca 500–5500 genů. Samostatně žijícím organismem s nejmenším genomem je Mycoplasma genitalium, jejíž genom má velikost 0,58 Mb a obsahuje 470 kódujících genů; naopak mezi organismy s největším počtem genů (až 50 000) patří rostliny.
První pokusy o sekvenování, tedy určení pořadí jednotlivých nukleotidů v DNA, byly započaty již v 60. letech minulého století, tedy jen několik let po objevení struktury DNA Jamesem Watsonem a Francisem Crickem v roce 1953. Nešlo však o sekvenování chemickou nebo enzymatickou cestou, ale za pomoci elektronového mikroskopu (1). Přibližně o 10 let později došlo v technologiích sekvenování DNA k významnému metodickému posunu. Laboratoř F. Sangera (2) zavedla a publikovala DNA sekvenační metodu využívající tzv. „2’,3’-dideoxy-“ analogy normálních stavebních jednotek deoxynukleosid-trifosfátů, které působí jako specifické inhibitory DNA polymerázy. Prvním takto osekvenoveným genomem byl bakteriofág phiX174 (3). Alternativní techniku DNA sekvenování na principu chemických reakcí štěpících terminálně značené báze s následnou elektroforézou značených DNA fragmentů v polyakrylamidovém gelu zavedli A. Maxam a W. Gilbert (4). Nové sekvenační technologie byly zpočátku schopny přečíst v jednom běhu pouze asi 100 bází, jejich postupným zdokonalováním, například automatizací detekce fluorescenčně značených molekul (5, 6) nebo zaváděním kapilárních sekvenátorů (7) je v současnosti standardně dosahováno přečtení až 1000 bází v jednom běhu. Tyto zdokonalené techniky umožnily rutinně využívat sekvenování pro analýzy fragmentů DNA (např. produktů PCR reakcí), a staly se tak běžnou diagnostickou metodou v medicínské praxi; sekvenování celých genomů však bylo stále velmi zdlouhavé a časově i finančně náročné. První kompletní genomy – genom bakterie Haemophilus influenzae následovaný genomem Mycoplasma genitalium byly osekvenovány v roce 1995 v „The Institute for Genomic Research“ (TIGR) založeném C. Venterem (8). Za účelem komerčního využití sekvenačních technologií v roce 1998 vznikla společnost „Celera Genomics“, která opět pod vedením C. Ventera pokračovala v genomovém sekvenování – např. sekvence mouchy octomilky byla publikována v roce 2000 (9). Použité technologie vycházely z metodického přístupu F. Sangera využívajícího dideoxynukleotid-trifosfáty (ddNTPs), avšak byly doplněny o přístupy umožňující provést analýzy delších úseků DNA. Jednou z možností bylo tzv. „shotgun“ sekvenování (10), kdy byla analyzovaná DNA náhodně naštěpena na krátké úseky, ty byly klonovány do vektoru a po té sekvenovány z obou konců za vzniku tzv. „mate pairs“. Získané úseky se vzájemně překrývaly, což umožnilo sestavení celé sekvence. Pro každý segment bylo nutno získat několik nezávislých čtení, které definovaly tzv. pokrytí sekvence („sequence coverage“).
Dlouho očekávaného cíle osekvenování lidského genomu započatého v roce 1990 bylo dosaženo v únoru 2001, kdy své výsledky publikovaly současně dva týmy: Venterův tým ze společnosti Celera (11) a International Human Genome Sequencing Consortium reprezentující 20 výzkumných skupin z USA, Velké Británie, Japonska, Francie, Německa a Číny (12), do kterého byl zapojen i J. Watson. Výsledky tohoto mezinárodního projektu („Human Genome Project“) jsou volně přístupné v databázi GenBank (www.ncbi.nlm.nih.gov/Genbank). Výsledky obou projektů založené dominantně na analýzách euchromatinových oblastí poskytly odhad celkového počtu lidských genů (odhadnuto 26–30 tisíc transkriptů kódujících proteiny) a nukleotidových polymorfismů (1,4–2,1 milionu SNP, tj. single-nucleotide polymorphism) i údaje o struktuře genomu (1,1% exony, 24% introny, 75% intergenové oblasti). Přestože zjištěné údaje musely být v následujících letech upřesňovány a doplňovány (v současné době předpokládáme existenci přibližně 4 milionů SNP a 24 tisíc protein-kódujících genů), mělo zpřístupnění sekvence lidského genomu velký vliv na další rozvoj molekulárně-biologických technologií a znamenalo začátek nové éry lidské genetiky.
Použité přístupy vycházející z automatizované Sangerovy metody, v současnosti reprezentované zejména např. přístroji řady ABI3730XL, se však dostaly na maximum kapacitních možností a další rozvoj sekvenování lidských genomů byl podmíněn vznikem nové výkonnější a levnější technologie. Prvním komerčním sekvenátorem nové generace využívajícím principu tzv. pyrosekvenování byl systém Roche/454 GS FLX uvedený na trh v roce 2005. O necelý rok později následoval Genome Analyzer od firmy Illumina (původně nazývaný Solexa) využívající reverzibilních terminátorů syntézy DNA. V létě 2007 byl firmou Applied Biosystems (nyní Life Technologies) představen třetí typ sekvenátoru nazvaný SOLiD, což znamená „Sequencing by Oligo Ligation and Detection“. Srovnání technických parametrů uvedených technologií je uvedeno v tabulce 1.
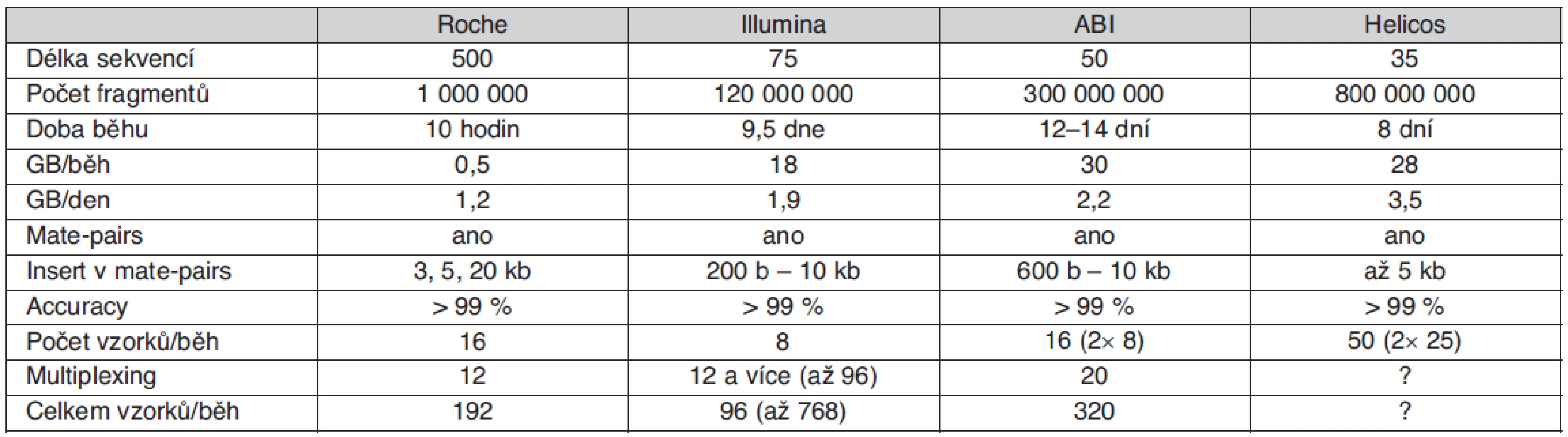
Kromě zmíněných, v současné době zřejmě nerozšířenějších systémů, však existuje i řada dalších perspektivních technologií. Nové možnosti analýz jednotlivých molekul DNA otevírá sekvenátor firmy Helicos Biosciences „HeliScope Single Molecule Sequencer“ dostupný od roku 2008, produkt firmy Pacific Biosciences umožňující tzv. SMRT (Single Molecule Real Time) sekvenování nebo tzv. Polony sequencing technology. Lze tedy předpokládat, že rychlý rozvoj sekvenačních technologií nové generace povede k možnostem sekvenování lidského genomu v ceně do 1000 USD již dříve avizované C. Venterem a v současné době intenzivně připravované např. firmou VisiGen Biotechnologies.
Technologické principy sekvenování nové generace
Technologie masivního paralelního sekvenování („next-generation sequencing“) musí řešit dvě zásadní otázky: fyzické oddělení jednotlivých fragmentů nukleových kyselin a vlastní čtení sekvence. První otázka je řešena emulzní PCR nebo amplifikací ve shlucích. Při emulzní PCR dochází k vytvoření tzv. mikroreaktorů, v nichž jsou uzavřeny všechny potřebné komponenty pro PCR (nukleotidy, enzym polymeráza, primery). Na mikrokuličky je navázaný fragment nukleové kyseliny. Při emulzní PCR dochází ke klonální amplifikaci – všechny molekuly DNA vytvořené v mikroreaktoru pochází z jediné molekuly templátu. Druhá otázka – čtení sekvence – je u dostupných zařízení řešena dvěma metodami: sekvenováním syntézou („sequencing by synthesis“) a sekvenováním založeném na ligaci („ligation based sequencing“).
Pro sekvenování genomů je často využíván již zmíněný „mate-pairs“ přístup, který dovoluje částečně odstranit problém repetitivních sekvencí. Při tomto postupu jsou vytvořeny fragmenty DNA známé délky, jejichž konce jsou následně sekvenovány. K (re)sekvenování kratších úseků DNA je celková kapacita paralelního sekvenování zbytečně velká, a proto je v tomto případě vhodné analyzovat v jednom běhu více vzorků. To je umožněno buď přímo uspořádáním přístroje, nebo multiplexováním vzorků s využitím tzv. tagů – krátkých sekvencí, které jsou součástí adaptérů. Kombinace obou přístupů dovoluje analyzovat až několik stovek vzorků najednou.
Technologie firmy Roche/454 Genome Sequencer FLX System (obr. 1)
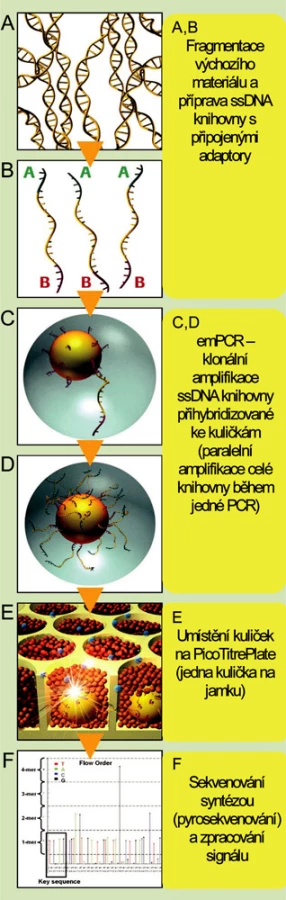
Jde o první komerčně dostupné zařízení pro masivní paralelní sekvenování uvedené na trh v roce 2005. Pro amplifikaci vzorku je využívána metoda emulzní PCR, ke čtení sekvence je použito pyrosekvenování. Sekvenační reakce probíhá ve speciálních destičkách složených z 3,2 milionů jamek o průměru 29 μm. V porovnání s ostatními tento systém produkuje nejdelší celistvé sekvence (v průměru 500 bází). Na jedné destičce je běžně sekvenován asi jeden milion fragmentů, během jednoho desetihodinového běhu je tak generováno až 0,5 gigabáze. Možnosti přístroje Genome Sequencer FLX vhodně doplňuje nová technologie pro obohacení vzorků pro resekvenování o sekvence a oblasti, které jsou pro daný účel potřebné, tzv. Sequence Capture (technologie je nyní dostupná pro lidské vzorky). Tato technologie je založená na hybridizaci komplementárních sekvencí na mikročipech s vysokou hustotou záznamu, odmytí nenavázané DNA a získání zachycených DNA sekvencí.
Technologie Illumina Genome Analyzer IIx (obr. 2)
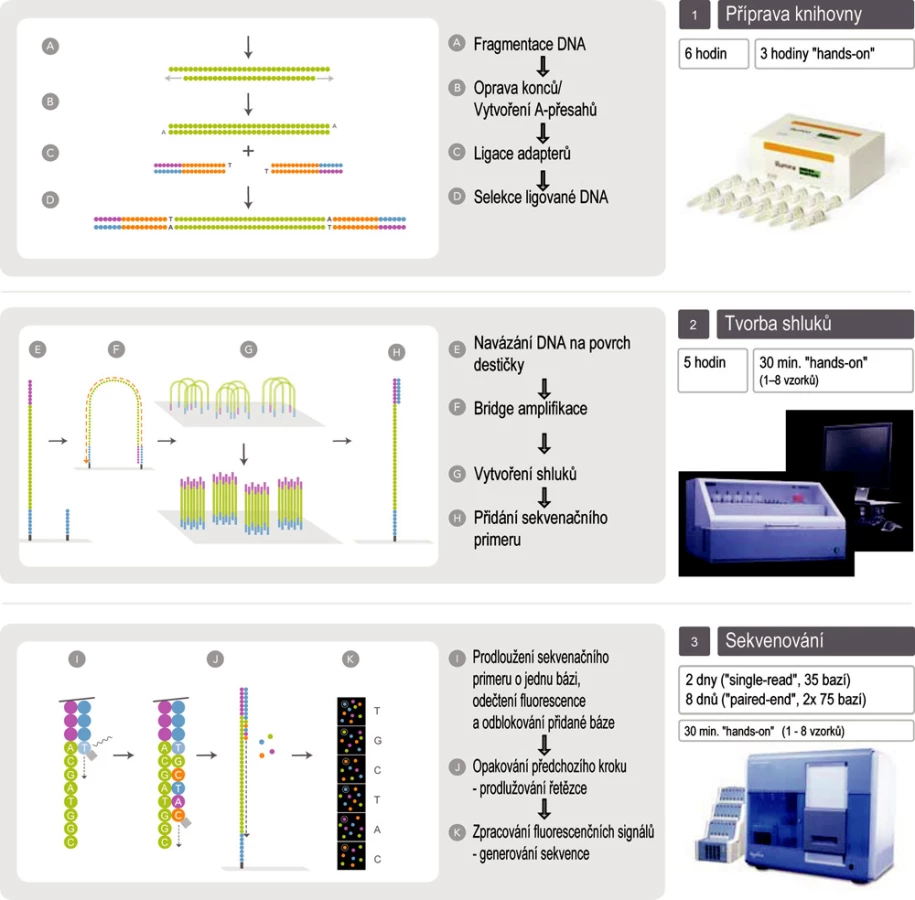
Využívá metodu amplifikace ve shlucích, ke čtení sekvencí sekvenování syntézou s detekcí pomocí reverzibilně terminačních fluorescenčně značených nukleotidů. Amplifikace i sekvenování probíhá na stejném místě – na povrchu speciální destičky (flow-cell). Fragmenty nukleových kyselin s adaptéry navázanými na obou koncích jsou náhodně zachyceny na povrchu destičky, amplifikovány a sekvenovány buď pouze z jednoho konce (single-reads), nebo z obou konců (paired-end reads). Během jednoho běhu je možné získat sekvenci až 120 milionů fragmentů o délce až 2× 75 bází (i více), systém tedy může vyprodukovat až 18 gigabází během 9,5 dne (1,9 gigabáze/den).
Technologie Applied Biosystems/Life Technologies SOLiD 3 (obr. 3)

K amplifikaci je použita metoda emulzní PCR, pro sekvenování metoda sekvenování ligací. Metoda sekvenování ligací využívá hybridizaci krátkých fluorescenčně značených sond, které mají definovány první dva nukleotidy. K přečtení kompletní sekvence je zapotřebí proces hybridizace a následné ligace zopakovat celkem 5×, každá pozice v sekvenci je pak charakterizována dvěma fluorescenčními signály, čímž je zajištěna vyšší spolehlivost určení báze. Systém je schopen přečíst sekvenci až 300 milionů fragmentů o délce 2× 50 bází (mate-pairs), v jednom běhu trvajícím 12–14 dní tak generuje až 30 gigabází.
Technologie HeliScope – Helicos Genetic Analysis System
Tento systém je výjimečný v tom, že nevyžaduje amplifikaci vzorku. Metodou pro čtení sekvence je sekvenování syntézou s reverzibilně terminačními fluorescenčně značenými nukleotidy. Podobně jako u Genome Analyzeru firmy Illumina jsou fragmenty nukleových kyselin nejprve zachyceny na povrchu speciálního substrátu a pak přímo sekvenovány bez předchozí amplifikace. Citlivost zařízení dovoluje detekci fluorescence emitované jedním fluorescenčně značeným nukleotidem. Běžná délka získané sekvence je 30–35 bází, během jednoho běhu je analyzováno až 800 miliónů fragmentů. Za osm dní tak systém vyprodukuje až 28 gigabází sekvence.
Využití sekvenačních technologií nové generace a jejich možné uplatnění v medicíně
Sekvenační technologie nové generace schopné denně produkovat jedním přístrojem cca 1–2 gigabáze sekvenčních dat poskytují obrovské možnosti pro rozvoj molekulární medicíny. Přestože v současnosti jsou tyto technologie výsadou vysoce specializovaných genomických laboratoří, předpokládá se, že za 5 let budou rutinně využívány v biomedicínském výzkumu a za 10 let i v běžné genetické diagnostice. V poslední době byly nové technologie použity v řadě projektů mapujících první individuální lidské genomy a tyto snahy nadále intenzivně pokračují. Prvním konkrétním výsledkem bylo publikování pilotní analýzy 1% lidského genomu v rámci ENCODE projektu v červnu 2007 (13). Zanedlouho potom byl pracovníky Institutu C. Ventera v Rockville publikován první tzv. individuální genom, tedy kompletní diploidní sekvence konkrétního člověka – Craiga Ventera (14). S využitím klasické Sangerovy „di-deoxy“ technologie bylo identifikováno 2,81 miliardy nukleotidů se 7,5 násobným pokrytím. Ve Venterově genomu bylo detekováno 4,1 milionu variant, z toho 3,2 milionu SNP, 292 tisíc malých inzercí a delecí a mnoho dalších změn, přičemž 44 % těchto změn bylo heterozygotních. V dubnu 2008 publikoval analogické výsledky druhého kompletního individuálního genomu již s využitím sekvenátorů nové generace tým odborníků z Baylor College v Texasu, který analyzoval DNA Jamese D. Watsona (15). Sekvenování tohoto diploidního genomu se 7,4násobným pokrytím trvalo dva měsíce a identifikovalo 3,3 milionu SNP, z nichž více než 10 tisíc způsobuje záměnu aminokyseliny v proteinovém produktu genu, a může mít tedy významný dopad na fenotyp. Porovnání prvních dvou individuálních genomů významných genetiků Ventera a Watsona získaných různými metodickými přístupy ukázalo významnou shodu získaných dat a prokázalo plnou využitelnost sekvenačních technologií nové generace. Oba genomy mají společných 1,68 milionu SNP a liší se 7648 aminokyselinovými záměnami, které pravděpodobně významně přispívají k fenotypovým rozdílům mezi oběma jedinci.
Krátce po zveřejnění kompletních analýz prvních genomů se začaly hromadit další nové poznatky získané celogenomovým sekvenováním, které zřejmě jednoznačně znamenají počátek éry sekvenování individuálních lidských genomů. V květnu 2008 byly publikovány výsledky mapování strukturní variability osmi lidských genomů pocházejících z Evropy (dva jedinci), Asie (dva jedinci) a Afriky (čtyři jedinci z nigerského etnika Yoruba) (16); na konci roku 2008 následovalo zveřejnění sekvence prvního kompletního diploidního genomu pocházejícího z Asie (anonymní muž z Číny) (17) získané technologiemi masivního paralelního sekvenování s 36násobným pokrytím umožňujícím detailní osekvenování 99,97 % genomu. Současně se začaly rozvíjet také analýzy genomů specifických pro určitá onemocnění. Typickým zástupcem těchto aktivit je mezinárodní Cancer Genome Project, jehož cílem je zmapování genomů jednotlivých typů nádorových buněk (Cancer Genome Atlas). Zajímavé výsledky již přineslo osekvenování genomu buněk akutní myeloidní leukémie (AML) s cílem definovat nádorově specifické somatické mutace (18). Srovnání genomu cytogeneticky normálních AML buněk se zdravými kožními buňkami téhož pacienta odhalilo deset genů mutovaných v AML buňkách, z nichž osm bylo nově asociovaných s AML a pouze dva již byly v minulosti popsány (FLT3 a NPM1). Příklad AML tedy jednoznačně ukazuje potenciál nových sekvenčních technologií pro hledání kandidátních genů relevantních pro patogenezi závažných onemocnění i nových nádorových markerů využitelných v onkologické diagnostice.
Jedním z ambiciózních aktuálně probíhajících projektů je projekt „1000 genomes – A Deep Catalog of Human Genetic Variation“ organizovaný mezinárodním konsorciem s dominantním podílem Sangerova institutu v Hinxtonu, National Institute of Health v Bethesdě a Genomickým institutem v Pekingu. Cílem je detailně charakterizovat genetickou variabilitu genomů na základě informací získaných sekvenováním cca 1200 lidí z různých částí světa a využít těchto poznatků v medicíně. První výsledky projektu prezentované v prosinci 2008 charakterizovaly SNP a tzv. „copy number variations“ (CNV) u čtyř jedinců. Data získávaná sekvenačními analýzami se šestinásobným pokrytím jsou průběžně aktualizována a elektronicky přístupná v databázích NCBI a EBI.
Rychlý rozvoj sekvenačních technologií v posledních letech jednoznačně nastartoval rozvoj personální genomiky umožňující sekvenování individuálních genomů pro potřeby prediktivní medicíny i sekvenování genomů patologicky změněných lidských buněk vyvolávajících nádorová a jiná onemocnění. Sekvenátory nové generace však mohou být využity i pro mnoho dalších aplikací. Významné uplatnění mají například v metagenomice, která se zabývá genomickými analýzami patogenů a umožňuje definovat tzv. mikrobiom člověka. Dále jsou tyto technologie využitelné pro analýzy DNA-proteinových interakcí včetně detekce vazebných DNA sekvencí proteinů po chromatinové imunoprecipitaci – tzv. „CHIP-seq“ a mohou být velmi přínosné při hledání malých nekódujících RNA, zejména microRNA (19).
Sekvenování lidského genomu je i přes dosažené úspěchy stále záležitostí velmi nákladnou, nicméně reálná cena za sekvenci kompletního genomu se stále výrazně snižuje. Původní rozpočet na projekt lidského genomu dokončeného v roce 2001 činil 3 miliardy USD, osekvenování Venterova genomu v roce 2007 stálo „jen“ 70 milionů USD. V současné době je cena sekvenování lidského genomu klasickou Sangerovou metodou přibližně 10 milionů USD, avšak při použití nové technologie Roche/454 se potřebná finanční částka i doba potřebná k získání kompletní sekvence mnohonásobně sníží (20). Použití dalších technologií (Illumina Genome Analyzer, Applied Biosystems – SOLiD) umožňuje ještě další redukci finanční náročnosti sekvenování a v současnosti se cena pohybuje v řádu stovek tisíc Kč za genom. Velký tlak na redukci nákladů na sekvenování a nová technická řešení je vyvíjen v souvislosti s probíhajícím projektem 1000 genomů a lze předpokládat, že v horizontu několika let bude možno sekvenovat lidské genomy za řádově desítky tisíc Kč.
V souvislosti s brzkou technickou a finanční dostupností sekvenování genomů však vyvstává řada otázek týkajících se možností využití získaných informací i etických problémů s tím spojených. Ještě řadu let pravděpodobně nebudeme schopni plně interpretovat získaná sekvenční data a vyhodnotit relevanci zjištěného genotypu k fenotypu jedince, navíc komplikovanou faktory epigenetickými a environmentálními. Přesto však bude možnost sekvenování lidských genomů znamenat začátek nové éry molekulární medicíny a umožní zásadní změny v diagnostice závažných onemocnění včetně onkologických.
Zkratky
AML – akutní myeloidní leukémie
CNV – copy number variations
ddNTPs – dideoxynukleotid-trifosfáty
TIGR – The Institute for Genomic Research
SNP – single-nucleotide polymorphism
SOLiD – Sequencing by Oligo Ligation and Detection
Práce byla podpořena granty IGA MZ ČR NR9293-3/2007, IGA MZ ČR NS10439-3/2009 a výzkumným záměrem MŠMT MSM0021622430.
Autoři děkují dr. O. Holeňovi, dr. P. Žákovi, dr. O. Menssingovi a dr. R. Zelenkovi za poskytnutí obrazového materiálu a cenné připomínky.
Adresa pro korespondenci:
RNDr. Šárka Pospíšilová, PhD.
Centrum molekulární biologie a genové terapie
Interní hematoonkologická klinika LF MU a FN Brno
Černopolní 9, 625 00 Brno
fax: +420 532 234 623, e-mail: sarka.pospisilova@fnbrno.cz
Sources
1. Moudrianakis EN, Beer M. A selective reagent for the study of base sequence in nucleic acids. Nature 1964; 204 : 685–686.
2. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 1977; 74 : 5463–5467.
3. Sanger F, Air GM, Barrell BG, et al. Nucleotide sequence of bacteriophage phi X174 DNA. Nature 1977; 265 : 687–695.
4. Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA 1977; 74 : 560–564.
5. Smith LM, Sanders JZ, Kaiser RJ, et al. Fluorescence detection in automated DNA sequence analysis. Nature 1986; 321 : 674–679.
6. Ansorge W, Sproat B, Stegemann J, et al. Automated DNA sequencing: ultrasensitive detection of fluorescent bands during electrophoresis. Nucleic Acids Res 1987; 15 : 4593–4602.
7. Bashkin JS, Bartosiewicz M, Roach D, et al. Implementation of a capillary array electrophoresis instrument. J Capillary Ellectrophoresis 1996; 3 : 61–68.
8. Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995; 269 : 496–512.
9. Adams MD, Celniker SE, Holt RA, et al. The genome sequence of Drosophila melanogaster. Science 2000; 287 : 2185–2195.
10. Weber JL, Myers EW. Human whole-genome shotgun sequencing. Genome Res 1997; 7 : 401–409.
11. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science 2001; 291 : 1304–1351.
12. Lander ES, Linton LM, Birren B, et al. International Human Genome Sequencing Consortium: Initial sequencing and analysis of the human genome. Nature 2001; 409 : 860–921.
13. The ENCODE Project Consortium: Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007; 447 : 799–816.
14. Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PloS Biol 2007; 5 : 254–286.
15. Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008; 452 : 872–877.
16. Kidd JM, Cooper GM, Donahue WF, et al. Mapping and sequencing of structural variation from eight human genomes. Nature 2008; 453 : 56–64.
17. Wang J, Wang W, Li R, et al. The diploid genome sequence of an Asian individual. Nature 2008; 456 : 60–65.
18. Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008; 456 7218 : 66–72.
19. Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet 2008; 24 : 133–141.
20. Pettersson E, Lundeberg J, Ahmadian A. Generations of sequencing technologies. Genomics 2009; 93 : 105–111.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Oboustranný spontánní pneumotorax – chybný léčebný postup
- Sekvenování lidského genomu – technologie nové generace aneb budeme rutinně sekvenovat lidské genomy?
- Eozinofily v gastrointestinálním traktu
- O lidském stárnutí a dlouhověkosti – 2. vnitřní podmínky