
Diabetes mellitus 2. typu jako subklinický zánět
Type 2 diabetes mellitus as a subclinical inflammation
Type 2 diabetes mellitus develops as a combination of genetic background and external factors. The disease development is caused by increased oxidative stress under various metabolic factors and in parallel by complex inflammatory reaction without clinical signs. The resulting subclinical inflammation is a consequence of defensive anti-inflammatory reactions. Such Type 2 diabetes conception brings various possibilities in the treatment and prevention.
Key words:
type 2 diabetes, oxidative stress, inflammation, anti-inflammatory mechanisms.
Authors:
Jan Škrha
Authors‘ workplace:
Univerzita Karlova v Praze, 1. lékařská fakulta, III. interní klinika VFN
Published in:
Čas. Lék. čes. 2010; 149: 277-281
Category:
Review Article
Overview
Diabetes mellitus 2. typu se vyvíjí v rámci kombinace genetické vlohy a faktorů zevního prostředí. Rozvoj onemocnění je podmíněn zvýšeným oxidačním stresem vyvolaným různými metabolickými faktory a současně komplexní zánětlivou reakcí, která nemá klinické projevy. Výsledný subklinický zánět je následkem selhávání ochranných protizánětlivých reakcí. Tato koncepce vzniku diabetu 2. typu přináší různé možnosti v terapii a prevenci.
Klíčová slova:
diabetes mellitus 2. typu, oxidační stres, zánět, protizánětlivé mechanismy.
Diabetes mellitus se stává v posledních dvaceti letech celospolečenským problémem pro svůj masový výskyt, a tím i rostoucí nároky na léčbu komplikací. V České republice se počet diabetiků v uplynulých 35 letech ztrojnásobil (z cca 235 tisíc v roce 1975 na cca 780 tisíc koncem roku 2009). Přibývá zejména diabetiků 2. typu, i když trvale narůstá i skupina diabetiků 1. typu. Pro vznik obou typů diabetu je zapotřebí genetická vloha, i když se u obou typů liší, kdežto další faktory přistupují ze zevního prostředí. Na rozvoji diabetu 2. typu se významně podílí stále se zhoršující nepoměr mezi nadměrným energetickým příjmem a tomu neodpovídajícím výdejem energie. Proto existuje významná asociace mezi nadváhou či obezitou a manifestací diabetu. Zvýšená konzumace energeticky bohatých potravin, sedavý způsob života, nedostatek fyzické aktivity, ale vedle toho i přibývající stres jsou typickými faktory moderního života, které u geneticky predisponovaného jedince podmíní dříve či později rozvoj diabetu.
V současné době byla shromážděna řada poznatků na molekulární úrovni, které dokládají, k jakým změnám v organismu při rozvoji diabetu dochází. Postupně se tak daří skládat mozaiku patofyziologie a biochemie diabetu. Tomu nasvědčují i výsledky experimentálních prací, které současně ukazují na některá možná řešení vedoucí nejen k léčbě diabetického syndromu, ale i k jeho prevenci. Úloha faktorů zánětu u diabetu 2. typu byla nedávno shrnuta v přehledném článku (1). Předmětem tohoto sdělení je stručný přehled základních mechanismů, které se podílejí na rozvoji diabetu 2. typu.
Lipidy jako aktivátory zánětu
Energeticky vydatná strava a současně snížená fyzická aktivita jako typické atributy moderního způsobu života vedou k postupnému narůstání hmotnosti s ukládáním tuku. Jeho hromadění ve viscerální lokalizaci vede ke zvýšeným nárokům na sekreci inzulínu, neboť narůstá inzulínová rezistence. U jedince se zvyšuje jak bazální inzulinémie, tak postprandiální koncentrace hormonu. Objemná tuková tkáň, která je současně i významným zdrojem hormonálních aktivit a cytokinů, podléhá dysregulaci projevující se jednak v nadměrném uvolňování substrátů (zvýšený výdej volných mastných kyselin), jednak v porušené expresi hormonů i cytokinů. Dochází k ektopickému ukládání tuku v játrech, pankreatu, svalech, ale i endotelu. S tím dále narůstá inzulínová rezistence a nároky na sekreci inzulínu se dále stupňují. V této fázi se uplatňují zejména volné mastné kyseliny, jejichž koncentrace v plazmě je zvýšená. Jsou příčinou fenoménu označovaného jako lipotoxicita. Ta se projevuje na buněčné úrovni v různých orgánech. V B-buňce pankreatu způsobuje postupné selhávání její sekreční schopnosti. Toto stadium nevede ke klinickým problémům, v běžné klinické praxi zůstává nepovšimnuto a zatím vzácně se označuje jako diabetes lipidus (2).
Na různých úrovních buněk i orgánů je v této fázi možné pozorovat změny. V objemné tukové tkáni dochází k lokálnímu zánětu, hypertrofické adipocyty podmiňují aktivaci imunitních buněk, zejména makrofágů ale též i T-lymfocytů (3). Tyto změny jsou výraznější ve viscerálním než v podkožním tuku (4). Postupně však též dochází k zániku jednotlivých tukových buněk a k uvolnění pro-zánětlivých cytokinů či chemokinů, které „přitahují“ další makrofágy, a tím zvyšují lokální zánětlivou reakci.
Mastné kyseliny vedou ke zvýšené produkci reaktivních forem kyslíku, které se vytvářejí zejména v mitochondriích. Dále se aktivují některé izoenzymy proteinkinázy C, což se promítá do abnormální fosforylace serinu namísto tyrosinu s následnou porušenou aktivitou substrátu inzulínového receptoru (IRS) a rozvojem inzulínové rezistence (obr. 1). Současně se aktivuje NADPH oxidáza a dále se stupňuje tvorba reaktivních forem kyslíku, zejména superoxidového radikálu. Zvyšuje se genová exprese prozánětlivých cytokinů, IL-1, IL-6, IL-18, adhezivních molekul a aktivuje se nukleární faktor NFκB. Stoupá koncentrace inhibičního faktoru migrace makrofágů, který se považuje za významný prozánětlivý cytokin. K těmto reakcím dochází po požití stravy bohaté na nasycené tuky (5, 6) a také v experimentu po infuzi neesterifikovaných volných mastných kyselin (7). Zánětlivé změny jsou výrazněji prokazatelné u obézních osob nebo diabetiků 2. typu než u neobézních a zdravých jedinců (8–10). Tento subklinický zánět, který jinak nemá žádné klinické projevy, vede ke vzestupu rezistence na inzulín v játrech (11).
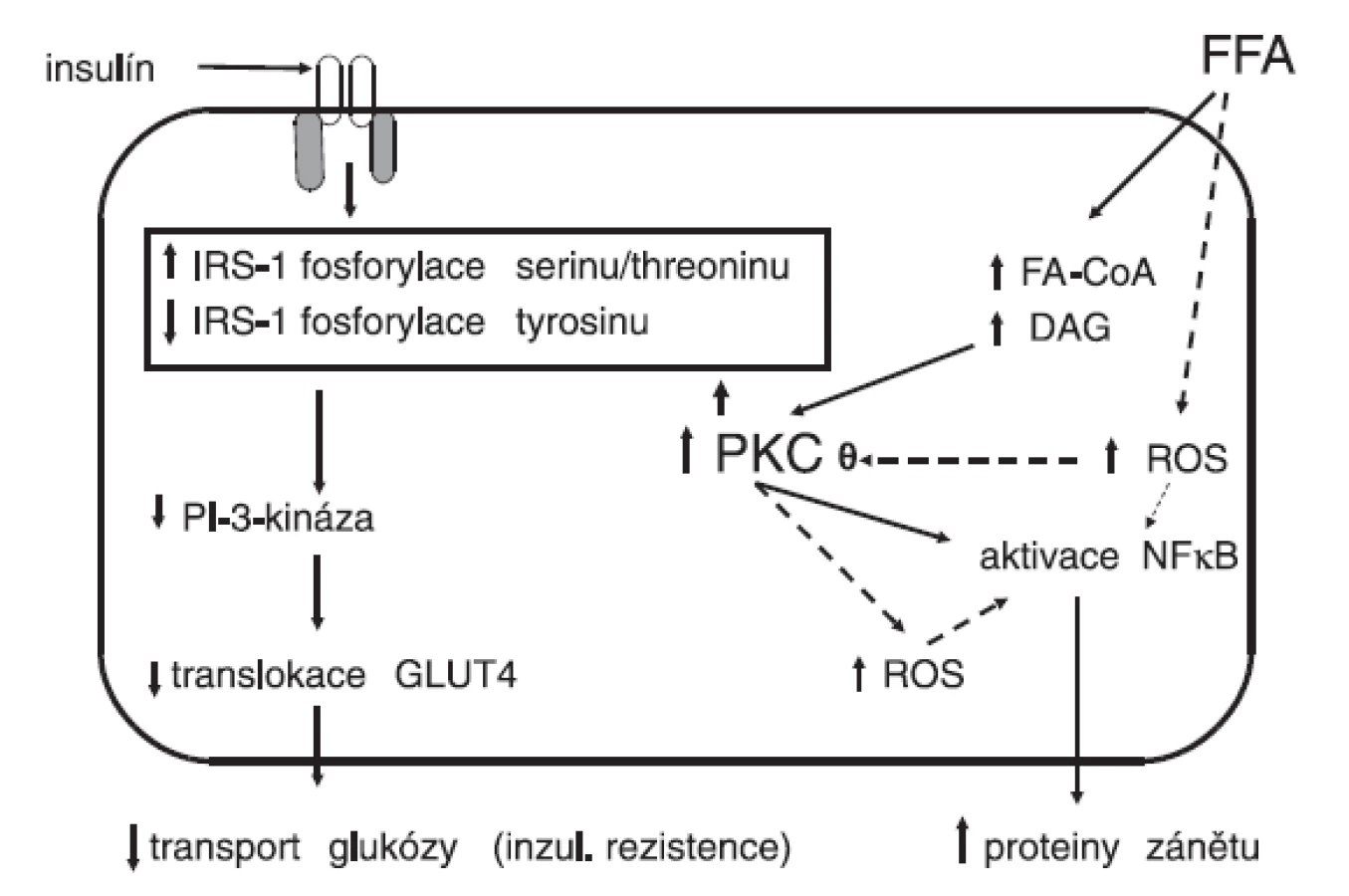
Řadu dokladů přinášejí experimentální práce na zvířatech. Podání neesterifikovaných mastných kyselin vede po interakci s lipopolysacharidovým receptorem TLR4 (toll like receptor 4) k aktivaci NFκB, který je hlavní signalizační molekulou při rozvoji zánětlivé odpovědi (12, 13). Zablokování této reakce ať již na úrovni receptoru TLR4, nebo NFκB znamená ochranu před kaskádou aktivovaných zánětlivých cytokinů a následně se zamezí rozvoji jaterní i systémové inzulínové rezistence (14, 15).
Také na úrovni B-buňky se odehrávají zánětlivé děje s aktivací IL-1, který podmiňuje toxicitu volných mastných kyselin vůči B-buňce (16). Mastné kyseliny snižují tvorbu PPARγ v ostrůvcích, a tím též omezují ochranné buněčné protizánětlivé mechanismy (17). Tato lipotoxicita kombinovaná s účinkem NFκB a stresem postihujícím endoplazmatické retikulum pak vyústí v apoptózu B-buněk (17–19).
Glukóza jako aktivátor zánětu
Všechny uvedené změny, které byly původně vyvolány hypertrofií adipocytů, a to především ve viscerální oblasti, podmínily jednak rozvoj inzulínové rezistence (jaterní i periferní), jednak způsobily lipotoxickým účinkem postupný zánik B-buněk. U jedince, který má genetickou vlohu k diabetu, pak dochází k manifestaci diabetu, neboť snižující se množství B‑buněk a jejich zhoršující se sekreční schopnost nemohou zajistit fyziologickou glykoregulaci, glykémie začne stoupat. Kombinace poruchy lipidů a metabolismu glukózy se promítá do fáze označované jako diabetes mellipidus (20). Tuto situaci lze v praxi prokázat obvykle v rámci záchytu diabetu, i když se uvedený pojem zatím moc nepoužívá. Nicméně přiléhavě vystihuje kombinovanou poruchu lipidů a glukózy.
Organismus je pak vystaven chronické hyperglykémii, která vede také k patologickým změnám. Glukóza je totiž schopná podléhat autooxidaci vedle schopnosti glykovat proteiny. Zvyšuje se pak dále tvorba reaktivních forem kyslíku a aktivuje se komplex pro-zánětlivých cytokinů především jako následek aktivace faktoru NFκB. Dlouhodobá expozice hyperglykémii vyvolává u B-buněk desenzitizaci a další zhoršení sekrece. Na buněčné úrovni se uplatňuje fenomén glukotoxicity (1).
Hyperglykémie podmiňuje zvýšenou expresi genů pro proteiny zánětu, zejména pro IL-6, IL-8, monocytární chemoatraktant protein-1 (MCP-1) a také pro cytoadhezivní molekuly (21). Aktivace zánětlivé reakce je ještě potencována oxidačním stresem, který je způsoben zvýšenou tvorbou superoxidového radikálu účinkem NADPH oxidázy (22). Stupňuje se tvorba mediátorů zánětu aktivací některých kináz (p38, JNK a tyrosinkináz), transkripčních faktorů, nukleárního faktoru NFκB a polymerázy poly-ADP-ribózy (23, 24). Tyto změny se zesilují při dekompenzaci diabetu, ale také při zvýšeném požívání sladkých pokrmů. Naopak výrazná dietní opatření s restrikcí sacharidů a současnou konzumací ovoce a zeleniny způsobují pokles zánětlivé reakce (25).
Akutní efekt glukózy se též podařilo doložit při hyperglykemickém clampu, kdy stoupala koncentrace IL-6 a TNF-α vlivem jejich zvýšené genové exprese (26, 27). Stimulace cytokinů může být proto významně zvýšena již v samotném začátku diabetu 2. typu, a to v době, kdy ještě nebyl diagnostikován. Někdy dosahují glykémie poměrně vysokých hodnot, aniž by byly provázeny klinickou symptomatologií. Bylo prokázáno, že stimulace pro-zánětlivých cytokinů indukuje apoptózu B-buněk, k níž tudíž může docházet již velmi brzy vlivem chronické hyperglykémie. Při inhibici receptoru pro IL-1 účinkem antagonisty IL-1RA se apoptóza B-buněk sníží (28). Z těchto nálezů vyplývá, že proti sobě účinkuje systém pro-zánětlivých a protizánětlivých faktorů a hyperglykémie posouvá tuto rovnováhu v neprospěch protizánětlivých faktorů. Následkem je převaha zánětlivé reakce s negativním efektem na B-buňky (29).
Protizánětlivě působící opatření
Zatímco zmnožení tukové tkáně, energeticky bohatá strava, volné mastné kyseliny a chronická hyperglykémie vedou ke stimulaci zánětlivé reakce, působí některá opatření zcela opačně. Zvýšený obsah vlákniny v dietě způsobuje pokles koncentrace CRP a IL-6 v plazmě, jak bylo prokázáno v klinických studiích (30, 31). Podobně působí výrazná restrikce energie v potravě, tedy např. redukční diety.
Složitější je efekt fyzické aktivity, resp. cvičení. Zatímco akutně a intenzivně probíhající cvičení vede ke vzestupu mediátorů zánětu, zejména IL-6, IL-8 a IL-15, opakované či pravidelné cvičení podněcuje spíše vzestup protizánětlivých mediátorů, např. IL-10 (30, 32). Tyto nálezy vedly k představě, že uvolnění mediátorů zánětu je provázeno současně zvýšenou expresí protizánětlivých faktorů (33). Znamená to, že faktory vyvolávající zánětlivou reakci zároveň podněcují organismus k obranné reakci, a tedy ke stimulaci ochranných mechanismů. Tento fenomén je dobře znám z působení oxidačního stresu vyvolaného glukózou nebo lipidy, který zvyšuje aktivitu antioxidačních mechanismů. Vzestup aktivity scavengerových enzymů pak vyvažuje zvýšenou produkci reaktivních forem kyslíku. Výsledný efekt je závislý na funkční kapacitě ochranných mechanismů, zda se jim podaří negativní působení oxidačního stresu utlumit či omezit. Vzhledem k tomu, že vystupňovaný oxidační stres je zároveň aktivátorem prozánětlivých cytokinů, je zřejmé, že dochází ke generování zánětu, který nemá klinický korelát či symptomatologii, na rozdíl od klasicky pojatého zánětu.
Psychosociální vlivy
Zkušenosti z klinické praxe opakovaně doložily význam psychických změn pro rozvoj diabetu 2. typu. Velké psychické vypětí, trauma, ale i dlouhodobé deprese se často dávají do souvislosti s manifestací diabetu (34). Současné poznatky svědčí o zvýšené zánětlivé aktivitě. Byla prokázána jednak u akutní i chronické spánkové deprivace a u depresí. Zkrácení spánku vede ke snížení inzulínové senzitivity (zhoršení inzulínové rezistence) a glukózové tolerance (35). Současně bylo zjištěno zvýšení CRP a pro-zánětlivých cytokinů včetně aktivace NFκB (36, 37).
Deprese je dalším významným stimulátorem zánětlivé reakce, kdy byl pozorován vzestup CRP, IL-1 a IL-6 (38). Naopak léčba deprese vedla k poklesu TNF-α a CRP (39). Vztah mezi diabetem 2. typu a depresí je oboustranný, takže i u manifestního diabetu byl prokázán zvýšený sklon k rozvoji depresí (40, 41).
Diabetes mellitus 2. typu jako selhání protizánětlivých mechanismů
Vznik onemocnění, jakým je diabetes 2. typu, lze chápat jako dlouhodobé selhávání ochranných mechanismů zapojených jak do oxidačního stresu, tak do zánětlivé reakce (obr. 2). Jde velmi pravděpodobně o geneticky podmíněné změny enzymů a cytokinů (např. v podobě polymorfismů), jejichž kombinace může vyústit do méně účinného obranného systému s následným rozvojem onemocnění. Vedle tohoto efektu pozorovaného u polymorfismů antioxidačních enzymů (superoxiddismutázy, katalázy aj.) fungujících jako scavengery byly popsány například snížené koncentrace endogenního antagonisty receptoru pro IL-1 (IL-1RA) u diabetu 2. typu (42). Podávání rekombinantního IL-1RA vedlo ke zlepšení glykémií a funkce B-buněk, kdežto působení inzulínu zůstalo nezměněno (43, 44). Také dlouho známý efekt vyšších dávek salicylátů na snížení glykémie byl nedávno doložen studiemi o úloze protizánětlivé reakce v ochraně funkce B-buněk (45).
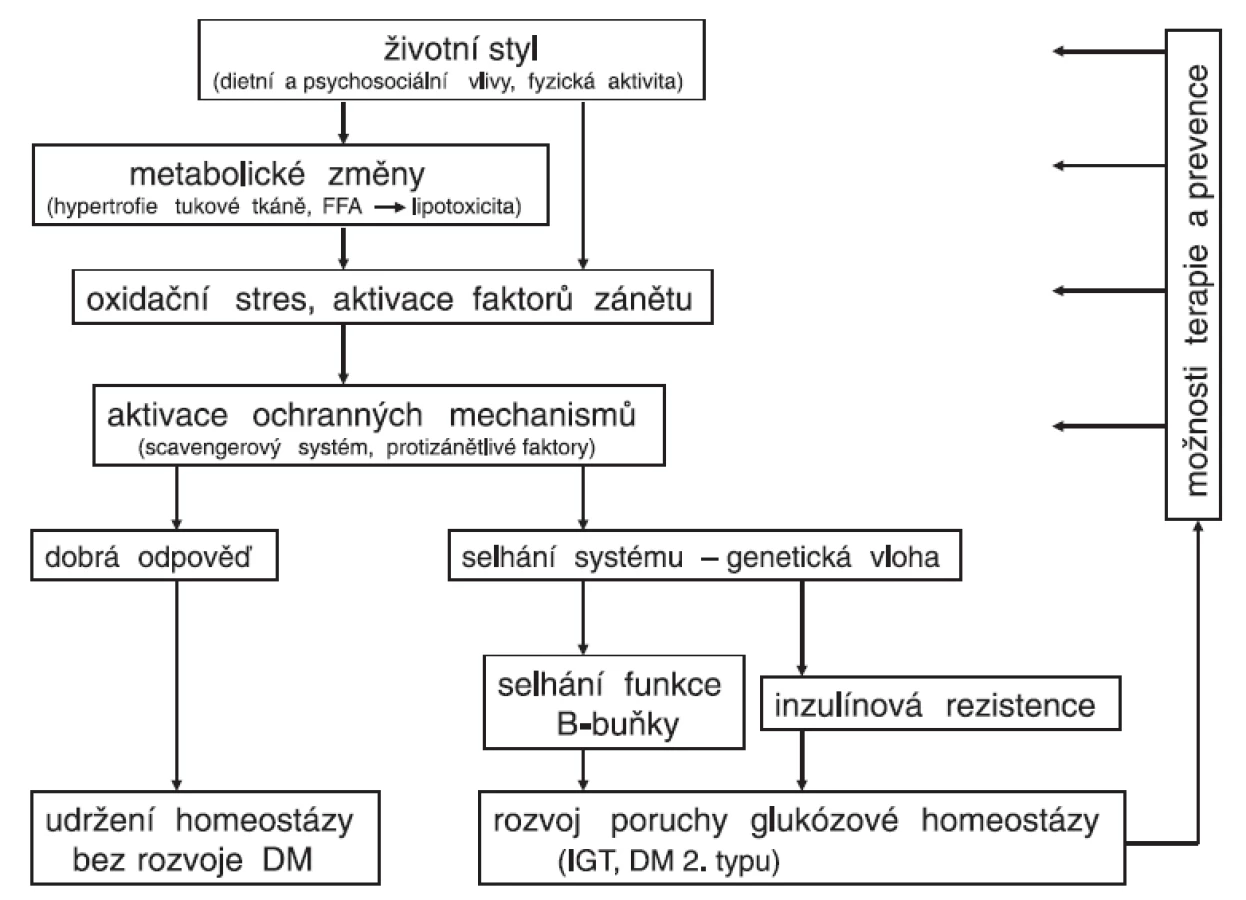
Vystupňovaná prooxidační/prozánětlivá reakce vyvolaná zejména zevními vlivy (nadměrný energetický příjem v kombinaci se sníženým výdejem) nevede u geneticky predisponovaných jedinců k patřičné odezvě v obranných mechanismech. Nedojde tedy k dostatečné stimulaci systému chránícího před zvýšeným oxidačním stresem a současně ani před aktivovanou zánětlivou reakcí. Následkem je pak porucha v obou „součástech“ patogeneze diabetu 2. typu, tedy selhání B-buňky (stimulací apoptózy) s postupnou progresí poklesu sekrece inzulínu a dále snižující se inzulínovou senzitivitou. Tento pohled vysvětluje, proč u některých jedinců při stejných podmínkách nedojde k rozvoji onemocnění, kdežto u jiných, geneticky predisponovaných, vede stejná stimulace různě rychle k onemocnění.
Závěr
Současný pohled na patogenezi diabetu 2. typu vytváří jednotící princip, který se v posledních letech dostává do popředí u řady onemocnění. Nakonec i rozvoj cévních komplikací je v tomto světle obdobný. Jistě půjde do budoucna o to, zda uvedené patogenetické mechanismy zároveň umožní nastolit nové cesty jak k terapii tohoto masově narůstajícího onemocnění, tak zejména k jeho prevenci.
Zkratky
- CRP – C-reaktivní protein
- IRS – substrát inzulínového receptoru
- NADPH – redukovaný nikotinamidadenindinukleotid fosfát
- NFkB – nukleární faktor kappa B
- PPARgamma – receptor gamma aktivovaný peroxisomálními proliferátory
- TLR4 – toll like receptor 4
Adresa pro korespondenci:
prof. MUDr. Jan Škrha, DrSc.
III. interní klinika 1. LF UK a VFN
U Nemocnice 1, 128 08 Praha 2
e-mail: jan.skrha@lf1.cuni.cz
Sources
1. Kolb H, Mandrup-Poulsen T. The global diabetes epidemic as a consequence of lifestyle-induced low-grade inflammation. Diabetologia 2010; 53 : 10–20.
2. Škrha J. Biochemie a patofyziologie. In: Diabetologie, eds. J. Škrha a spol. Praha: Galén 2009; 33–75.
3. Kintscher U, Hartge M, Hess K. et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscl Thromb Vasc Biol 2008; 28 : 1304–1310.
4. Hamdy O, Porramatikul S, Al Ozairi E. Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev 2006; 2 : 367–373.
5. Aljada A, Mohanty P, Ghanim H, et al. Increase in intranuclear nuclear factor kappa B and decrease in inhibitor kappaB in mononuclear cells after a mixed meal: evidence for a proinflammatory effect. Am J Clin Nutr 2004; 79 : 682–690.
6. Esposito K, Nappo F, Giugliano F, et al. Meal modulation of circulating interleukin 18 and adiponectin concentrations in healthy subjects and in patients with type 2 diabetes mellitus. Am J Clin Nutr 2003; 78 : 1135–1140.
7. Tripathy D, Mohanty P, Dhindsa S, et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 2003; 52 : 2882–2887.
8. Blackburn P, Despres JP, Lamarche B, et al. Postprandial variations of plasma inflammatory markers in abdominally obese men. Obesity 2006; 14 : 1747–1754.
9. Nappo F, Esposito K, Cioffi M, et al. Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: role of fat and carbohydrate meals. J Am Coll Cardiol 2002; 39 : 1145–1150.
10. Patel C, Ghanim H, Ravishankar S, et al. Prolonged reactive oxygen species generation and nuclear factor-kappaB activation after a high-fat, high-carbohydrate meal in the obese. J Clin Endocrinol Metab 2007; 92 : 4476–4479.
11. Cani PD, Neyrinck AM, Fava F, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxemia. Diabetologia 2007; 50 : 2374–2383.
12. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate imunity and fatty acid-induced insulin resistance. J Clin Invest 2006; 116 : 3015–3025.
13. Milanski M, Degasperi G, Coope A, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 2009; 29 : 359–370.
14. Poggi M, Bastelica D, Gual P, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007; 50 : 1267–1276.
15. Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF‑kappaB. Nat Med 2005; 11 : 183–190.
16. Aarnes M, Schonberg S, Grill V. Fatty acids potentiate interleukin-1 beta toxicity in the beta-cell line INS-1E. Biochem Biophys Res Commun 2002; 296 : 189–193.
17. Kim EK, Kwon KB, Koo BS, et al. Activation of peroxisome proliferator-activated receptor gamma protects pancreatic beta-cells from cytokine-induced cytotoxicity via NF kappaB pathway. Int J Biochem Cell Biol 2007; 39 : 1260–1275.
18. Grunnet LG, Aikin R, Tonnesen MF, et al. Proinflammatory cytokines activate the intrinsic apoptotic pathway in the beta cells. Diabetes 2009; 58 : 1807–1815.
19. Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001; 50 : 69–76.
20. McGarry JD. Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002; 51 : 7–18.
21. Haubner F, Lehle K, Munzel D, Schmid C, Birnbaum DE, Preuner JG. Hyperglycemia increases the levels of vascular cellular adhesion molecule-1 and monocyte-chemoatractant-protein-1 in the diabetic endothelial cell. Biochem. Biophys Res Commun 2007; 360 : 560–565.
22. Dasu MR, Devaraj S, Jialal I. High glucose induces IL-1beta expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab 2007; 293: E337–E346.
23. Devaraj S, Venugopal SK, Singh U, Jialal I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase C-α and ß. Diabetes 2005; 54 : 85–91.
24. Piconi I, Quagliaro L, Da Ros R, et al. Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: the role of poly(ADP-ribose) polymerase. J Thromb Haemost 2004; 2 : 1453–1459.
25. Esposito K, Marfella R, Ciotola M, et al. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA 2004; 292 : 1440–1446.
26. Esposito K, Nappo F, Marfella R, et al. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 2002; 106 : 2067–2072.
27. Kempf K, Rose B, Herder C, et al. The metabolic syndrome sensitizes leukocytes for glucose-induced immune gene expression. J Mol Med 2007; 85 : 389–396.
28. Maedler K, Sergeev P, Ris F, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002; 110 : 851–860.
29. Boni-Schnetzler M, Thorne J, Parnaud G, et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab 2008; 93 : 4065–4074.
30. Herder C, Peltonen M, Koenig W, et al. Anti-inflammatory effect of lifestyle changes in the Finnish Diabetes Prevention Study. Diabetologia 2009; 52 : 433–442.
31. North CJ, Venter CS, Jerling JC. The effects of dietary fibre on C-reactive protein, an inflammation marker predicting cardiovascular disease. Eur J Clin Nutr 2009; 63 : 921–933.
32. Mathur N, Pedersen BK. Exercise as a mean to control low-grade systemic inflammation. Mediators Inflamm 2008 : 109502. dot:10.1155/2008/109502
33. Silva LA, Silveria PC, Pinho CA, Tuon T, Dal Pizzol F, Pinho RA. N-acetylcystein supplementation and oxidative damage and inflammatory response after eccentric exercise. Int J Sport Nutr Exerc Metab 2008; 18 : 379–388.
34. Knol MJ, Twisk JW, Beekman AT, Heine RJ, Snoek FJ, Pouwer F. Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 2006; 49 : 837–845.
35. Tasali E, Leproult R, Spiegel K. Reduced sleep duration or duality: relationships with insulin resistance and type 2 diabetes. Prog Cardiovasc Dis 2009; 51 : 381–391.
36. Meier-Ewert HK, Ridker PM, Rifai N, et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol 2004; 43 : 678–683.
37. Irwin MR, Wang M, Ribeiro D, et al. Sleep loss activates cellular inflammatory signaling. Biol Psychiatry 2008; 64 : 538–540.
38. Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 2009; 71 : 171–186.
39. Tousoulis D, Drolias A, Antoniades C, et al. Antidepressive treatment as a modulator of inflammatory process in patients with heart failure: effects on proinflammatory cytokines and acute phase protein levels. Int J Cardiol 2009; 134 : 238–243.
40. Maraldi C, Volpato S, Penninx BW, et al. Diabetes mellitus, glycemic control, and incident depressive symptoms among 70 - to 79-year-old persons: the health, aging, and body composition study. Arch Intern Med 2007; 167 : 1137–1144.
41. Pulkki-Raback L, Elovainio M, Kivimaki M, et al. Depressive symptoms and the metabolic syndrome in childhood and adulthood: a prospective cohort study. Health Psychol 2009; 28 : 108–116.
42. Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007; 356 : 1517–1526.
43. Berchtold LA, Larsen CM, Vaag A, et al. IL-1 receptor antagonism and muscle gene expression in patients with type 2 diabetes. Eur Cytokine Netw 2009; 20 : 81–87.
44. Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1-receptor antagonist treatment in type 2 diabetes mellitus. Diabetes Care 2009; 32 : 1663–1668.
45. Koska J, Ortega E, Bunt JC, et al. The effect of salsalate on insulin action and glucose tolerance in obese non-diabetic patients: results of a randomised double-blind placebo controlled study. Diabetologia 2009; 52 : 385–393.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
Most read in this issue
- Neurinom akustiku – vestibulární schwannom – osobní pohled na nejmodernější postupy v jeho léčbě
- Kalcitonin a jeho úloha v regulaci kalciofosfátového metabolismu
- Diabetes mellitus 2. typu jako subklinický zánět
- Karcinom pankreatu – naše zkušenosti se vztahem k diabetes mellitus