
Farmakogenetika v léčbě kardiovaskulárních chorob aneb léčba podle guidelines či podle potřeb nemocného?
Pharmacogenetics in cardiovascular diseases therapy – treatment according guidelines or according the individual requirement?
In clinical practice the individual response to a drug represents a great problem. Many of us experienced a situation when the drug effect declined or drug-related toxic reaction occurred. The typical example is the variation of optimal warfarin dosage. The main cause of interindividual differences in the drug response is the polymorphism in drug resorption and elimination systems, enzymes responsible for drug biotransformation, target receptors or signal molecules. The individualized drug therapy requires physicians to be oriented in main causes of pharmacokinetic and pharmacodynamic variations and be able to predict accurately the individual drug response.
Key words:
polymorphism, clopidogrel, warfarin, betablockers, drug metabolism, transport protein, pharmacogenetics.
Authors:
Jan Bultas
Authors‘ workplace:
Univerzita Karlova V Praze, 3. lékařská fakulta, Ústav farmakologie
Published in:
Čas. Lék. čes. 2010; 149: 476-481
Category:
Review Article
Overview
V praxi se občas setkáváme s tím, že u některých nemocných léčba určitým lékem selhává a naopak u jiného se objeví nečekaně toxická reakce. Typickým příkladem je, že u některých nemocných nedosáhneme účinného nastavení antikoagulace warfarinem ani při dávkách nad 15 mg denně a naopak u jiných se setkáváme s krvácením již při dávce 1,5 mg. Jedním z hlavních důvodů odlišné terapeutické odpovědi jsou velké interindividuální rozdíly ve vstřebávání, metabolismu a ve vylučování léku, stejně jako ve vlastní odpovědi na lék na úrovni efektoru. Podkladem těchto rozdílů bývá polymorfismus enzymů, transportních systémů zajišťujících osud a účinek léků v organismu i receptorů, enzymů či signálních molekul ovlivňujících vlastní účinek léčiva. Dříve, než zaškrtneme své „recipe“, je dobře být orientován o tom, jak farmakogenetické odchylky ovlivňují individuální odpověď na léčbu.
Klíčová slova:
polymorfismus, klopidogrel, warfarin, betablokátory, izoenzymy CYP, transportní proteiny, farmakogenetika.
ÚVOD
Málokterý obor prodělal v posledních dvou desetiletích takový rozvoj jako kardiologie. Velký podíl na zlepšení naší léčebné strategie mělo zavedení medicíny založené na důkazech a převedení výsledků do praxe ve formě doporučených postupů. Skutečně, nedávná analýza výsledků dodržování těchto zásad ukázala, že důsledné aplikování těchto poznatků – například v léčbě akutních koronárních příhod – vede k poklesu mortality zhruba na třetinu (RR 0,31, 95% Cl: 0,17–0,57), farmakoterapie se pak na tomto poklesu podílí neméně polovinou (1). Studie dokumentovala, že čím důslednější byla léčba, tím větší prospěch přinesla nemocnému. Z tohoto pohledu je pochopitelný tlak na dodržování doporučených léků a plošné aplikování doporučených dávek, na všech kongresech jsme k této praxi od předsednických stolů vyzýváni.
Nicméně nic není tak jednoduché, jak se na prvý pohled zdá – ne každý lék je vhodný pro každého a ještě méně platí, že doporučená dávka je pro každého nemocného tou dávkou optimální. My všichni včetně našich nemocných se navzájem významně lišíme. Díky polymorfismu se lišíme na úrovni resorpce léku, jeho biotransformace, bioeliminace, transportu a distribuce – tedy v procesech ovlivňujících farmakokinetiku. Méně si však uvědomujeme, že se lišíme i ve vlastní odpovědi na lék – tedy v odpovědi farmakodynamické. Příkladem jsou interindividuální a dokonce intraindividuální rozdíly na úrovni receptoru (tedy jeho exprese, afinity k ligandu, poměru aktivace a inhibice), na úrovni aktivity sdružených regulačních pochodů, či v odpovědi efektoru na vlastní stimulaci. Podobně, působíme-li farmakologicky na enzym je odpověď modifikována opět expresí, afinitou k substrátu, vlastní katalytickou aktivitou apod. Tyto rozdíly ve farmakogenetické výbavě vedou k odlišné odpovědi na léčbu, zejména však k rozdílné odpovědi na danou dávku léčiva. Z těchto důvodů se stále častěji i v kardiologii prosazuje individiální pohled na pacienta i na jeho léčbu, čili prosazuje se personalizovaná medicína beroucí ohled na genotyp i fenotyp léčeného.
V řadě případů již individuální rozdíly v odpovědi na léčbu respektujeme. Půl století zkušeností s warfarinem nás naučilo plně respektovat významné rozdíly v dávkování, někdo potřebuje dávku 1,5 mg denně, jiný 15 mg denně, i zde stojí v pozadí farmakogenetika. V případě warfarinu tyto rozdíly nikoho nepřekvapí, obdobná situace však je například u lipofilních blokátorů ß-adrenergních receptorů, u antiarytmik – zejména propafenonu, a jak ukážeme, též u některých protidestičkových léků. Bohužel odlišnosti v farmakogenomické výbavě neurčují pouze terapeutický efekt, významně je ovlivněn i tolerance léku. Podívejme se proto, jaké jsou příčiny, jaký je klinický dopad a jak těmto interindividuálním rozdílům v účinku léku čelit.
Příčiny interindividuálních rozdílů v odpovědi na lék
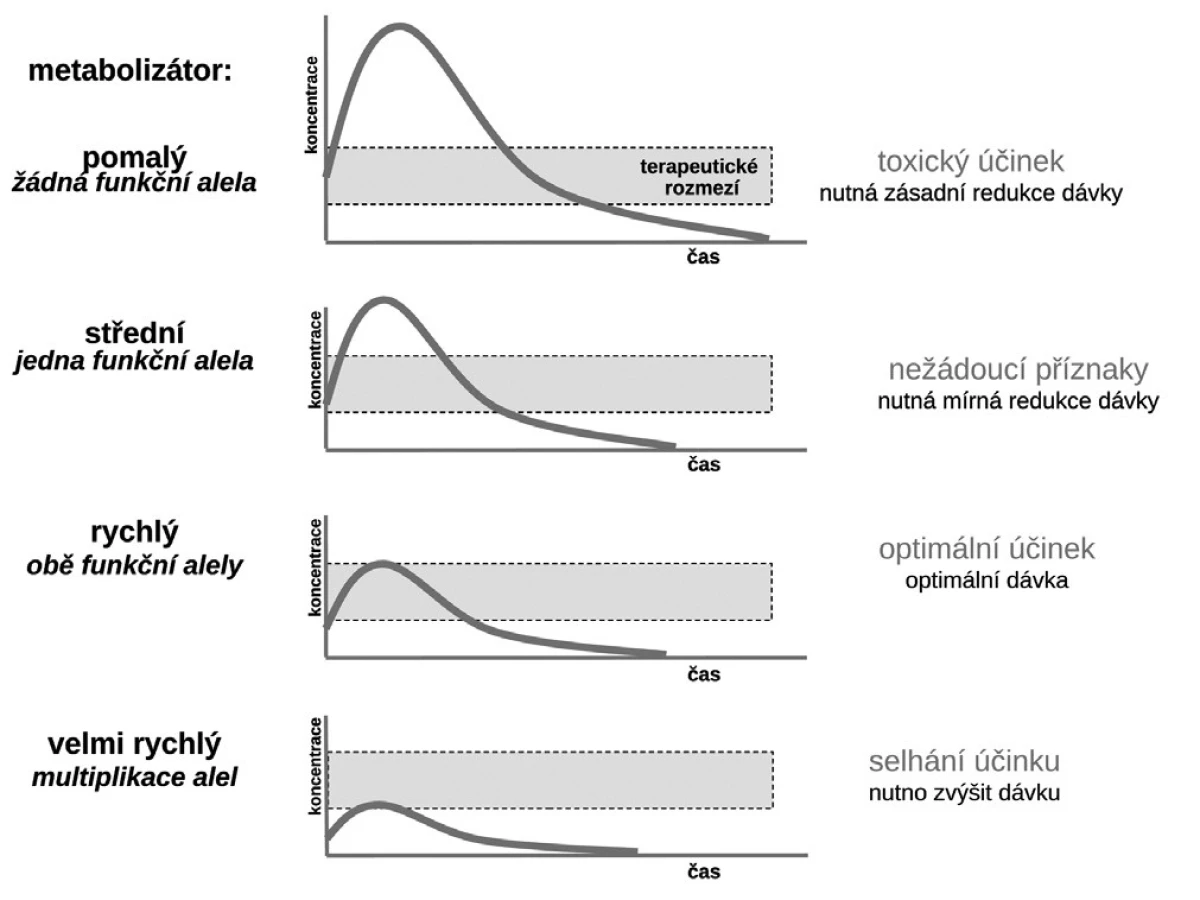
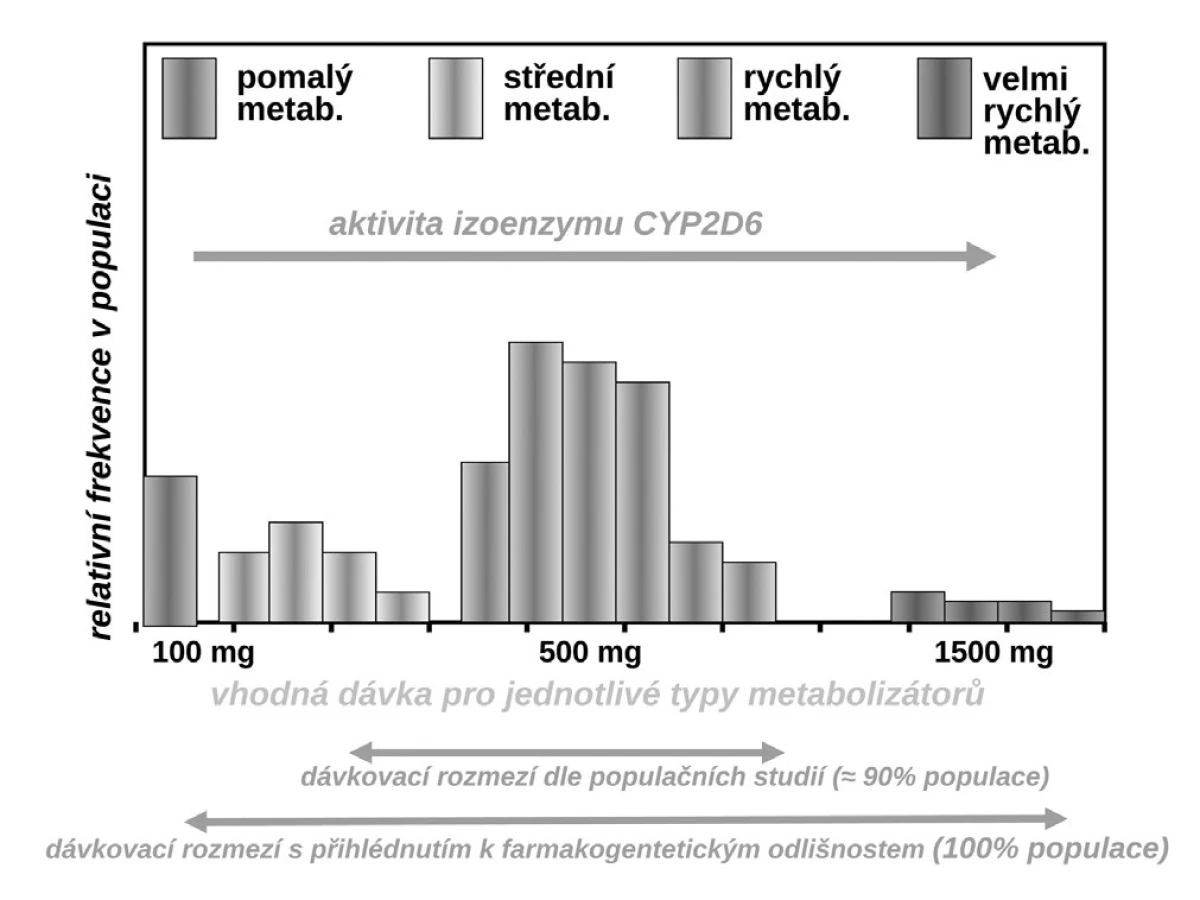
Budeme-li mluvit o interindividuálních rozdílech v odpovědi na lék, shrňme krátce naše znalosti o základních transportních a biotransformačních systémech. Prvý systém, se kterým se lék po perorálním podání setká, je systém transportních membránových proteinů umožňující jak vstup do enterocytu, tak jeho eliminaci (obr. 1a). Již ve vlastním enterocytu molekula podléhá biotransformaci zejména izoenzymy systému oxidáz CYP. Obdobné transportní proteiny umožňují přestup z enterocytu do kapilár portálního oběhu či z krve zpět do enterocytu. Stejná situace se opakuje na úrovni hepatocytu s tím rozdílem, že hepatální mikrozomy jsou v biotransformaci a bioeliminaci aktivnější, velká část zejména lipofilních léčiv bývá přímo eliminována do žluči (obr. 1b). Frakce léčiva, která se objeví v systémové krvi, může dosahovat jen několika mála procent, či se může blížit 100 %. Dalším klíčovým orgánem eliminace a inaktivace léčiv jsou ledviny, zejména epitelie renálních tubulů. I ty jsou osazeny výkonnými transmembranózními transportními systémy i metabolickými systémy (obr. 1c). Na všech úrovních, tj. enterocytu, hepatocytu, epitelií tubulárního systému ledvin, endotelií cév či neuroglií, probíhá intenzivní přeměna cizorodých látek (xenobiotik), tedy i léků s cílem jejich inaktivace a eliminace. Většina těchto systémů (s významnou výjimkou klíčového izoenzymu CYP3A4), je polymorfních a významná část populace pak patří na úrovni daného enzymu k rychlým (při obou funkčních alelách), středním (při jedné funkční alele) či pomalým metabolizátorům (při obou afunkčních alelách). Při multiplikaci alel, s nimiž se setkáváme u CYP2D6, bývá ultrarychlý metabolizátor (obr. 2). Analogický vztah je v oblasti transportních systémů. Tyto rozdíly pak stojí v pozadí za skutečně významnými a často život ohrožujícími odchylkami na úrovni farmakokinetiky léků. Většina léků působí aktivaci či inhibici konkrétního receptoru či enzymu. Též na této úrovni se setkáváme s významnými polymorfismy měnícími odpověď na lék (obr. 1d). Nicméně, vzhledem k provázanosti zpětnovazebnými kontrolami a dalšími kontrolními mechanismy, se tyto polymorfismy na úrovni receptorů a enzymů uplatní významněji až při postižení systému na více etážích.



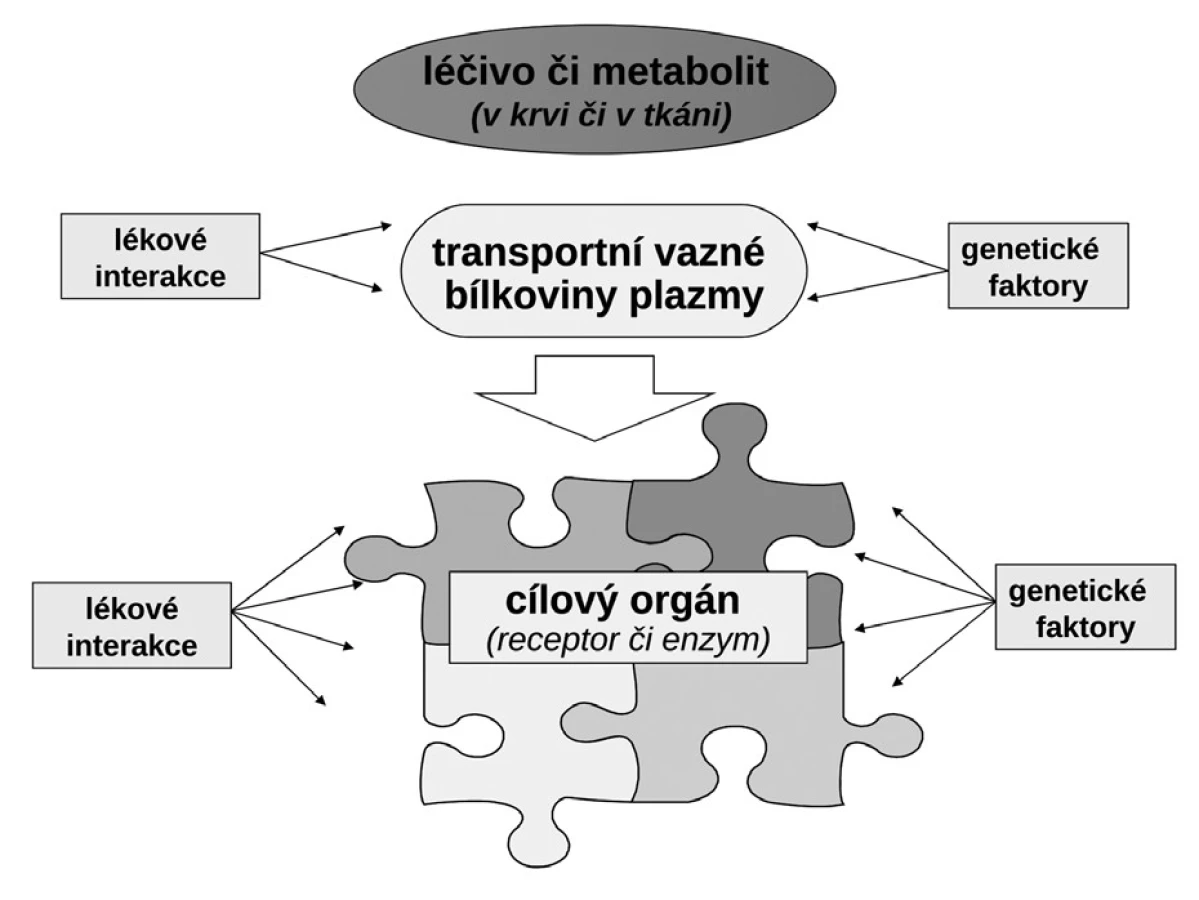
Interindividuální rozdíly na úrovni polymorfismu transportních a metabolických systémů
Prvým příkladem, kdy polymorfismus ovlivní expozici léčivem s přímým dopadem na prognózu je vztah expozice digoxinu a efluxní pumpy glykoproteinu P (P-gp). Digoxin má úzké terapeutické okno, již o málo vyšší koncentrace může navodit závažné poruchy rytmu. Asi u čtvrtiny populace bývá přítomen polymorfismus genu řídícího expresi P-gp ve střevě C3435T spojený s nízkou aktivitou P-gp. Tato nízká aktivita vede ke zvýšenému vstřebávání digoxinu se vzestupem jeho koncentrace a s rizikem zahlcení kardiomyocytu kalciem a zvýšeného rizika vzniku život ohrožujících arytmií (2). Stanovování hladiny digoxinu 2–3 týdny po nasazení digoxinu a úprava dávky podle zjištěných hodnot by měla být pravidlem.
Druhým, daleko komplikovanějším příkladem polymorfismu ovlivňujícím prognózu nemocných, je soubor polymorfismů zodpovědných za resorpci a bioaktivaci klopidogrelu. Klopidogrel, inhibitor destičkových ADP receptorů P2Y12, je důležitý lék snižující riziko aterotrombotické příhody – infarktu myokardu, iktu či úmrtí. Opakovaně bylo doloženo, že část nemocných nereaguje na klopidogrel optimálně, či je dokonce k léčbě rezistentní. Příčin je více, společným jmenovatelem je však nedostatečná hladina klopidogrelu potřebná k účinné inhibici trombocytů. S touto situací, při podávání doporučených dávek, se setkáváme u čtvrtiny až třetiny populace.
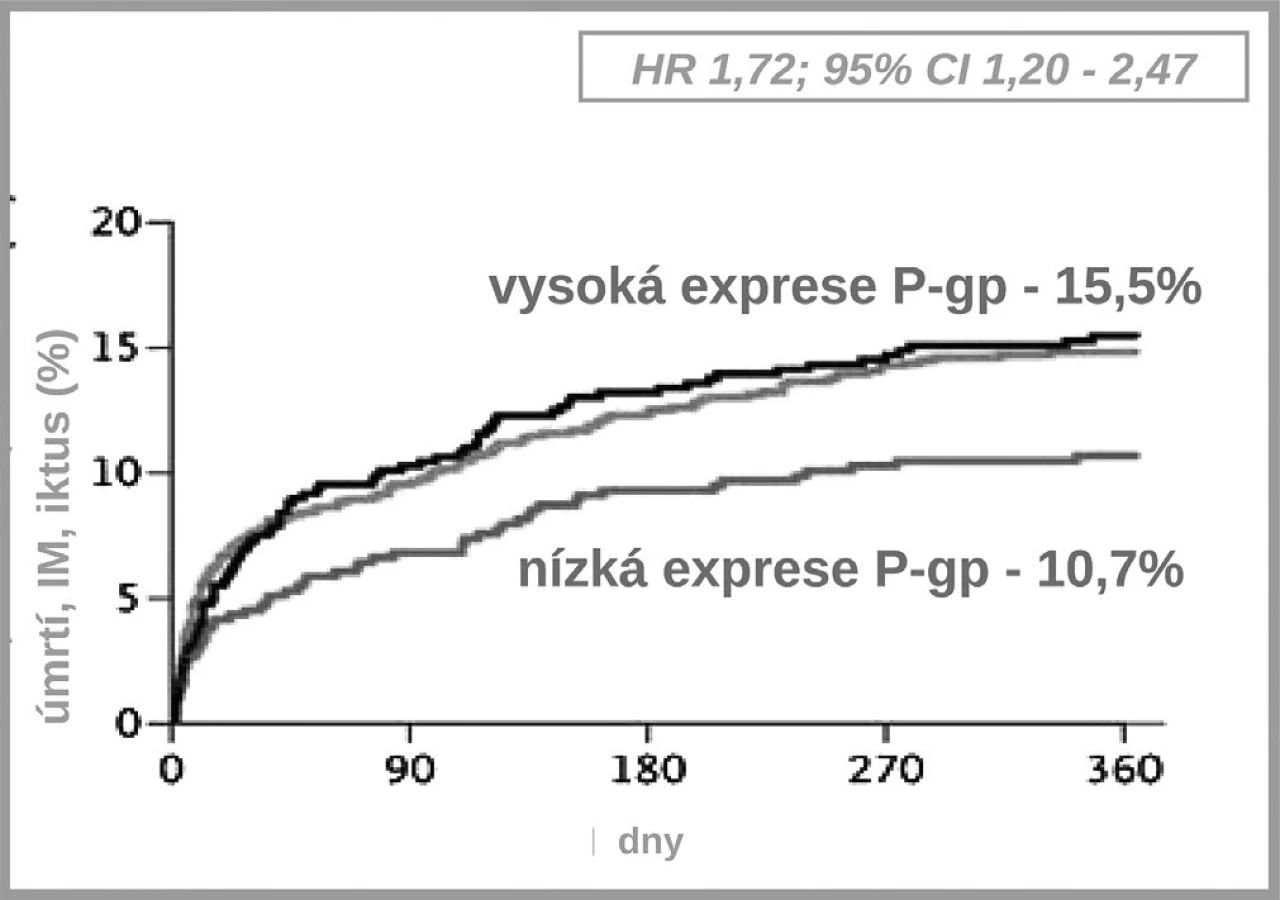
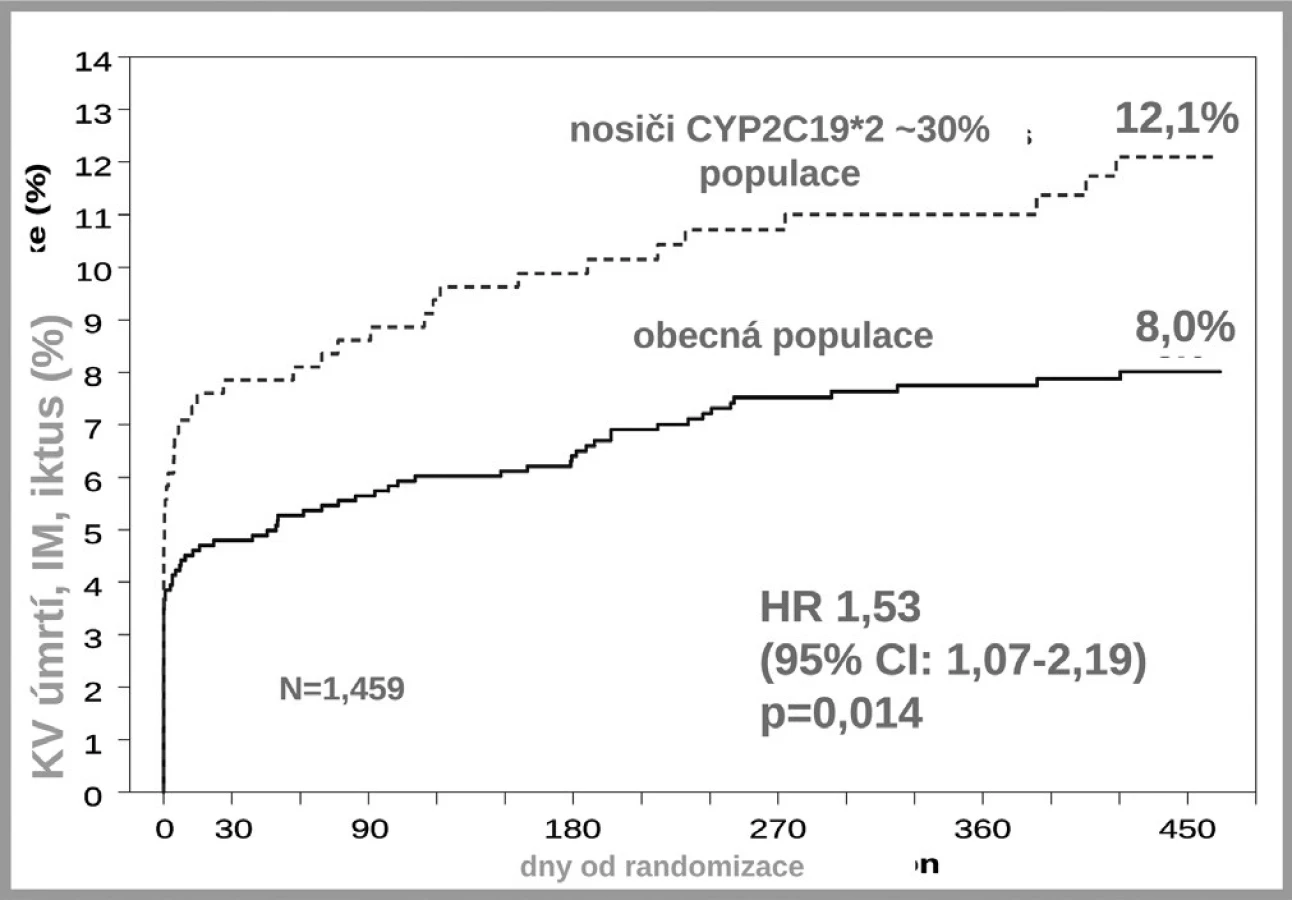
Jak ukázuje obrázek 3, v bioaktivaci klopidogrelu je několik klíčových kroků. Prvým je resorpce proléčiva v enterocytu. Ta může být významně ovlivněna aktivitou transportéru – glykoproteinu P. Asi u čtvrtiny populace, nosičů genotypu 3435TT, se setkáváme s vysokou expresí P-gp a s významně nižší dostupností klopidogrelu. Jak ukázala řada studií, tato vyšší exprese přináší nižší dostupnost klopidogrelu, doporučená dávka je nedostatečná a výsledkem je vyšší riziko příhody. Analýza registru nemocných s akutními koronárními příhodami FAST-MI nalezla vzestup rizika příhody o 72 % (HR 1,72; 95% CI 1,20–2,47) pro nemocné s genotypem 3435TT (3) (obr. 4). Zcela stejné zvýšení rizika, tj. o 72 % (HR 1,72; 95% CI 1,22 –2,44) nalezla recentní post-hoc analýza studie TRITON-TIMI 38 (4).
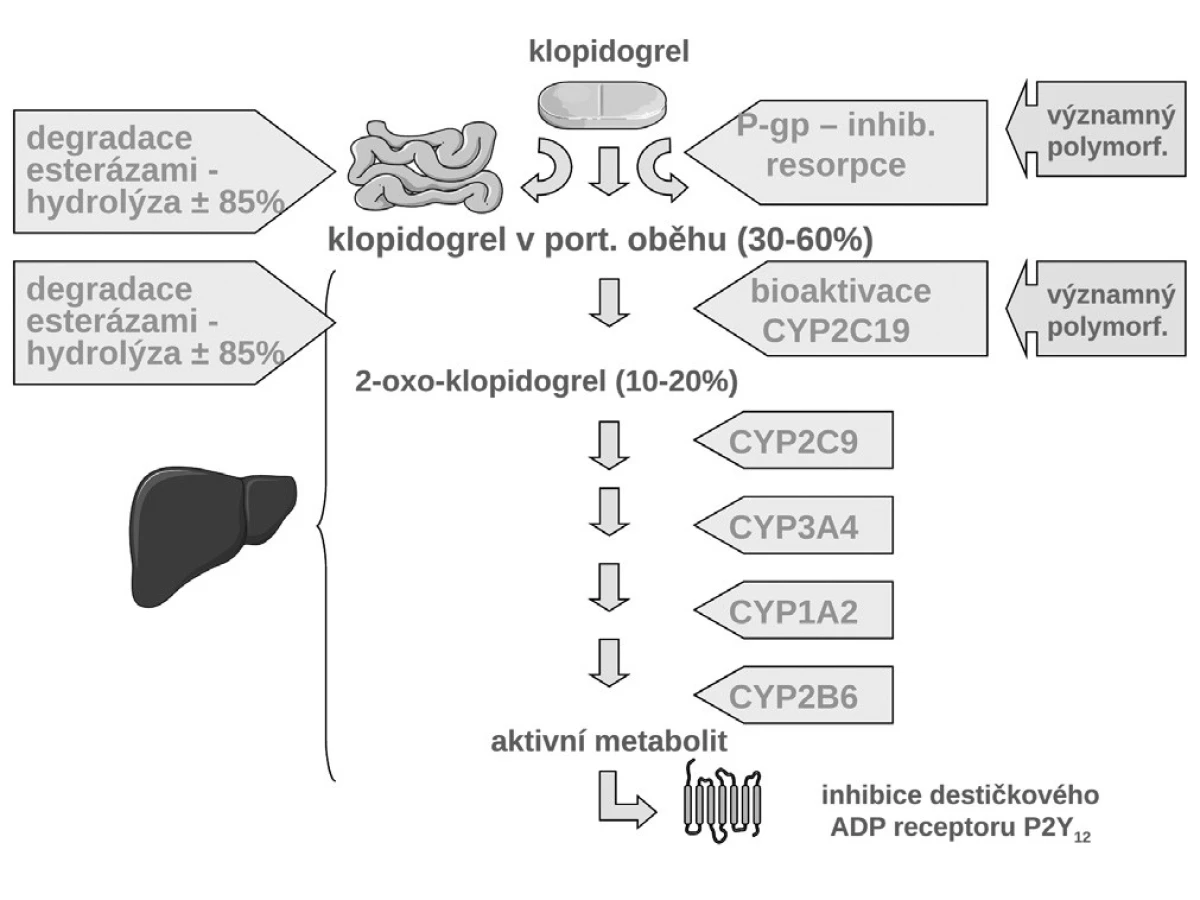
Druhým důvodem nedostatečné inhibice trombocytů po doporučených dávkách klopidogrelu je nedostatečná biokonverze klopidogrelu na vlastní aktivní metabolit. Klopidogrel, stejně jako ostatní thienopyridinové inhibitory ADP receptorů, je podáván pro lepší resorpci jako proléčivo. Za fyziologické situace je asi 85 % proléčiva degradováno esterázami a pouze 15 % je aktivováno na účinný metabolit. V této skutečnosti je kámen úrazu, přeměna na aktivní metabolit neproběhne vždy tak, jak bychom očekávali – podkladem může být polymorfismus konvertujících enzymů, či inhibice konverze některými léky. Enzymů nutných k bioaktivaci je více, klíčovým však je polymorfní oxidáza CYP 2C19. U nositelů alely CYP2C19*2 nebo CYP2C19*3, probíhá konverze velmi pomalu, významně větší část proléčiva je degradována na neúčinné metabolity a efekt klopidogrelu selhává. Jak ukázala farmakogenetická analýza studie TRITON-TIMI-38, u nemocných s alespoň jednou defektní alelou pro tento izoenzym, vyskytující se u 25–30 % indoevropské populace, se objevilo o polovinu (HR 1,53; 95% CI 1,07–2,19) více závažných kardiovaskulárních příhod (KV úmrtí, infarkt či iktus); trombóz po implantaci stentu bylo dokonce 3× více než u zbytku populace s funkčním izoenzymem (5) (obr. 5). Tento nález byl potvrzen recentní analýzou registru myokardiálních infarktů ve Francii (3). Na druhé straně vedle polymorfismů snižujících aktivitu CYP2C19 jsou známy polymorfismy zvyšujících aktivitu oxidázy. Jeden z nejčastějších, objevující se asi u třetiny populace, je polymorfismus CYP2C19*17. V analýze studie CURE zvýšila přítomnost tohoto genotypu efekt léčby klopidogrelem, u nositelů tohoto typu zvyšujících konverzi klopidogrelu na aktivní metabolit byl rozdíl ve výskytu závažných kardiovaskulárních příhod po akutní koronární příhodě mezi placebem a klopidogrelem plných 45 %, naproti tomu u nosičů ostatních typů pouze 15 % (6). Dá se tak soudit, že většina účinku klopidogrelu na pokles mortality/morbidity byla koncentrována do skupiny velmi rychlých metabolizátorů na úrovni izoenzymu CYP2C19.
Jak čelit těmto významným rozdílům v účinku klopidogrelu na úrovni farmakogenetiky? Jednou z možností je farmakogenetické testování. To je sice schůdné, na vybraných pracovištích se provádí, nicméně pro praxi je realizace v současné době obtížná. Další cestou je zdvojnásobení dávky klopidogrelu, dosáhneme tak účinné inhibice aktivace a agregace trombocytů u většiny nemocných. Konečně se nabízí užití blokátorů destičkových ADP receptorů nezávislých na farmakogenetických rozdílech v aktivitě P-gp a izoenzymu CYP2C19. I tato cesta je schůdná, meta-analýza účinku čtyř nových inhibitorů – prasugrelu, ticagreloru, cangreloru a elinogrelu na pokles mortality u nemocných po koronární intervenci o 17% (RR 0,83; 95% CI 0,75–0,92; p < 0,001) a pokles výskytu trombóz v oblasti stentu byl plných 40 % ve srovnání s klopidogrelem v obvyklém dávkování (7). Výskyt krvácení se paralelně zvýšil o 23 %, což reflektuje efekt u většího spektra nemocných, farmakodynamický účinek je totiž u všech léků srovnatelný.
Mluvíme-li o protidestičkových lécích, pak nelze opominout, že farmakogenetické rozdíly ovlivňují i výskyt nežádoucích účinků. Uveďme jen jeden příklad z analýzy studie PRAGUE-8 u nemocných po koronární angiografii léčených typicky acetylsalicylovou kyselinou, ale neužívajících klopidogrel (8). Při sledování řady trombocytárních polymorfismů měly vztah k výskytu krvácivých komplikací pouze dvě varianty, obě na úrovni cyklo-oxygenázy-1 (COX-1(-842A>G a 50C>T, p = 0,013). Oba polymorfismy ovlivňují syntézu eikosanoidů – tromboxanu A2 a řady prostanoidů. Autoři uzavírají, že farmakogenetické testování může zvýšit bezpečnost intervenčních výkonů.
Příkladů dokumentujících klinickou závažnost polymorfismů měnících farmakokinetické vlastnosti systému můžeme nalézt stovky, řada z nich je velmi závažných. Vzpomeňme jen často užívané antiarytmikum – propafenon. Ten je metabolizován stejně jako řada jiných antiarytmik velmi polymorfním izoenzymem CYP2D6. Tato oxidáza, transformující asi čtvrtinu běžně užívaných léků včetně lipofilních ß-blokátorů, má největší počet klinicky významných polymorfismů. Část populace (kolem 5–10 %) jsou pomalí metabolizátoři – ti běžné dávky propafenonu nestačí degradovat, lék se kumuluje a objevují se toxické příznaky. Většina, tj. 85–90 % indoevropské populace, jsou středními či rychlými metabolizátory a pro ty běžné léčebné dávky vyhovují. Naopak pro velmi rychlé metabolizátory s multiplikací genu je dávka nedostatečná a léčba selhává. Z populačního hlediska sice standardní dávka (300–600 mg propafenonu denně) vyhovuje, pro prvý typ nemocných by však bylo potřeba volit dávku kolem 100 mg denně, naopak pro druhý extrém dávku o řád vyšší (9).
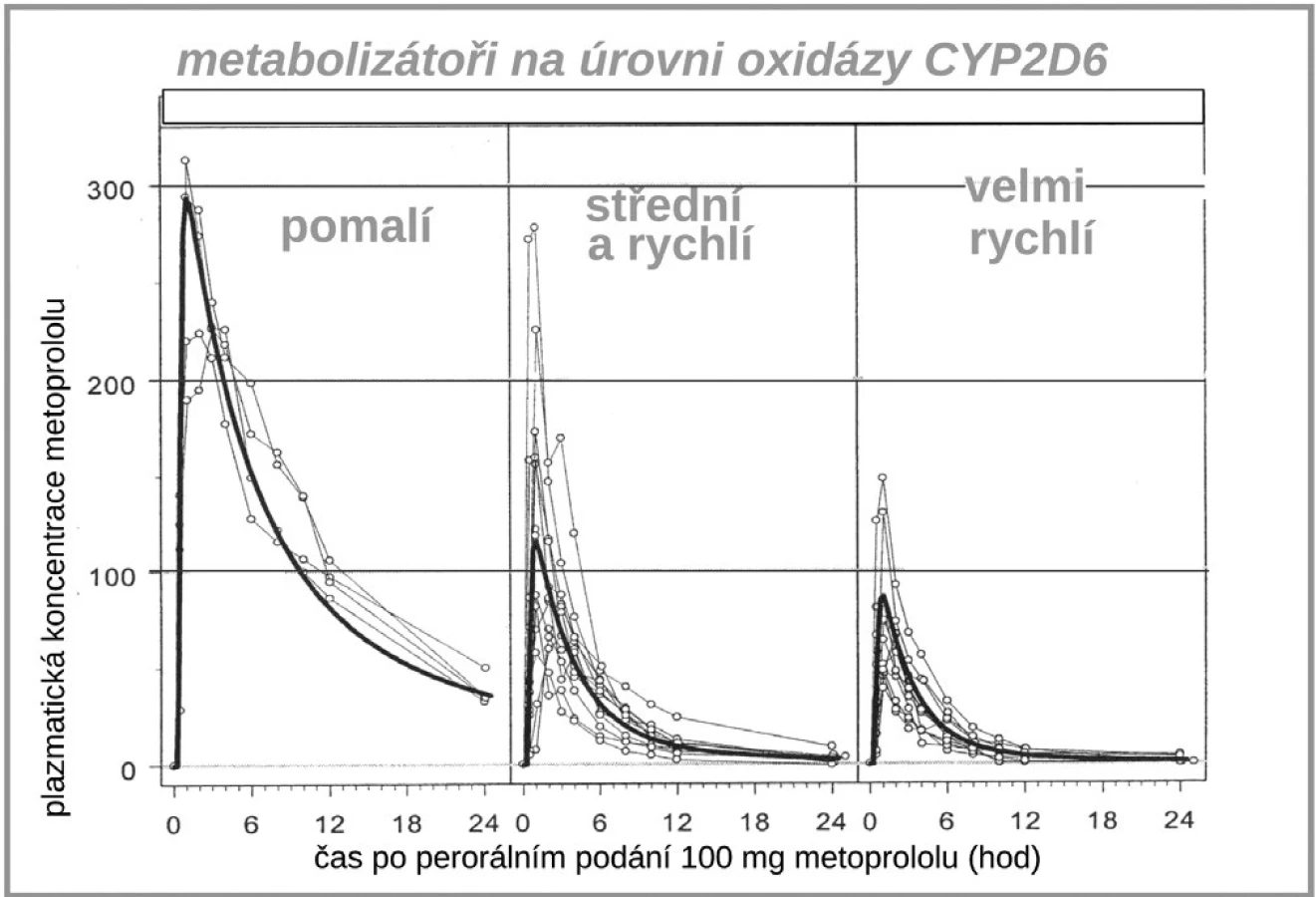
Dalším zajímavým příkladem, kdy farmakogenetika významně ovlivňuje farmakokinetické vlastnosti léku je vztah lipofilních ß-blokátorů, tj. metoprololu, karvedilolu či nebivololu k polymorfismu již diskutovaného izoenzymu CYP2D6. Díky rozdílné aktivitě této oxidázy se setkáváme s velmi variabilní hladinou lipofilních ß-blokátorů (10, 11). V populaci se objevují až desetinásobné interindividuální rozdíly koncentrace metoprololu, nebivololu či karvedilolu, zatímco u hydrofilních blokátorů (např. bisoprololu či betaxololu) jsou rozdíly jen dvoj - až trojnásobné (obr. 6). U pomalých metabolizátorů s velmi vysokou expozicí lipofilních ß-blokátorů je též několikanásobně zvýšen výskyt nežádoucích účinků (12). Naštěstí u ß-blokátorů si lehce s léčebnou dávkou poradíme, stačí se orientovat podle srdeční frekvence, horší to je u výše popsaného dopadu polymorfismu CYP2D6 na expozici antiarytmiky.
U pomalých metabolizátorů CYP2D6 se můžeme setkat také s opačným fenoménem. Tento enzym je nutný k transformaci proléčiva kodeinu na analgeticky působící morfin. Podobná situace je u tramadolu, i zde je nutná biotransformace k zajištění analgetického efektu. U pomalých metabolizátorů účinek těchto léků bohužel selhává, nevytvoří se dostatečná hladina aktivního metabolitu. Uvědomíme-li si, že z čistě genetické příčiny asi 10 % nemocných metabolizuje buď překotně, či nedostatečně řadu významných lékových skupin, vedle ß-blokátorů či antiarytmik též psychofarmaka, znamená to, že asi milion osob v České republice můžeme považovat z hlediska vhodného dávkování léků transformovaných CYP2D6 za jaksi „zapomenutou“ populaci. Podobná situace je i u jiných transportních a metabolických systémů.
Interindividuální rozdíly na úrovni polymorfismu cílových struktur (receptorů, enzymů apod.)
Druhou a stejně významnou oblastí jsou změny dané nikoli změněnými farmakokinetickými vlastnostmi, tedy odlišnou expozicí léku či aktivního metabolitu, ale rozdílnou odpovědí organismu na léčivo. Řada cílových struktur – zejména receptorů a enzymů – je polymorfních, tj. odpovídají a působení léku s větší či s menší intenzitou (obr. 1d). Tak se v populaci objevuje velká mozaika jedinců s různou citlivostí k léku.
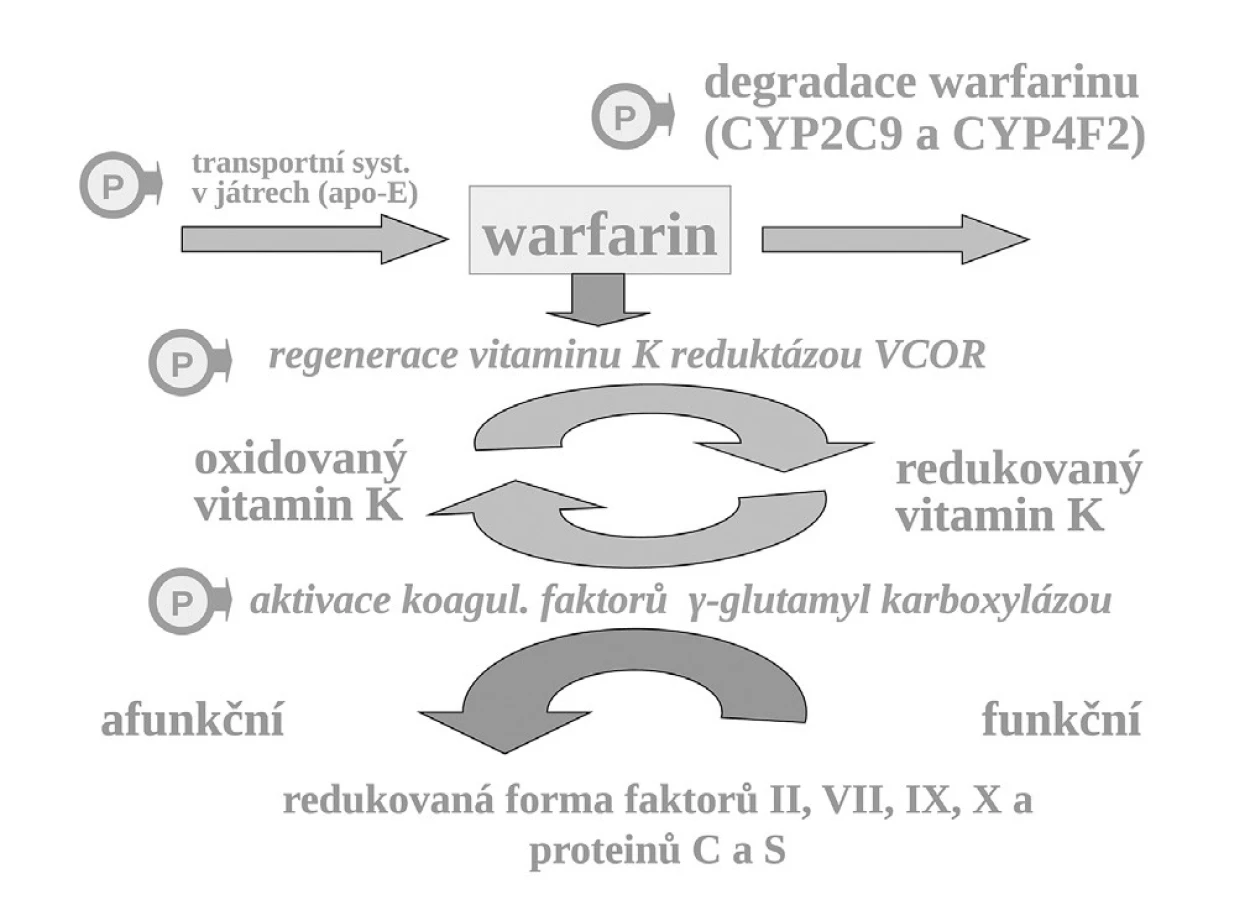
Příkladem polymorfismu na úrovni změněné vnímavosti i změněné expozice je léčba antikoagulanciem warfarinem. K aktivaci koagulačních faktorů je potřeba redukovaného vitaminu K, který přechází do formy oxidované a tato redukovaná forma je regenerována pomocí vitamin K-reduktázy (VKOR). Vlastní redukce koagulačních faktorů je zprostředkována enzymaticky – γ-glutamyl karboxylázou (GGCX) (obr. 7).
Warfarin, stejně jako ostatní antivitaminy K, inhibuje regeneraci oxidovaného vitaminu K blokádou tohoto enzymu. VKOR, resp. jeho podjednotka C1 je polymorfní a asi u čtvrtiny populace je více citlivá k inhibici antivitaminy K. Nositelé alelické varianty VKORC1AA konstituují výslednou subpopulaci warfarin-senzitivní a na druhém pólu stejná část jedinců s variantou VKORC1BB je k působení warfarinu méně citlivých, tj. subpopulace warfarin-rezistentní. Nositelé smíšeného genotypu VKORC1AB, tvořících asi 50 % populace, jsou středně citliví. Takto máme vedle sebe tři skupiny s rozdílnou odpovědí na lék. Nezávisle na přítomnosti dalších polymorfismů ovlivňujících účinek warfarinu, pro genotyp senzitivní se účinná dávka pohybuje kolem 3 mg denně, pro rezistentní kolem 6 mg denně (13). Vedle polymorfismu VCORC1 hraje v citlivosti k warfarinu též aktivita γ-glutamyl karboxylázy, výsledná citlivost k warfarinu však je polymorfismem významně méně ovlivněna (14). Rovněž další polymorfismy, např. v apolipoproteinu E (apo-E), kontrolující transport vitaminu K a warfarinu v játrech, mají klinicky relativně malý význam.
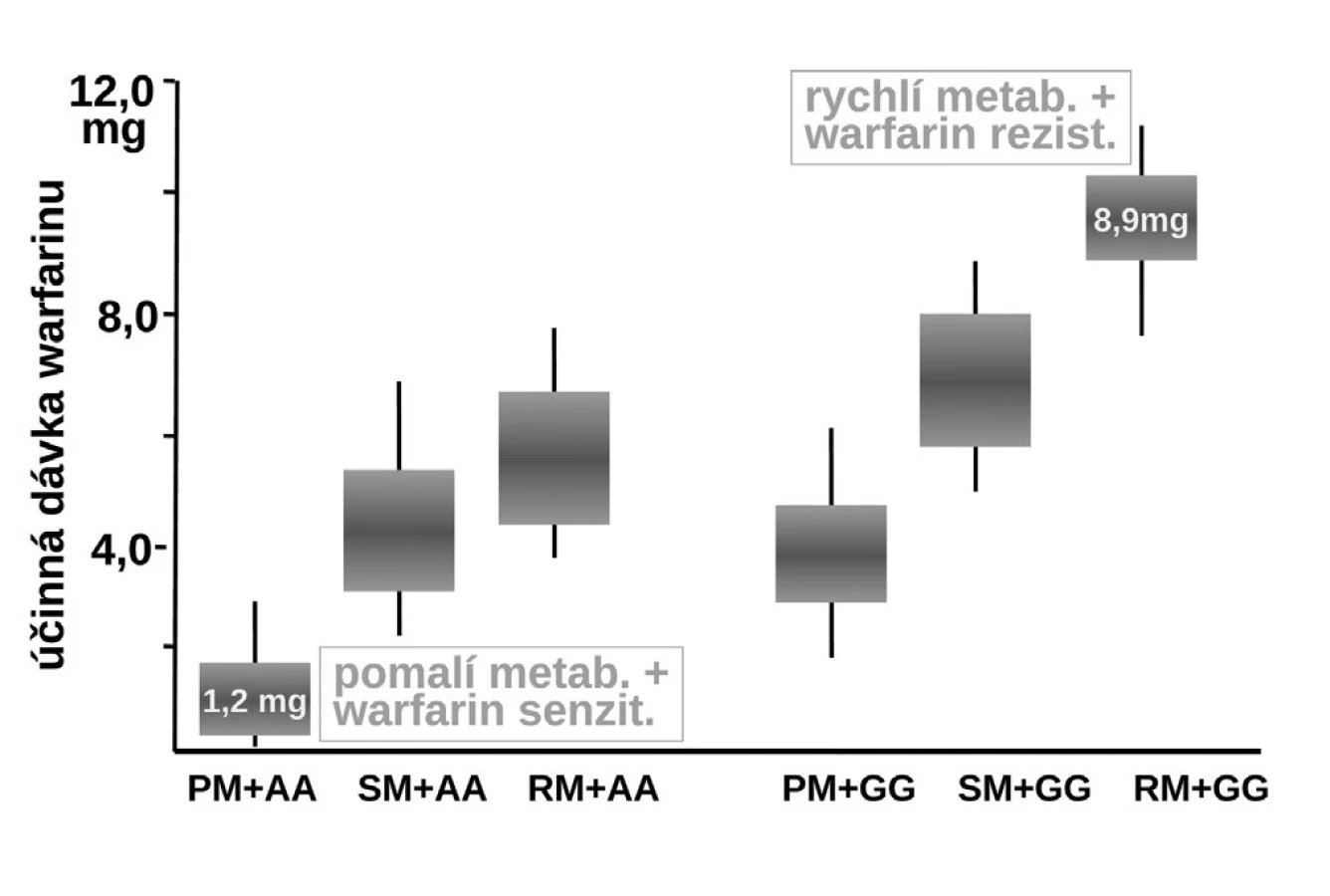
Naproti tomu, zcela zásadně ovlivňuje efekt warfarinu jeho metabolismus. Warfarin je transformován a inaktivován izoenzymem CYP2C9 a CYP4F2. Oba izoenzymy mohou být polymorfní. Hlavní význam má však oxidáza CYP2C9, která u části populace může být téměř afunkční – typ CYP2C9*1*1 – pomalí metabolizátoři či naopak hyperaktivní, s genotypem CYP2C9*2*2 nebo CYP2C9*1*3, čili rychlí metabolizátoři. U prvých stačí k plnému antikoagulačnímu účinku dávky warfarinu nízké, pro druhý extrém je potřeba volit dávky vysoké. Sejde-li se však současně genotyp pomalého metabolizátora a současně genotyp „senzitivní“, pak odpovídající léčebná dávka se pohybuje kolem 1–2 mg denně či může dokonce klesnout i pod 1 mg warfarinu denně (obr. 8). Naopak druhým extrémem je koincidence „rezistentního“ genotypu a současně rychlého metabolizátora. Zde musíme zvýšit dávku až k 10 či 15 mg warfarinu denně (15–17). Ve výsledné dávce warfarinu se uplatní k řadě dalších faktorů, význam mají lékové interakce a příjem vitaminu K v potravě. Nicméně genetické faktory zahrnující VKORC1 a izoenzymy CYP2C9 a CYP5F2 vysvětlují asi dvě třetiny interindividuálních rozdílů v odpovědi na warfarin.
Podobné rozdíly v účinnosti jiných léčiv nejsou v kardiologii naštěstí tak běžné, nicméně zmiňme jen krátce několik zajímavých polymorfismů s praktickým dopadem. Prvým jsou polymorfismy adrenergního receptoru ß a α, které ovlivňují odpověď na blokátory těchto receptorů. Pro pochopení vztahu nutno uvést základní schéma kontroly. Presynaptický receptor α tlumí systém inhibicí výdeje katecholaminů, naopak postsynaptické receptory ß1 a ß2 jsou aktivační. Hyperaktivita receptoru aktivačního je kompenzována recipročním útlumem systému díky stimulací presynaptického receptoru inhibičního. Objeví-li se však souhra dvou polymorfismů na tomto receptoru, jednoho snižujícího činnost presynaptického receptoru α a druhého zvyšujícího aktivitu postsynaptického ß, dojde ke klinicky významné hyperaktivaci sympatiku, zhoršení prognózy kardiovaskulárních chorob a ke zvýšenému uplatnění ß-blokátorů. Takovou situací je například kombinace presynaptického receptoru α2c (Del32-325) s nižší inhibiční aktivitou a postsynaptického ß1 (Arg389Gly) s vyšší stimulační aktivitou (18). Podobné polymorfismy v aktivitě systému renin-angiotenzin-aldosteron – například na úrovni angiotenzinogenu, angiotenzin konvertujícího enzymu, na úrovni receptoru AT1, receptoru MAS či receptorů kininových – ovlivňují nejen výskyt hypertenze, průběh diabetu či prognózu řady kardiovaskulárních onemocnění, ale i odpověď na léčbu inhibitory ACE, sartany či inhibitory reninu. Konečně též v léčbě dyslipidémií se uplatňují polymorfismy jak v odpovědi na hypolipidemika, tak ve výskytu nežádoucích účinků. Zde však jsou vztahy výrazně složitější a jejich rozbor přesahuje možnosti této práce.
Stále tak platí slova klasika: „Léčíme chorobu, o které víme velmi málo, podáváme lék, o kterém víme ještě méně, a to vše u pacienta, o kterém nevíme téměř nic.“ Bohužel, ještě po 200 letech tato slova platí – v převážné většině o vlastním nemocném toho skutečně víme žalostně málo. Jeho farmakogenetická výbava nám dosud zůstává zpravidla neodhalena. Přitom, jak je tomu například u zmíněného klopidogrelu či u warfarinu, farmakogenetické testování je již rutinně dostupné, jen je musíme u indikovaných nemocných využít. Pomalu se začínají naplňovat slova žijícího „klasika“ – kardiologa profesora Eugena Braunwalda, který před 10 lety právě uvedení poznatků farmakogenomiky do praxe pokládá za největší výzvu medicíny.
Zkratky
GGCX – γ-glutamyl karboxyláza
P-gp – glykoprotein P
VKOR – vitamin K-reduktázy
Adresa pro korespondenci:
prof. MUDr. Jan Bultas, CSc.
Farmakologický ústav 3. LF UK
Ruská 87, 100 00 Praha 10
e-mail: jbult@lf1.cuni.cz
Sources
1. Chew DP, et al. Six-month survival benefits associated with clinical guideline recommendations in acute coronary syndromes, Heart published online June 7, 2010.
2. Hoffmeyer S, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA 2000; 97 : 3473–3478.
3. Simon T, et al. French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators: Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360(4): 363–375.
4. Mega JL, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. The Lancet, Early Online Publication, 29 August 2010.
5. Mega JL, et al. Cytochrome P-450 Polymorphisms and Response to Clopidogrel. N Engl J Med 2009; 360(4): 411–413.
6. Paré G, et al. Effects of CYP2C19 genotype on clopidogrel treatment in CURE and ACTIVE. N Eng J Med 2010; Early Online Publication August 2010.
7. Bellemain-Appaix A, et al: New P2Y12 inhibitors versus clopidogrel in percutaneous coronary intervention. J Am Coll Cardiol 2010; Early Online Publ., 30 August 2010.
8. Motovska Z, et al. Platelet gene polymorphisms and risk of bleeding in patients undergoing elective coronary angiography: Agenetic substudy of the PRAGUE-8 trial, Atherosclerosis 2010, journal homepage: www.elsevier.com/locate/athero sclerosis.
9. Jazwinska-Tarnawska E, et al. The influence of CYP2D6 polymorphism on the antiarrhythmic efficacy of propafenone in patients with paroxysmal atrial fibrillation during 3 months propafenone prophylactic treatment. Int J Clin Pharmacol Ther 2001; 39(7): 288–292.
10. Seeringer A, et al. Enantiospecific pharmacokinetics of metoprolol in CYP2D6 ultra-rapid metabolizers and correlation with exercise-induced heart rate. Eur J Clin Pharmacol 2008; 64(9): 883–888.
11. Kirchheiner J, et al. Impact of the ultrarapid metabolizer genotype of cytochrome P450 2D6 on metoprolol Pharmacokinetics and Pharmacodynamics*Impact of the ultrarapid metabolizer genotype of Cytochrome P450 2D6 on metoprolol Pharmacokinetics and Pharmacodynamics, Clinical Pharmacology & Therapeutics 76, 302–312.
12. Wuttke H, et al. Increased frequency of cytochrome P450 2D6 poor metabolizers among patients with metoprolol-associated adverse effects. Clin Pharmacol Ther 2002; 72(4): 429–437.
13. Moyer TP, et al. Warfarin sensitivity genotyping: a review of the literature and summary of patient experience. Mayo Clin Proc 2009; 84(12): 1079–1094.
14. King CR, et al. Gamma-glutamyl carboxylase and its influence on warfarin dose. Thromb Haemost 20105; 104(4).
15. Yang L, et al. Impact of VKORC1 gene polymorphism on interindividual and interethnic warfarin dosage requirement - A systematic review and meta analysis. Thromb Res., Early Online Publication 2009 Nov 24.
16. Moyer TP, et al. Warfarin sensitivity genotyping: a review of the literature and summary of patient experience. Mayo Clin Proc 2009; 84(12): 1079–1094.
17. Wang B, et al. Genetic Polymorphism of the Human Cytochrome P450 2C9 Gene and Its Clinical Significance. Curr Drug Metab 2009; 10(7): 781–834.
18. Small KM, et al. Synergistic polymorphisms of beta1 - and alpha2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med 2002; 347(15): 1135–1142.
19. Sconce EA, et al. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood 2005; 106(7): 2329–2333.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Spasmolytic Effect of Metamizole
Most read in this issue
- Význam TDM, fenotypizace a genotypizace pro správné dávkování léčiv
- Dědičné trombofilie – doporučení k provádění genetických testů v klinické praxi
- Farmakogenetické aspekty současné medikamentózní léčby
- Farmakogenetika v léčbě kardiovaskulárních chorob aneb léčba podle guidelines či podle potřeb nemocného?