
Morfologie a etiopatogeneze aneuryzmatu břišní aorty
Morphology and etiopathogenesis of the abdominal aortic aneurysm
The paper summarizes the latest research on the abdominal aorta aneurysm etiopathogenesis and compares normal aorta morphology with changes in the aortic aneurysm wall. The role of risk factors, especially hemodynamic and genetic, is discussed in detail. Special attention is paid to inflammatory processes including cytokines and matrix degrading proteases that contribute to the development of aneurysm. The role of thrombus and the current results of research into biomarkers indicating the risks and progression of the disease are analysed. Finally, a review of pharmacomodulation of the aortic aneurysm using statins, antibiotics, antihypertensive and nonsteroidal antiinflammatory drugs is presented.
Key words:
abdominal aortic aneurysm, histology, etiopathogenesis, biomarkers, pharmacomodulation. Eb.
:
Lada Eberlová 1,2; Zbyněk Tonar 1; Věra Křížková 1; Jitka Kočová 1; Marie Korabečná 3; Vladislav Třeška 4; Jiří Moláček 4; Karel Houdek 4; Ludmila Boudová 5; Ondřej Topolčan 6; Jindra Vrzalová 6; Martin Pešta 6; Vlastimil Kulda 7; Lukáš Nedorost 1; Jiří Valenta 2
:
Ústav histologie a embryologie Lékařské fakulty UK, Plzeň
1; Ústav anatomie Lékařské fakulty UK, Plzeň
2; Ústav biologie Lékařské fakulty UK, Plzeň
3; Chirurgická klinika FN, Plzeň
4; Šiklův ústav patologie FN, Plzeň
5; Centrální laboratoř pro imunoanalýzu, 2. interní klinika FN, Plzeň
6; Ústav lékařské chemie a biochemie Lékařské fakulty UK, Plzeň
7
:
Čas. Lék. čes. 2012; 151: 55-63
:
Review Article
Příspěvek shrnuje současný stav poznání etiopatogeneze aneuryzmatu abdominální aorty. Vychází z morfologie normální aorty a ze změn, které postihují její jednotlivé vrstvy. Sumarizuje známé rizikové faktory a s ohledem na úlohu a postižení významných složek cévní stěny se blíže věnuje vlivům hemodynamickým a genetickým. Zvláštní pozornost věnuje zánětlivým procesům ve stěně aneuryzmatu včetně cytokinů a matrix-degradujících proteáz s prokázaným vztahem k rozvoji aneuryzmatu. Rozebírá i vliv trombu a současné výsledky hledání možných biomarkerů rizika a progrese onemocnění. V závěru shrnuje dosavadní zkušenosti s farmakomodulací aneuryzmatu s využitím antihypertenziv, statinů, antibiotik a nesteroidních antiflogistik.
Klíčová slova:
aneuryzma břišní aorty, histologie, etipatogeneze, biomarkery, farmakomodulace.
ÚVOD
Aneuryzma abdominální aorty (AAA) je závažné onemocnění, jehož incidence během posledních tří dekád vzrostla (1, 2). Možnými příčinami tohoto trendu jsou stárnutí populace, zdokonalení vyšetřovacích metod i zavedení screeningových programů. Prevalence AAA u mužů nad 65 let je u evropské populace 5–9 % (3), což je asi 8krát více než u stejné skupiny žen (4). V této skupině populace je příčinou 1–3 % úmrtí (5). Nejobávanější komplikací a často prvním příznakem onemocnění je ruptura, jejíž riziko se zvyšuje s průměrem aneuryzmatického vaku a rychlostí jeho růstu (6). Celková incidence ruptur AAA se v západních zemích pohybuje mezi 6–18 na 100 000 obyvatel ročně (7–9), mortalita ruptur se pohybuje mezi 80–90 % (8, 10). Asi třetina neléčených AAA končí rupturou (11). Přestože i malá aneuryzmata praskají, je elektivní zákrok běžně indikován u pacientů s AAA ≥ 5,5 cm, kdy riziko ruptury obvykle překračuje riziko perioperační mortality (12–14).
Pravé aneuryzma je definované jako permanentní, lokalizovaná dilatace všech vrstev cévní stěny se zvětšením průměru na alespoň 1,5 násobek normy. Obvyklá klasifikace aneuryzmat vychází z jejich etiologie, lokalizace a tvaru. Tepenná aneuryzmata se vyskytují v celém arteriálním stromu, extrakraniální aneuryzmata postihují predominantně infrarenální aortu. Tato část aorty je anatomicky rozdílná, je tenčí a obsahuje relativně méně elastinu (15). Tvarem je většina AAA fuziformních (vřetenovitých), tedy postihujících celý obvod stěny.
Nepravé aneuryzma (pulzující hematom) je v podstatě hematom komunikující s luminem cévy a opouzdřený adventicií nebo okolními vazivovými tkáněmi. Zvláštním, od ruptury AAA odlišným akutním postižením aorty, je disekce, při níž následkem ruptury intimy proniká krev mezi vnitřní a zevní vrstvu medie s vytvořením nepravého kanálu, falešného lumen. Histologický podklad disekce je nejasný, uvádějí se změny elastických i svalových vláken. Disekce je častější u osob s Marfanovým syndromem, u kterých vzniká ve 20–40 %, může ale vzniknout i u osob bez známek tohoto syndromu. Klasickým podkladem je Erdheimova cystická medionekróza, avšak ani ta se nevyskytuje ve všech případech disekcí. V oblasti břišní aorty vzniká izolovaná disekce vzácně, zpravidla je součástí disekce hrudní aorty DeBakeyho typu I nebo IIIb.
STAVBA NORMÁLNÍ AORTY
Aorta je tepna elastického typu, jejíž vrstvy odpovídají obecné stavbě tepny: tunica intima, tunica media a tunica adventitia.
Tunica intima je u lidské aorty silná 80–140 μm, je tvořena jednou vrstvou endotelových buněk a s věkem narůstající vrstvou subendotelovou. Endotelové buňky jsou kotveny v lamina basalis a jsou mezi sebou spojeny spoji typu zonula occludens nebo nexus (tight a gap junctions). V cytoplazmě endotelu se nacházejí Weibelova-Paladeho tělíska obsahující von Willebrandův faktor (koagulační faktor VIII). Ten je spolu s transmembránovými proteiny CD 31 a CD 34 používán k imunohistochemické detekci endotelových buněk. Jemné kolagenní fibrily a elastická vlákna subendotelu mají převážně podélnou orientaci, z buněčných komponent zde nacházíme buňky hladké svaloviny (VSMC), případně makrofágy. Membrana elastica interna je tvořena fenestrovanou vrstvou elastinu, která v mikroskopickém obraze splývá s elastickými membránami medie.
Tunica media je nejsilnější částí aortální stěny, u dospělého člověka se její tloušťka pohybuje mezi 500–700 μm. Stavba medie je přizpůsobena velikým mechanickým nárokům a má výrazně lamelární strukturu, hlavními stavebními komponentami jsou rozvětvené a fenestrované elastické membrány a buňky hladké svaloviny. Na příčném řezu se střídají lamely elastinu s interlamelárním prostorem (obr. 1A). Elastická lamela se sousedním interlamelárním prostorem pak tvoří lamelární jednotku. Aorta dospělého člověka obsahuje 40–70 lamelárních jednotek o tloušťce asi 11–15 μm. Do koncentrických, fenestrovaných blanek (membrán) se upínají buňky hladké svaloviny. VSMC jsou obklopeny laminou bohatou na fibronektin, která připomíná laminu basalis, a sítí kolagenních fibril. Kolagenní vlákna jsou tvořena kolagenem typu I, III a V. Interlamelární prostory tvoří asi 50 % tloušťky stěny aorty. Elastin tu tvoří pruhy, které vybíhají ze sousedních elastických lamel a zabíhají do membránovývh záhybů VSMC. Interlamelární matrix obsahuje mikrofibrily silně se barvící na fibrilin 1 a kolagen IV (16).
VSMC jsou velmi heterogenní buněčnou populací. S ohledem na jejich fenotyp a funkci můžeme vymezit dva základní typy: kontraktilní a syntetický. Kontraktilní typ udržuje napětí cév a mechanické vlastnosti obecně. Buňky syntetického typu VSMC obsahují dobře vyvinuté granulární endoplazmatické retikulum a nacházíme je zejména ve vyvíjejících se regenerujících a zánětlivě změněných tepnách. Jsou schopné proliferace a ovlivňují svůj fenotyp s ohledem na poškození nebo mechanické stimuly (17, 18), čímž přispívají ke změnám mechanických vlastností cévy. Buňky hladké svaloviny produkují komponenty extracelulární matrix a mohou také v reakci na růstové faktory produkovanými endoteliemi migrovat do intimy. Zevní třetinu medie penetrují adventiciální vasa vasorum (19). Nejzevnější elastická lamela, lamina elastica externa, odděluje medii od adventicie.
Tunica adventitia je silná 200–500 μm. Silná vlákna kolagenu typu I a vlákna elastická mají spíše longitudinální uspořádání. Dále obsahuje nervi et vasa vasorum, fibroblasty a makrofágy.
MORFOLOGIE ANEURYZMATICKÉ AORTY
Pravé AAA vzniká nejčastěji jako komplikace dilatační formy aterosklerózy. Aneuryzmatická degenerace zahrnuje několik vzájemně propojených procesů: chronický zánět spojený s neovaskularizací, progresivní destrukci elastinu a úbytek buněk hladké svaloviny z medie.
Mikroskopickému obrazu dominuje ztráta elastických lamel, jejich náhrada kolagenem a setření vrstev cévní stěny (obr. 1B). Ta může vykazovat všechny typy aterosklerotické degenerace (20–23).
- Typ 1: adaptivní ztluštění proteoglykany prosycené intimy, výskyt ojedinělých pěnových buněk (makrofágů s fagocytovanými kapénkami lipidů).
- Typ 2: denzní ložiska pěnových buněk, tukové vakuoly se objevují i v některých VSMC intimy.
- Typ 3: extracelulární depozita lipidů (preateromy) vznikající lýzou pěnových buněk.
- Typ 4: vznik ateromu, tj. denzního ložiska extracelulárních lipidů krytých vrstvou pěnových buněk a intimou. V ateromu mohou být přítomny krystaly cholesterolu, intra - i extracelulární depozita kalcia nebo fibróza. Následující typy lézí jsou alternativami dalšího vývoje tohoto stadia.
- Typ 5: reparativní zmnožení fibromuskulárního krytu (čepičky), které vede ke zvýšené rigiditě stěny a výraznějšímu uzávěru cévy.
- Typ 6: přítomnost eroze, hematomu nebo trombu v lézi typu IV nebo V.
- Typ 7: Nodulární kalcifikace stěny, která může přecházet až v osteoidní metaplazii.
- Typ 8: převaha fibrotizace uvnitř fibrokalcifikovaného plaku.


Zatímco jsou u aterosklerózy zánětlivé buňky lokalizovány primárně v poškozené intimě, zánětlivý infiltrát se u AAA vyskytuje obvykle transmurálně s převahou v medii a adventicii (25). Distribuce infiltrátu je v rozmezí rozptýlených buněk až po denzní fokální agregace, místy až lymfatických foliklů. Ve stěnách ruptur nacházíme výraznější zánětlivý infiltrát než ve stěnách asymptomatických aneuryzmat: husté infiltráty medie s B - a T-lymfocyty, buňky plazmatické, transmurální výskyt makrofágů (25, 26). Proliferující vasa vasorum medie jsou obvykle dilatovaná a naplněná krví, často jsou přítomny hemoragie. Ve stěně AAA dochází k úbytku kontraktilních elementů a posunu k sekrečnímu fenotypu VSMC (27). Tento jev je spojen se změnami mechanických vlastností a snížením pevnosti cévní stěny, může ale přinášet i pozitivní aspekty – jako stabilizaci ateromového plátu danou zvýšenou fibroprodukcí.
Dalším výrazným rysem aneuryzmatické stěny je neovaskularizace. Hustota vasa vasorum obvykle pozitivně koreluje s denzitou přítomných leukocytů, zvláště makrofágů (27). Holmes et al. (28) prokázal 15násobné zvýšení neovaskularizace stěny AAA ve srovnání s normální a trojnásobné v komparaci s aterosklerotickou aortou. Novotvorba cév je důležitým rysem progrese AAA (29). Je potencována nejen zánětlivou infiltrací, ale také oxidativním stresem a hypoxií stěny ztluštělé často i nástěnným trombem (29, 30). Cévy prorůstající do medie jsou zdrojem výživy a kyslíku, ale také cytokinů, růstových faktorů, proteáz a jiných prozánětlivých působků, jejich stěna je nekompletní, fragilní a má zvýšenou permeabilitu (31).
Přibližně u 75 % AAA je přítomen intraluminální trombus (32). Je obvykle strukturovaný a podle jeho složení lze rozlišit luminální, střední a abluminální vrstvu (33, 34). V trombu AAA byly v luminální vrstvě popsány okrsky erytrocytů a fibrinu s trombocyty, dále polymorfonukleáry, T-lymfocyty a makrofágy (35). Střední vrstva je tvořena zejména fibrinem s minimem buněčných elemetů, vrstva abluminální je acelulární, vykazující známky fibrinolýzy (34).
ETIOPATOGENEZE AAA
Studium kroků patogeneze AAA vychází ze zkoumání tkání pokročilé aneuryzmatické degenerace vzorků operovaných pacientů nebo experimentálních animálních modelů. Specifická etiologie zůstává nejasná. Obecně je akceptováno, že na vznik a růst AAA mají vliv rizikové faktory spolu s genovým polymorfismem imunoregulačních genů, metaloproteináz a cytokinů. Jako iniciační se předpokládá léze stěny v kombinaci s rizikovými faktory. Většina AAA je tzv. nespecifických a vznikají jako komplikace dilatační formy aterosklerózy. Specifickými agens mohou být: trauma, akutní infekce (salmonelóza, brucelóza), chronický zánět (TBC, lues), neinfekční aortitis (Behćetova, Takayasu, revmatoidní arteritis) nebo vrozené poruchy tvorby pojiv (Marfanův, Ehlersův-Danlosův syndrom typ IV).
RIZIKOVÉ FAKTORY VZNIKU AAA
Statisticky nejvýznamnějším rizikovým faktorem je pozitivní rodinná anamnéza (36), která zvyšuje riziko výskytu AAA u příbuzných prvého stupně téměř dvanáctinásobně (37) a je spojena i s rychlejší progresí onemocnění (38). Dalším velmi významným rizikovým faktorem je kouření. Prevalence AAA u dlouhodobých kuřáků je 5krát vyšší než u nekuřáků (39), u kuřáků byl prokázán i rychlejší růst aneuryzmatu (40). Následují pohlaví a věk. Incidence asymptomatického AAA roste s věkem, maxima dosahuje u mužů stáří 65 let (3). Jako další rizikové faktory vzniku AAA byly prokázány zvýšený diastolický tlak, CHOPN a hyperlipidémie (41).
Rizikovými faktory progrese AAA jsou ženské pohlaví, kouření a CHOPN (42). Na druhou stranu byl prokázán překvapivě protektivní účinek diabetu (43, 44), také nižší prevalence AAA u afroamerické rasy (44).
HEMODYNAMICKÉ VLIVY VZNIKU A PROGRESE AAA
Z mechanického hlediska dochází k dilataci nebo ruptuře cévy poté, co napětí ve stěně překročí mez její pevnosti. Napětí stěny je ovlivněno mnoha faktory, je výstupem dynamických interakcí mezi prouděním krve a mechanikou stěny. Jeho výšku lze základně odhadnout z Laplaceova zákona, který vyjadřuje jeho závislost na transmurálním krevním tlaku, poloměru cévy a tloušťce stěny a pro válcovitý útvar reprezentující cévu má tvar:
σ = (P × r)/t,
kde σ – napětí stěny, P – transmurální tlaková diference, r – vnitřní poloměr cévy, t – tloušťka stěny.
Z uvedeného lze pro vznik a rozvoj AAA odvodit jak rizikovost arteriální hypertenze, tak i protektivní úlohu přítomnosti intraluminálního trombu. Je také zřejmé, že dilatace a zeslabení stěny uzavírají bludný kruh vedoucí k postupné progresi mechanické zátěže aneuryzmatické stěny.
Hemodynamické síly se generují z kinetické energie při průtoku krve cévami. Můžeme je popsat ve třech složkách:
- WSS (wall shear stress) – smykové napětí – způsobené tečnou (tangenciální) silou vyvolanou pohybem krve podél osy cévy;
- hydrostatický tlak – vyvolaný silou působící kolmo na cévní stěnu a
- RWS (relative wall stress), napětí stěny vyvolané silami spojenými s cyklickými tlakovými a objemovými změnami cévní stěny.
Hemodynamické podmínky se v průběhu aorty významně mění, od laminárního proudění u kořene aorty k měnícímu se smykovému napětí a vířivému proudění u její bifurkace. Také poměr elastin/kolagen se ve stěně aorty distálně snižuje (15). Snížená elasticita v kombinaci se zvýšeným tlakem způsobeným odrazy pulzových vln od aortální bifurkace a následných tepen mohou zvyšovat napětí stěny a její náchylnost ke vzniku AAA (45).
Ztráta elastinu vede primárně k elongaci a tortuozitě. V důsledku toho vzniká vířivé proudění, abnormální zátěž stěny vede k poškození endotelu (46), což predisponuje ke vzniku trombu. Přítomnost vulnerabilních aterosklerotických lézí riziko vzniku trombu zvyšuje.
VLIV GENETICKÝCH FAKTORŮ
OMIM databáze (Online Mendelian Inheritance in Man) popisuje AAA jako polygenní, multifaktoriální onemocnění, které se může vyskytovat izolovaně nebo familiálně. Lokusy pro familiální AAA jsou spojeny s polymorfizmy na chromozomech 19q13, 4q31 a 9q21.
I přes nesporné důkazy heritability AAA nebyl dosud nalezen žádný konkrétní gen, který by patogenezi AAA osvětlil. Jako první vyslovil hypotézu dědičnosti AAA Clifton (47) v roce 1977, když popsal výskyt ruptury původně asymptomatického AAA u tří bratří. Familiální výskyt potvrdily i mnohé další práce (48, 49). Výstupy studií analyzujících typ dědičnosti se shodují na autozomálním typu dědičnosti (50, 51), vyloučen nebyl ani typ polygenní (50, 52).
Cílem hledání genetických vlivů u nefamiliálních AAA je identifikace polymorfizmů v genech, jejichž exprese přispívá ke vzniku nebo progresi AAA. U pacientů s AAA byly prokázány polymorfizmy řady genů. Ty, které ovlivňují zánětlivou reakci, budou zmíněny v odstavci věnovaném roli zánětu. Dalšími sledovanými byly geny modulující krevní tlak – ACE (53), AGTR-1 (54), metabolismus homocysteinu – MTHFR (55), oxidativní stres – eNOS (56) nebo imunitní reakci – HLA 2. třídy (57).
Genotyp predisponující ke vzniku nefamiliálního typu AAA není dosud znám. Ačkoliv byly nalezeny mnohé spojitosti mezi polymorfizmem konkrétních genů a AAA, lze předpokládat, že polymorfizmus v jednotlivých genech má na vývoj AAA jen omezený vliv a že k jeho vzniku predisponují až kombinace těchto polymorfizmů s environmentálními faktory (58).
ROLE ZÁNĚTU
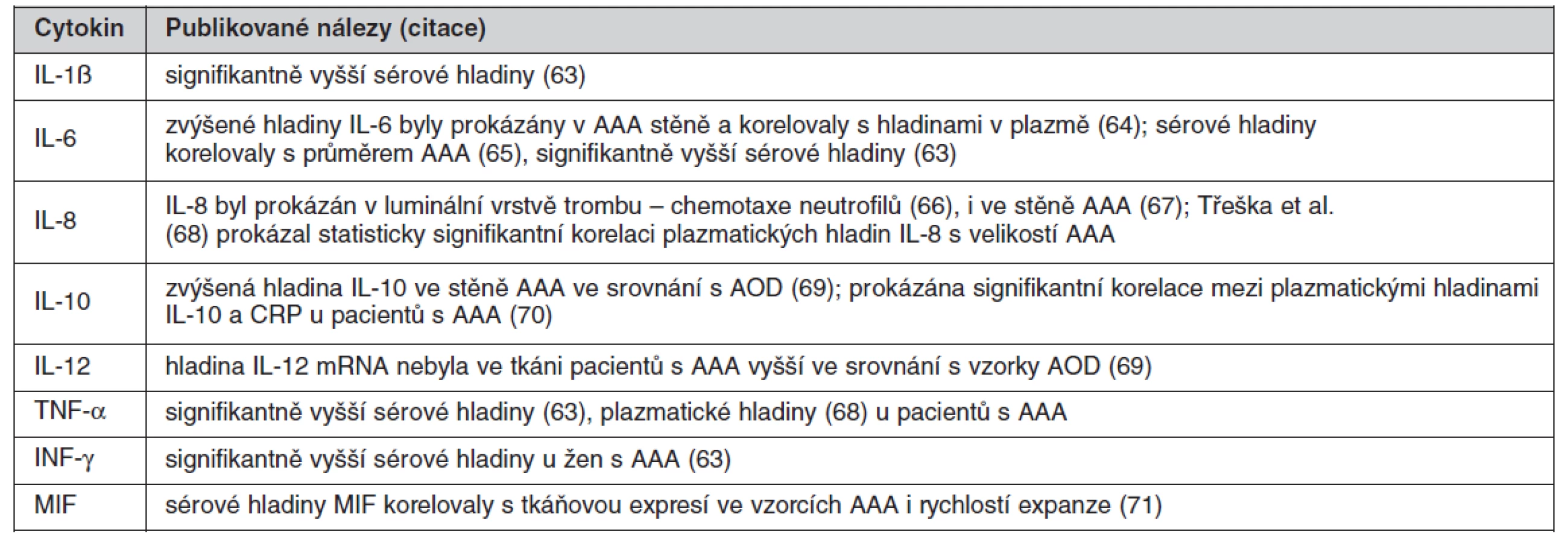
Jak již bylo popsáno, význačným histologickým rysem stěny AAA je extenzivní transmurální infiltrace lymfocyty a makrofágy. Příčina chronického zánětu není známa, zvažována je autoimunitní reakce (59) i vliv Chlamydia pneumoniae (60). Komponenty vzniklé degradací extracelulární matrix jako elastin, laminin a fibronektin pak mohou působit chemotakticky (61). Zánětlivé buňky jsou zdrojem cytokinů, které indukují expresi a zvyšují aktivitu proteáz a jejich tkáňových inhibitorů. Z řady sledovaných cytokinových polymorfizmů byl u pacientů s AAA prokázán signifikantní polymorfizmus pouze u genu pro IL-10 (62).
Přehled cytokinů zvýšených u pacientů s AAA ukazuje tabulka 1.
PROTEÁZY A JEJICH INHIBITORY V AAA
Ačkoliv není etiopatogeneze AAA objasněna, je zřejmé, že matrix metaloproteinázy (MMP) hrají v remodelaci stěny zásadní význam. Jejich zdrojem mohou být jak rezidentní buňky tkáně, tak i zánětlivého infiltrátu. MMP patří do skupiny endopeptidáz, v současnosti je jich známo více než 20 a štěpí různorodé substráty. Nejvýznamnějšími elastázami byly v cévní stěně prokázány MMP-2, -7, -9, -12, MMP-1, -8 a -13 mají zejména kolagenázový účinek (72). Aktivita MMP je regulována v několika stupních:
- transkripcí a translací neaktivních prekurzorů,
- následnou aktivační proteolýzou,
- interakcí s jejich inhibitory (TIMP).
Kromě zvýšené exprese některých MMP (tab. 2a, b) byly u pacientů s AAA prokázány polymorfizmy v promotorech genů pro některé MMP, např. MMP-9 (73), -2, -3 a -12 (74). Jejich kauzalita v patogenezi AAA nebyla ale prokázána.


Další skupinou enzymů zapojených v procesu remodelace jsou serinové proteázy, katepsiny. Ve stěně AAA byly prokázány zvýšené hladiny katepsinů K, L, S (91, 92). Endogenním inhibitorem katepsinů je cystatin C, u kterého byla prokázána slabě negativní korelace plazmatických hladin s velikostí i rychlostí růstu AAA (93, 94).
Plazmin je aktivátorem jak cysteinových, tak i serinových a MMP proteáz. Je tvořen štěpením plazminogenu, tento proces je regulován aktivátory a inhibitory plazminogenu. Tkáňový aktivátor plazminogenu (tPA) zajišťuje proteolýzu v krvi, typ urokinázový (uPA) pak ve tkáni. Hlavním inhibitorem aktivátorů plazminogenu je inhibitor typu 1 (PAI-1). Poté, co se plazmin dostane do cirkulace, je okamžitě inaktivován antiplazminem za vzniku plazmin-antiplazminových (PAP) komplexů. Plazmatické hladiny PAP a tPA byly prokázány jako statisticky signifikantní markery expanze AAA (95–97). Křížková et al. prokázala korelaci mezi plošným podílem PAI-1 pozitivních elementů stěny AAA a jeho klinickou klasifikací (98).
V souvislosti s elastolýzou byla intenzivně zkoumána i elastáza. Lindholt et al. (99) zkoumal plazmatické hladiny elastázy (elastáza-α1-antitrypsinových komplexů) – P-elastázy, kotininu a FEV1 u kuřáků s AAA velikosti 3–5 cm. Výstupy ukázaly negativní korelaci mezi FEV1 a hladinami P-elastázy, dále pozitivní korelaci mezi hladinami P-elastázy a expanzí AAA. Nebyla ale prokázána žádná korelace mezi FEV1 a růstem AAA, což by mohlo poukazovat na významnou účast dalších proteáz ve stěně AAA.
Intraluminální infuze prasečí elastázy je používána pro indukci aneuryzmatické dilatace u zvířecích modelů (100).
ROLE TROMBU
Význam trombu pro vznik a rozvoj aneuryzmatu je také intenzivně zkoumán. Z hemodynamického hlediska trombus snižuje napětí v cévní stěně, na druhé straně zvětšení tloušťky cévní stěny vede k hypoxii, která indukuje zvýšenou neovaskularizaci a zánět. Studie ukazují, že stěna pod trombem je tenčí a vykazuje zvýšené množství makrofágů, Tc a Th lymfocytů a zvýšenou apopotózu VSMC (101). Trombus je také důležitým zdrojem proteáz – MMP-2, -9, plazminogenu a jeho aktivátoru (u-PA) (102, 103), biologicky nejaktivnější se jeví jeho luminální vrstva (66). Rychlost růstu AAA koreluje s rychlostí růstu trombu (104), velikost trombu pozitivně koreluje s rizikem ruptury (105). Objem trombu byl větší u pacientů s rupturou, avšak poměr průměr AAA/objem trombu byl stejný bez ohledu na to, zda došlo či nedošlo k ruptuře (106).
CHLAMYDIA PNEUMONIAE
Význam chlamydiové infekce byl zkoumán i v souvislosti s aterosklerózou (107). Jednoznačný vliv tohoto agens na vznik nebo progresi aneuryzmatu břišní aorty nebyl dosud prokázán (108). V anglické i dánské kohortě byl sice titr IgA proti Chlamidia pneumoniae ≥ 20 ověřen jako nezávislý prediktor zvýšené expanze AAA (109, 110). Na druhou stranu Falkensammer et al. (111) neprokázal rozdíl titrů protilátek IgA a Ig G proti Chlamidia pneumoniae u pacientů s AAA a věkem odpovídající kontrolní skupiny, i když u některých pacientů s vyšším titrem IgA byl sledován rychlejší růst AAA. Navíc v žádném ze vzorků stěny AAA operovaných pacientů nebyla prokázána přítomnost DNA specifické pro chlamydie. V neposlední řadě i výstupy studií sledujících vliv antibiotik na vývoj AAA nevykazují jednoznačné výsledky.
BIOMARKERY AAA
Biomarker je definován jako měřitelná struktura (buňka, protein, sekvence DNA, metabolický produkt), která reprezentuje biologický proces v organismu (112). Jeden nebo více biomarkerů může být indikátorem onemocnění jako takového, nebo například zvýšeného rizika jeho progrese. Za zástupný biomarker progrese je považována rychlost růstu aneuryzmatu, která odráží závažnost degenerativního procesu ve stěně, a může tudíž ukazovat i na zvýšené riziko ruptury. Nalezení senzitivních a specifických biomarkerů by mohlo pomoci indikovat optimální intervaly kontrolních vyšetření pacientů, také ale rozkrýt mechanismy patogeneze, jejichž znalost by pak mohla být využita pro účinnou farmakologickou inhibici růstu AAA. Obecnou slabinou dosavadních výstupů výzkumu je malokohortovost studií a nespecifičnost zatím známých biomarkerů.
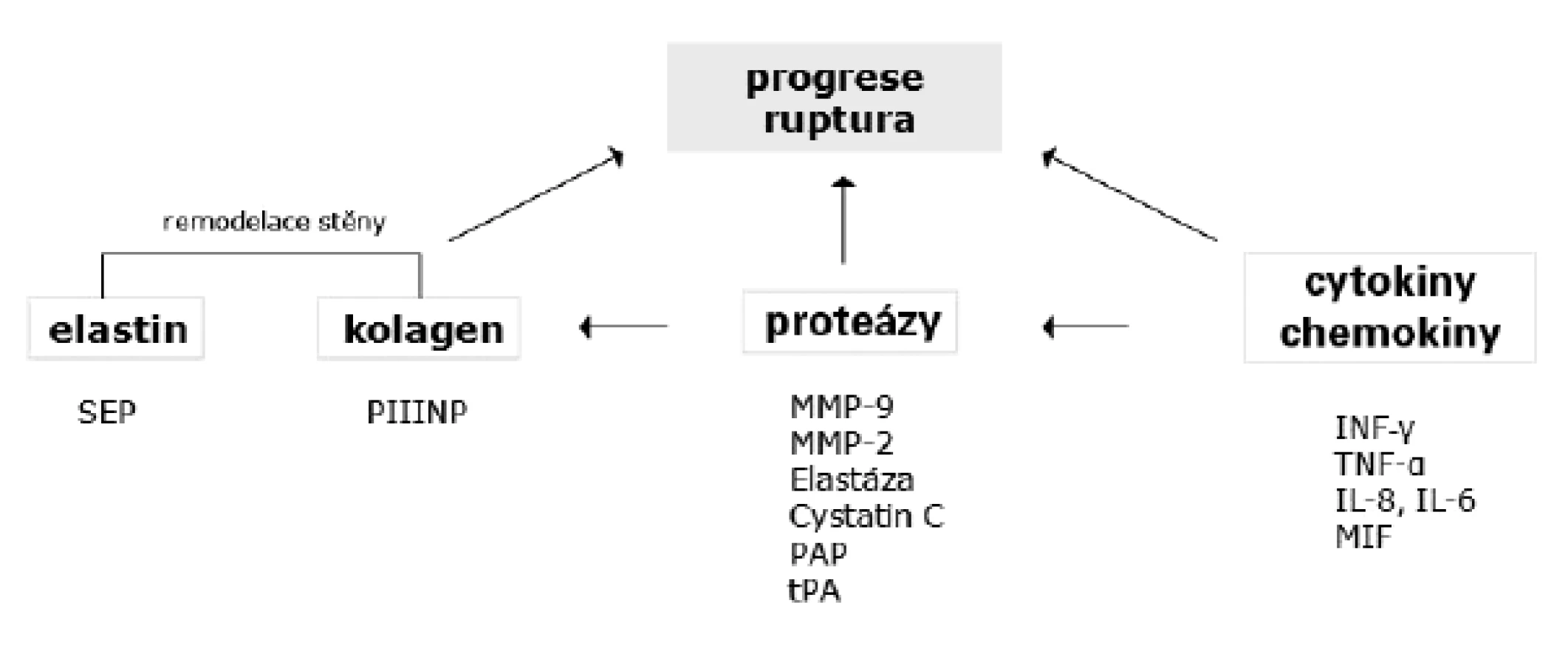
Statisticky nejvýznamnějšími markery progrese AAA jsou zvýšené hladiny peptidů elastinu (SEP), plazmin-antiplazminových komplexů (PAP), matrix metaloproteinázy-9 (MMP-9) a INF-γ (113).
Vývoj AAA je jednoznačně spojen s alterací pojivové tkáně. Jedním ze základních znaků AAA je degradace a fragmentace elastinu. Novotvorba elastinu je u dospělého člověka minimální a poškozený elastin je nahrazován kolagenem. Elastin degradační produkty působí leukotakticky, stimulují také produkci cytokinů (61). Sérová hladina peptidů elastinu v několika studiích korelovala s rychlostí růstu AAA (114, 115). Jednalo se ale o studie malokohortové, širší aplikace detekce SEP by navíc vyžadovala standardizaci metody ELISA (115).
Úbytek kolagenu ve stěně AAA může predisponovat k ruptuře. Metabolický obrat kolagenu lze sledovat měřením prokolagenových propeptidů. Satta et al. (116) prokázal zvýšenou hladinu prokolagen III N-terminálního propeptidu (PIIINP) v séru pacientů s AAA, jeho hladina se s růstem aneuryzmatu zvyšovala. Obdobného výsledku dosáhl i Třeška et al. (117) při vyšetřování vzorků plazmy, nebyla ale nalezena žádná korelace mezi tkáňovou a plazmatickou hladinou PIIINP, což omezuje jeho využití jako cirkulujícího biomarkeru.
Přehled potencionálních biomarkerů progrese AAA ukazuje obrázek 2.
FARMAKOLOGICKÉ OVLIVNĚNÍ RŮSTU AAA
Většina aneuryzmat zachycených screeningem jsou aneuryzmata malá (průměr < 5 cm) a nevyžadují okamžitý chirurgický zákrok. Roční incidence ruptury aneuryzmatu velikosti do 5,5 cm je menší než 1 % (118), mortalita elektivních výkonů se pohybuje kolem 7 % (119), tedy převyšuje riziko ruptury u malých AAA. Průměrný růst malých AAA je 0,3 cm za rok (120). Pokud by se podařilo růst aneuryzmatu snížit například o 50 %, trval by nárůst AAA ze 4 na 5,5 cm 11 let namísto 5 roků. Právě v ovlivnění růstu malých aneuryzmat se otevírá prostor pro farmakologickou intervenci. Zatím se zdá perspektivních jen několik léčiv: ACE inhibitory, doxycyklin, NSA (121). Jejich podávání by spolu s antiagregační terapií a statiny včetně úpravy životosprávy a kuřácké abstinence, mohlo být součástí léčby malých AAA. Zatím chybí ale dostatečně rozsáhlé, zaslepené prospektivní studie, které by pomohly lékovou strategii standardizovat. V současnosti zahrnuje konzervativní postup léčbu antihypertenzivy (zejména ACE inhibitory a betablokátory) a hypolipidemiky (statiny) (122).
Antihypertenziva
Klinicky nejprobádanější lékovou skupinou jsou v této souvislosti ß-blokátory. Ačkoliv studie animálních modelů prokázala statisticky významný vliv propanololu (123) i ACE inhibitorů (124) na expanzi AAA, výstupy klinických studií se liší a jen některé ukazují na statisticky významné snížení růstu AAA při léčbě ß-blokátory (125) nebo ACE inhibitory (126). Hackam et al. sledoval v rozsáhlé studii 15 326 pacientů s AAA po dobu 10 let. Pacienti, kteří užívali ACE inhibitory, měli signifikantně nižší riziko ruptury. Žádný statisticky významný efekt nebyl ale prokázán u ß-blokátorů, blokátorů receptoru pro angiotenzin, blokátorů kalciových kanálů nebo thiazidových diuretik. Výsledky naznačují, že příčinou účinku nejspíš není ovlivnění hemodynamických faktorů. Účinnost ACE inhibitorů lze zčásti vysvětlit tím, že váží zinek, který je kofaktorem zinek-dependentních metaloproteináz.
Statiny
Statiny jsou skupinou léků s pleiotropním účinkem. Kromě toho, že snižují hladinu LDL-cholesterolu v plazmě, působí protizánětlivě inhibicí MMP (127). Účinek statinů na zpomalení růstu AAA byl prokázán na animálních modelech (128). Rozsáhlejší prospektivní studie, které by porovnaly vývoj AAA bez použití statinů, jsou obtížně realizovatelné, jelikož značná část pacientů s diagnostikovaným aneuryzmatem je statiny léčena pro přidružená kardiovaskulární onemocnění. K dispozici jsou tedy výstupy retrospektivních studií, z nichž některé ukazují na zpomalení růstu AAA při současném podávání statinů (129) nebo snížení potřeby elektivních výkonů i výskytu ruptur (130).
Tetracykliny a makrolidy
Vliv doxycyklinu na vývoj AAA byl zkoumán na animálních modelech (131). Také malokohortová, randomizovaná, dvojitě zaslepená studie následně prokázala statisticky signifikantní, inhibiční účinek tříměsíčního podávání doxycyklinu u pacientů s malým AAA. Avšak vyšší titry protilátek proti Chlamydia pneumoniae zůstaly u části pacientů léčbou neovlivněny.
Jiná malokohortová studie sledovala účinek Roxitromycinu podávaného po dobu 4 týdnů (132). V následujících dvanácti měsících došlo, v porovnání s placebem léčenou skupinou, ke 40% redukci expanze AAA, avšak do dvou let byl prokazatelný rozdíl jen u 5 % pacientů.
Nesteroidní antiflogistika
Franklin et al. (133) prokázal snížení růstu AAA a exprese PGE2, IL-1 a -6 u pacientů léčených indometacinem.
ZÁVĚR
Lze předpokládat, že se se zdokonalováním zobrazovacích metod i zaváděním screeningových programů bude incidence AAA i nadále zvyšovat. Etiopatiogeneze tohoto onemocnění není zatím známa. Zpomalení progrese růstu AAA a oddálení elektivního výkonu skýtá značný potenciál jak pro snížení mortality, tak i pro úspory léčebných výdajů (134–136). Vedle farmakologického ovlivnění progrese tohoto onemocnění se nabízí cesta prevence a časného záchytu. Cílem genetického výzkumu je jednak lokalizovat a charakterizovat geny participující v patogenezi AAA, ještě efektivnějším se jeví vývoj testu pro dispenzarizaci pacientů s rizikem vzniku AAA. Velikou výzvou je také nalezení biomarkerů růstu a progrese AAA, které by umožnily časnou a cílenou léčbu potenciálně rizikových pacientů. Znalost morfologie a etiopatogeneze je na této cestě určující a limitující zároveň.
Zkratky
AAA – aneuryzma abdominální aorty
ACE – angiotenzin konvertující enzym
AGTR-1 – angiotenzin II typ1 receptor
Ao – aorta
AOD – aorto-okluzivní choroba
ECM – extracelulární matrix
eNOS – endoteliální syntáza oxidu dusnatého
FEV1 – jednovteřinová vitální kapacita
CHOBPN – chronická obstrukční plicní nemoc
MIF – faktor inhibující migraci makrofágů
MMP – metaloproteináza
MTHFR – methylentetrahydrofolátreduktáza
NSA – nesteroidní antiflogistika
PAI-1 – inhibitor plazminogenu typu 1
PAP – plazmin-antiplazminové komplexy
PIIINP – prokolagen III N-terminální propeptid
SEP – sérové peptidy prokolagenu
TIMP – tkáňový inhibitor metaloproteináz
tPA – tkáňový aktivátor plazminogenu
uPA – urokinázový aktivátor plazminogenu
VSMC – buňka hladké svaloviny
Tato práce vznikla v průběhu řešení projektu FAD č. 200647, TIP č.TI1/328 MPO a VZ MSM 0021620819.
ADRESA PRO KORESPONDENCI:
MUDr. Lada Eberlová
Ústav anatomie LF UK
Karlovarská 48, 301 00 Plzeň
e - mail: lada.eberlova@lfp.cuni.cz
Sources
1. Gillum RF. Epidemiology of aortic aneurysm in the United States. J Clin Epidemiol 1995; 48 : 1289–1298.
2. Eickhoff JH. Incidence of diagnosis, operation and death from abdominal aortic aneurysms in Danish hospitals: results from a nation-wide survey, 1977–1990. Eur J Surg 1993; 159 : 619–623.
3. Vardulaki KA, et al. Incidence among men of asymptomatic abdominal aortic aneurysms: estimates from 500 screen detected cases. J Med Screen 1999; 6 : 50–54.
4. Powell JT, Brady AR. Detection, management, and prospects for the medical treatment of small abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2004; 24 : 241–245.
5. Cowan JA Jr, et al. Epidemiology of aortic aneurysm repair in the United States from 1993 to 2003. Ann N Y Acad Sci 2006; 1085 : 1–10.
6. Lederle FA, et al. Rupture rate of large abdominal aortic aneurysms in patients refusing or unfit for elective repair. JAMA 2002; 287 : 2968–2972.
7. Bengtsson H, Bergqvist D. Ruptured abdominal aortic aneurysm: a population-based study. J Vasc Surg 1993; 18 : 74–80.
8. Heikkinen M, et al. Ruptured abdominal aortic aneurysm in a well-defined geographic area. J Vasc Surg 2002; 36 : 291–296.
9. Mealy K, Salman A. The true incidence of ruptured abdominal aortic aneurysms. Eur J Vasc Surg 1988; 2 : 405–408.
10. Kantonen I, et al. Mortality in ruptured abdominal aortic aneurysms. The Finnvasc Study Group. Eur J Vasc Endovasc Surg 1999; 17 : 208–212.
11. Darling RC, et al. Autopsy study of unoperated abdominal aortic aneurysms. The case for early resection. Circulation 1977; 56: II161–164.
12. Kent KC, et al. Screening for abdominal aortic aneurysm: a consensus statement. J Vasc Surg 2004; 39 : 267–269.
13. Šebesta P. Břišní aorta a pánevní tepny. In: Krajíček M, et al. Chirurgická a intervenční léčba cévních onemocnění. Praha: Grada Publishing 2007; 192.
14. Třeška V, et al. Aneuryzma břišní aorty. Praha: Grada Publishing 1999.
15. Wolinsky H, Glagov S. Structural Basis for the Static Mechanical Properties of the Aortic Media. Circ Res 1964; 14 : 400–413.
16. Dingemans KP, et al. Extracellular Matrix of the Human Aortic media: An Ultrastructural Histochemical and Immunohistochemical Study of the Adult Aortic Media. Anat Rec 2000; 258 : 1–14.
17. Stintzing S, et al. Differentiation patterning of vascular smooth muscle cells (VSMC) in atherosclerosis. Virchows Arch 2009; 455 : 171–185.
18. Shanahan CM, Weissberg PL. Smooth muscle cell phenotypes in atherosclerotic lesions. Curr Opin Lipidol 1999; 10 : 507–513.
19. Grabenwöger M, et al. Endothelialization of biosynthetic vascular prostheses after laser perforation. Ann Thorac Surg 1998; 66 : 110–114.
20. Stary HC. Natural History and Histological Classification of Atherosclerotic Lesions. An Update. Arterioscler Tromb Vasc Biol 2000; 20 : 1177–1178.
21. Stary HC. Slide atlas of atherosclerosis: Progression and regression. CD-ROM. New York: Parthenon Publishing 2002.
22. Virmani R, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2000; 20 : 1262–1275.
23. Tonar Z, et al. Kvantitativní popis aterosklerotických lézí na úrovni optické mikroskopie. Cor Vasa 2007; 49 : 95–101.
24. Kočová J. Overall staining of connective tissue and the muscular layer of vessels. Folia Morphologica 1970; 18 : 293–295.
25. Koch AE, et al. Human Abdominal Aortic Aneurysms Immunophenotypic Analysis Suggesting an Immune-mediated Response. American Journal of Pathology 1990; 137 : 1199–1213.
26. Třeška V, et al. Inflammation in the wall of abdominal aortic aneurysm and its role in the symptomatology of aneurysm. Cytokines, Cellular & Molecular Therapy 2002; 7 : 91–97.
27. Tonar Z, et al. Stereological tools for quantitative microscopy of the aortic wall with focus on the abdominal aortic aneurysm (2010). In: Méndez-Vilas A, Díaz J (Eds.). Microscopy: Science, Technology, Applications and Education. Applications in Biology and Medicine. Vol. 2. Formatex Research Centre, Badajoz, 926–935.
28. Holmes DR, et al. Medial neovascularization in abdominal aortic aneurysms: a histopathologic marker of aneurysmal degeneration with pathophysiologic implications. J Vasc Surg 1995; 21 : 761–771.
29. Choke E, et al. A review of biological factors implicated in abdominal aortic aneurysm rupture. Eur J Vasc Endovasc Surg 2005; 30 : 227–244.
30. Dalman RL. Oxidative stress and abdominal aneurysms: how aortic hemodynamic conditions may influence AAA disease. Cardiovasc Surg 2003; 11 : 417–419.
31. Buschmann I, Schaper W. Arteriogenesis versus angiogenesis: two mechanisms of vessel growth. News in Physiological Sciences 1999; 14 : 121–125.
32. Harter LP, et al. Ultrasonic evaluation of abdominal aortic thrombus. J Ultrasound Med 1982; 1 : 315–318.
33. Falk E. Dynamics in thrombus formation. Ann N Y Acad Sci 1992; 667 : 205–223.
34. Touat Z, et al. Renewal of mural thrombus releases plasma markers and is involved in aortic abdominal aneurysm evolution. Am J Pathol 2006; 168 : 1022–1030.
35. Adolph R, et al. Cellular content and permeability of intraluminal thrombus in abdominal aortic aneurysm. J Vasc Surg 1997; 25 : 916–926.
36. Tilson MD, Seashore MR. Human genetics of the abdominal aortic aneurysm. Surg Gynecol Obstet 1984; 158 : 129–132.
37. Johansen K, Koepsell T. Familial tendency for abdominal aortic aneurysms. JAMA 1986; 256 : 1934–1936.
38. Baird PA, et al. Sibling risks of abdominal aortic aneurysm. Lancet 1995; 346 : 601–604.
39. Vardulaki KA, et al. Quantifying the risk of hypertension, age, sex and smoking in patients with abdominal aortic aneurysm. Br J Surg 2000; 87 : 195–200.
40. Brady AR, et al. Abdominal Aortic Aneurysm Expansion: Risk Factors and Time Intervals for Surveillance. Circulation 2004; 110 : 16–21.
41. Blanchard JF, et al. Prevalence of and risk factors for abdominal aortic aneurysms in a population-based study: The TromsŅ Study. Am J Epidemiol 2001; 154 : 236–244.
42. Choke E, et al. Review of Biological Factors Implicated in Abdominal Aortic Aneurysm Rupture. Eur J Vasc Endovasc Surg 2005; 30 : 227–244.
43. Thompson A, et al. An analysis of drug modulation of abdominal aortic aneurysm growth through 25 years of surveillance. J Vasc Surg 2010; 52 : 55–61.
44. LaMorte WW, et al. Racial differences in the incidence of femoral bypass and abdominal aortic aneurysmectomy in Massachusetts: relationship to cardiovascular risk factors. J Vasc Surg 1995; 21 : 422–431.
45. Humphrey JD, Taylor CA. Intracranial and abdominal aortic aneurysms: similarities, differences, and need for a new class of computational models. Annu Rev Biomed Eng 2008; 10 : 221–246.
46. Nichols KB, Rodriguez AA. Comparison of and investigation into the size effects on the rotational dynamics of two spherical molecules: CCl4 and C60. J Phys Chem A 2005; 109 : 3009–3014.
47. Clifton MA. Familial abdominal aortic aneurysms. Br J Surg 1977; 64 : 765–766.
48. Salo JA, et al. Familial occurrence of abdominal aortic aneurysm. Ann Intern Med 1999; 130 : 637–642.
49. Baird PA, et al. Sibling risks of abdominal aortic aneurysm. Lancet 1995; 346 : 601–604.
50. Tilson MD, Seashore MR. Fifty families with abdominal aortic aneurysms in two or more first-order relatives. Am J Surg 1984; 147 : 551–553.
51. Majumder PP, et al. On the inheritance of abdominal aortic aneurysm. Am J Hum Genet 1991; 48 : 164–170.
52. Kuivaniemi H, et al. Familial abdominal aortic aneurysms: collection of 233 multiplex families. J Vasc Surg 2003; 37 : 340–345.
53. Pola R, et al. Abdominal aortic aneurysm in normotensive patients: association with angiotensin-converting enzyme gene polymorphism. Eur J Vasc Endovasc Surg 2001; 21 : 445–449.
54. Jones K, et al. The influence of 4G/5G polymorphism in the plasminogen activator inhibitor-1 gene promoter on the incidence, growth and operative risk of abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 2002; 23 : 421–425.
55. Strauss E, et al. Increased risk of the abdominal aortic aneurysm in carriers of the MTHFR 677T allele. J Appl Genet 2003; 44 : 85–93.
56. Fatini C, et al. eNOS G894T polymorphism as a mild predisposing factor for abdominal aortic aneurysm. J Vasc Surg 2005; 42 : 415–419.
57. Rasmussen TE, et al. Human leukocyte antigen class II immune response genes, female gender, and cigarette smoking as risk and modulating factors in abdominal aortic aneurysms. J Vasc Surg 2002; 35 : 988–993.
58. Sandford RM, et al. The genetic basis of abdominal aortic aneurysms: a review. Eur J Vasc Endovasc Surg 2007; 33 : 381–390.
59. Gregory AK, et al. Features of autoimmunity in the abdominal aortic aneurysm. Arch Surg 1996; 131 : 85–88.
60. Tambiah J, et al. Provocation of experimental aortic inflammation and dilatation by inflammatory mediators and Chlamydia pneumoniae. Br J Surg 2001; 88 : 935–940.
61. Hance KA, et al. Monocyte chemotactic activity in human abdominal aortic aneurysms: role of elastin degradation peptides and the 67-kD cell surface elastin receptor. J Vasc Surg 2002; 35 : 254–261.
62. Bown MJ, et al. The role of cytokine gene polymorphisms in the pathogenesis of abdominal aortic aneurysms: a case-control study. J Vasc Surg 2003; 37 : 999–1005.
63. Juvonen J, et al. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 1997; 17 : 2843–2847.
64. Dawson J, et al. Circulating cytokines in patients with abdominal aortic aneurysms. Ann N Y Acad Sci 2006; 1085 : 324–326.
65. Rohde LE, et al. Plasma concentrations of interleukin-6 and abdominal aortic diameter among subjects without aortic dilatation. Arterioscler Thromb Vasc Biol 1999; 19 : 1695–1699.
66. Houard X, et al. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc Res 2009; 82 : 532–541.
67. Middleton RK, et al. Characterisation of Interleukin-8 and monocyte chemoattractant protein-1 expression within the abdominal aortic aneurysm and their association with mural inflammation. Eur J Vasc Endovasc Surg 2009; 37 : 46–55.
68. Třeška V, et al. Cytokines as plasma markers of abdominal aortic aneurysm. Clin Chem Lab Med 2000; 38 : 1161–1164.
69. Davis VA, et al. Cytokine pattern in patients with abdominal aortic aneurysm. Vasc Endovascular Surg 2011; 45 : 63–68.
70. Muehling BM, et al. In vivo study on the expression pattern of resistin in patients with abdominal aortic aneurysm. Vasc Endovascular Surg 2011; 45 : 63–68.
71. Pan JH, et al. Macrophage migration inhibitory factor is associated with aneurysmal expansion. J Vasc Surg 2003; 37 : 628–635.
72. Loftus IM, Thompson MM. The role of matrix metalloproteinases in vascular disease. Vasc Med 2002; 7 : 117–133.
73. Jones GT, et al. Functional matrix metalloproteinase-9 polymorphism (C-1562T) associated with abdominal aortic aneurysm. J Vasc Surg 2003; 38 : 1363–1367.
74. Eriksson P, et al. Genotype-phenotype relationships in an investigation of the role of proteases in abdominal aortic aneurysm expansion. Br J Surg 2005; 92 : 1372–1376.
75. Irizarry E, et al. Demonstration of interstitial collagenase in abdominal aortic aneurysm disease. J Surg Res 1993; 54 : 571–574.
76. Knox JB, et al. Evidence for altered balance between matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation 1997; 95 : 205–212.
77. Davis V, et al. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 1998; 18 : 1625–1633.
78. Crowther M, et al. Increased matrix metalloproteinase 2 expression in vascular smooth muscle cells cultured from abdominal aortic aneurysms. J Vasc Surg 2000; 32 : 575–583.
79. Erdozain OJ, et al. Hypoxia in abdominal aortic aneurysm supports a role for HIF-1α and Ets-1 as drivers of matrix metalloproteinase upregulation in human aortic smooth muscle cells. J Vasc Res 2011; 48 : 163–170.
80. Elmore JR, et al. Expression of matrix metalloproteinases and TIMPs in human abdominal aortic aneurysms. Ann Vasc Surg 1998; 12 : 221–228.
81. Fontaine V, et al. Involvement of the mural thrombus as a site of protease release and activation in human aortic aneurysms. Am J Pathol 2002; 161 : 1701–1710.
82. Thompson RW, et al. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest 1995; 96 : 318–326.
83. Mao D, et al. Expression of collagenase-3 (MMP-13) in human abdominal aortic aneurysms and vascular smooth muscle cells in culture. Biochem Biophys Res Commun 1999; 261 : 904–910.
84. Tamarina NA, et al. Expression of matrix metalloproteinases and their inhibitors in aneurysms and normal aorta.Surgery. 1997; 122 : 264–271.
85. Elmore JR, et al. Expression of matrix metalloproteinases and TIMPs in human abdominal aortic aneurysms. Ann Vasc Surg 1998; 12 : 221–228.
86. Defawe OD, et al. TIMP-2 and PAI-1 mRNA levels are lower in aneurysmal as compared to athero-occlusive abdominal aortas. Cardiovasc Res 2003; 60 : 205–213.
87. Wilson WR, et al. Plasma matrix metalloproteinase levels do not predict tissue levels in abdominal aortic aneurysms suitable for elective repair. Vascular 2008; 16 : 248–252.
88. Hovsepian DM, et al. Elevated plasma levels of matrix metalloproteinase-9 in patients with abdominal aortic aneurysms: a circulating marker of degenerative aneurysm disease. J Vasc Interv Radiol 2000; 11 : 1345–1352.
89. Eugster T, et al. Aminoterminal propeptide of type III procollagen and matrix metalloproteinases-2 and -9 failed to serve as serum markers for abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 2005; 29 : 378–382.
90. Lindholt JS, et al. The plasma level of matrix metalloproteinase 9 may predict the natural history of small abdominal aortic aneurysms. A preliminary study. Eur J Vasc Endovasc Surg 2000; 20 : 281–285.
91. Abdul-Hussien H, et al. Collagen degradation in the abdominal aneurysm: a conspiracy of matrix metalloproteinase and cysteine collagenases. Am J Pathol 2007; 170 : 809–817.
92. Abdul-Hussien H, et al. The pathophysiology of abdominal aortic aneurysm growth: corresponding and discordant inflammatory and proteolytic processes in abdominal aortic and popliteal artery aneurysms. J Vasc Surg 2010; 51 : 1479–1487.
93. Shi GP, et al. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J Clin Invest 1999; 104 : 1191–1197.
94. Lindholt JS, et al. Cystatin C deficiency is associated with the progression of small abdominal aortic aneurysms. Br J Surg 2001; 88 : 1472–1475.
95. Lindholt JS, et al. Plasma levels of plasmin-antiplasmin-complexes are predictive for small abdominal aortic aneurysms expanding to operation-recommendable sizes. J Vasc Surg 2001; 34 : 611–615.
96. Lindholt JS, et al. Relationships between activators and inhibitors of plasminogen, and the progression of small abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 2003; 25 : 546–551.
97. Lindholt JS. Activators of plasminogen and the progression of small abdominal aortic aneurysms. Ann N Y Acad Sci 2006; 1085 : 139–150.
98. Kríková V, et al. Quantification of plasminogen activator inhibitor type 1 in the aortic wall. Int Angiol 2009; 28 : 44–49.
99. Lindholt JS, et al. Systemic levels of cotinine and elastase, but not pulmonary function, are associated with the progression of small abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 2003; 26 : 418–422.
100. Molacek J, et al. Optimization of the model of abdominal aortic aneurysm-experiment in an animal model. J Vasc Res 2009; 46 : 1–5.
101. Kazi M, et al. Influence of intraluminal thrombus on structural and cellular composition of abdominal aortic aneurysm wall. J Vasc Surg 2003; 38 : 1283–1292.
102. Sakalihasan N, et al. Activated forms of MMP2 and MMP9 in abdominal aortic aneurysms. J Vasc Surg 1996; 24 : 127–133.
103. Fontaine V, et al. Involvement of the mural thrombus as a site of protease release and activation in human aortic aneurysms. Am J Pathol 2002; 161 : 1701–1710.
104. Wolf YG, et al. Computed tomography scanning findings associated with rapid expansion of abdominal aortic aneurysms. J Vasc Surg. 1994; 20 : 529–535.
105. Stenbaek J, et al. Growth of Thrombus may be a Better Predictor of Rupture than Diameter in Patients with Abdominal Aortic Aneurysms. Eur J Vasc Endovasc Surg 2000; 20 : 466–469.
106. Hans SS, et al. Size and location of thrombus in intact and ruptured abdominal aortic aneurysms. J Vasc Surg 2005; 41 : 584–588.
107. Lindholt JS, et al. A review of Chlamydia pneumoniae and atherosclerosis. Eur J Vasc Endovasc Surg 1999; 17 : 283–289.
108. Lindholt JS, Shi GP. Chronic inflammation, immune response, and infection in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 2006; 31 : 453–463.
109. Lindholt JS, et al. Indicators of infection with Chlamydia pneumoniae are associated with expansion of abdominal aortic aneurysms. J Vasc Surg 2001; 34 : 212–215.
110. Lindholt JS, et al. Vascular surgical society of great britain and ireland: immunoglobulin A antibodies against chlamydia pneumoniae are associated with expansion of small abdominal aortic aneurysms and declining ankle blood pressure. Br J Surg 1999; 86 : 698.
111. Falkensammer B, et al. Lack of microbial DNA in tissue specimens of patients with abdominal aortic aneurysms and positive Chlamydiales serology. Eur J Clin Microbiol Infect Dis 2007; 26 : 141–145.
112. Becker RC. Emerging paradigms, platforms, and unifying themes in biomarker science. J Am Coll Cardiol 2007; 50 : 1777–1780.
113. Urbonavicius S, et al. Potential circulating biomarkers for abdominal aortic aneurysm expansion and rupture-a systematic review. Eur J Vasc Endovasc Surg 2008; 36 : 273–280.
114. Lindholt JS, et al. Serum-elastin-peptides as a predictor of expansion of small abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 1997; 14 : 12–16.
115. Lindholt JS, et al. Serum elastin peptides in the preoperative evaluation of abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 2001; 22 : 546–550.
116. Satta J, et al. Aminoterminal propeptide of type III procollagen in the follow-up of patients with abdominal aortic aneurysms. J Vasc Surg 1997; 25 : 909–915.
117. Treska V, Topolcan O. Plasma and tissue levels of collagen types I and III markers in patients with abdominal aortic aneurysms. Int Angiol 2000; 19 : 64–68.
118. Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms. The UK Small Aneurysm Trial Participants Lancet 1998; 352 : 1649–1655.
119. Filipovic M, et al. Elective surgery for aortic abdominal aneurysm: comparison of English outcomes with those elsewhere. J Epidemiol Community Health 2007; 61 : 226–231.
120. McCarthy RJ, et al. Recommendations for screening intervals for small aortic aneurysms. Br J Surg 2003; 90 : 821–826.
121. Cooper DG, et al. Role of medical intervention in slowing the growth of small abdominal aortic aneurysms. Postgrad Med J 2009; 85 : 688–692.
122. Linhart A, et al. Angiologie. In: Češka R. et al. Interna. Praha: Triton 2010; 189–190.
123. Ricci MA, et al. Effects of hypertension and propranolol upon aneurysm expansion in the Anidjar/Dobrin aneurysm model. Ann N Y Acad Sci 1996; 800 : 89–96.
124. Liao S, et al. Suppression of experimental abdominal aortic aneurysms in the rat by treatment with angiotensin-converting enzyme inhibitors. J Vasc Surg 2001; 33 : 1057–1064.
125. Biancari F, et al. Ten-year outcome of patients with very small abdominal aortic aneurysm. Am J Surg 2002; 183 : 53–55.
126. Hackam DG, et al. Angiotensin-converting enzyme inhibitors and aortic rupture: a population-based case-control study. Lancet 2006; 368 : 659–665.
127. Wilson WR, et al. HMG-CoA reductase inhibitors (statins) decrease MMP-3 and MMP-9 concentrations in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 2005; 30 : 259–262.
128. Steinmetz EF, et al.. Treatment with simvastatin suppresses the development of experimental abdominal aortic aneurysms in normal and hypercholesterolemic mice. Ann Surg 2005; 241 : 92–101.
129. Schouten O, et al. Statins are associated with a reduced infrarenal abdominal aortic aneurysm growth. Eur J Vasc Endovasc Surg 2006; 32 : 21–26.
130. Mosorin M, et al. The use of statins and fate of small abdominal aortic aneurysms. Interact Cardiovasc Thorac Surg 2008; 7 : 578–581.
131. Petrinec D, et al. Doxycycline inhibition of aneurysmal degeneration in an elastase-induced rat model of abdominal aortic aneurysm: preservation of aortic elastin associated with suppressed production of 92 kD gelatinase. J Vasc Surg 1996; 23 : 336–346.
132. Vammen S, et al. Randomized double-blind controlled trial of roxithromycin for prevention of abdominal aortic aneurysm expansion. Br J Surg 2001; 88 : 1066–1072.
133. Franklin IJ, et al. Vascular surgical society of great britain and ireland: non-steroidal anti-inflammatory drugs to treat abdominal aortic aneurysm. Br J Surg 1999; 86 : 707.
134. Thompson SG, et al. Multicentre Aneurysm Screening Study Group. Screening men for abdominal aortic aneurysm: 10 year mortality and cost effectiveness results from the randomised Multicentre Aneurysm Screening Study. BMJ 2009; 338: b2307.
135. Schmidt T, et al. Benefit, risks and cost-effectiveness of screening for abdominal aortic aneurysm. Rofo 2010; 182 : 573–580.
136. Lindholt JS, et al. Screening for abdominal aortic aneurysms: single centre randomised controlled trial. BMJ 2005; 330 : 750.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Prevalence of thrombophilic mutations of FV Leiden, prothrombin G20210A and PAI-1 4G/5G and their combinations in a group of 1,450 healthy middle-aged individuals in the Prague and Central Bohemian regions (results of FRET real-time PCR assay)
- Relapsing polychondritis
- Male hypogonadism and civilization diseases
- Morphology and etiopathogenesis of the abdominal aortic aneurysm