
Monogenně podmíněná obezita – současný stav molekulárně genetického výzkumu a význam v klinické praxi*
Monogenic obesity – current status of molecular genetic research and clinical importance
Obesity and its comorbidities represent one of the major health problems worldwide. A positive energy balance due to inappropriate life-style changes plays a key role in the current obesity epidemic. The influence of genetic factors is also significant – several studies concluded that genes contribute to the development of obesity by 40–70%. Genetic variability predisposes an individual to tendency or resistance to increase body weight in obesogenic environment. Polygenic type of inheritance is responsible in most of obese individuals. However, an intensive research of the past 20 years has led to an identification of several genes causing monogenic forms of obesity. To date, several monogenic genes (leptin, leptin receptor, prohormon convertase 1, proopiomelanocortin, melanocortin 4 receptor, single-minded homolog 1, brain-derived neurotrophic factor, neurotrophic tyrosine kinase receptor type 2) that are either involved in the neuronal differentiation of the paraventricular nucleus or in the leptin-melanocortin pathway are known to cause obesity. Mutation carriers apart from severe early onset obesity manifest with additional phenotypic characteristics as adrenal insufficiency, impaired immunity and impaired fertility. This review provides an overview of molecular-genetic and clinical research in the field of monogenic obesities including therapeutical approaches.
Keywords:
monogenic obesity – energy balance regulation – leptin – leptin receptor – proopiomelanocortin – melanocortin receptor – prohormon convertase – single-minded homolog 1 – brain-derived neurotrophic factor – neurotrophic tyrosine kinase receptor type 2
Authors:
Irena Aldhoon-Hainerová 1,2; Josef Včelák 1; Hana Zamrazilová 1
Authors‘ workplace:
Endokrinologický ústav, Praha
1; Klinika dětí a dorostu 3. LF UK a FNKV, Praha
2
Published in:
Čas. Lék. čes. 2014; 153: 200-206
Category:
Review Articles
Overview
Obezita a její komorbidity představují v současné době jeden z největších zdravotních problémů. V celosvětovém nárůstu prevalence obezity v posledních několika desetiletích hraje klíčovou roli pozitivní energetická bilance v důsledku nežádoucích změn životního stylu. Vliv genetických faktorů je rovněž podstatný – několik studií dospělo k závěru, že geny přispívají k rozvoji obezity ze 40–70 %. Genetická variabilita předurčuje jedince k náchylnosti či rezistenci navyšovat tělesnou hmotnost v interakci s obezitogenním prostředím. Ačkoliv se u naprosté většiny obézních jedinců jedná o tzv. polygenní typ dědičnosti, výzkum za posledních 20 let identifikoval nositele mutací genů způsobujících tzv. monogenně podmíněnou obezitu. Dosud bylo identifikováno jen několik genů (pro leptin, leptinový receptor, prohormon konvertázu 1, proopiomelanokortin, melanokortinový receptor 4. typu, single-minded homolog 1, brain-derived neurotrophic factor a receptor neurotrofické tyrozinové kinázy 2. typu), které se buď podílejí na neuronální diferenciaci hypothalamu, nebo jsou součástí leptino-melanokortinové signalizační osy. Nositelé mutací se kromě časně vzniklé obezity s hyperfagií dále vyznačují např. adrenální insuficiencí, poruchou imunity a fertility. Předkládaný přehledový článek rekapituluje aktuální stav molekulárně-genetického a klinického výzkumu v oblasti monogenně podmíněné obezity včetně terapeutických možností.
Klíčová slova:
monogenní obezita – řízení energetické bilance – leptin – leptinový receptor – proopiomelanokortin – melanokortinový receptor – prohormon konvertáza – single-minded homolog 1 – brain-derived neurotrophic factor – receptor neurotrofické tyrozinové kinázy 2. typu
ÚVOD
Obezita je klasické multifaktoriálně podmíněné onemocnění a celosvětově se řadí mezi nejzávažnější zdravotní problémy dneška. Diagnostikovat jednoznačnou příčinu je s ohledem na komplexnost onemocnění a individuální variabilitu vlivu jednotlivých faktorů mnohdy poměrně obtížné. Příčinou je nepochybně pozitivní energetická bilance v důsledku nepříznivých změn životního stylu. Ty vedou zejména u geneticky predisponovaných jedinců ke zmnožení tukových zásob. U naprosté většiny obézních jedinců můžeme kombinovaný vliv environmentálních a genetických faktorů označit za klíčový. Z genetického hlediska je tento typ obezity možné označit jako polygenně dědičný – na vzniku a rozvoji obezity se podílí řada genů, ovšem s malým efektem na tělesnou hmotnost. Jejich účinek je však v závislosti na přítomnosti pozitivních či negativních environmentálních faktorů potencován nebo naopak zeslabován. Takto postižení jedinci vykazují index tělesné hmotnosti (body mass index – BMI) v pásmu nadváhy až obezity s nástupem v podstatě kdykoliv během života, často v návaznosti na kritická životní období (puberta, menopauza, změna zaměstnání, změna rodinného stavu, úraz, operace).
V průběhu posledních 20 let se podařilo odhalit několik genů, jejichž jediná mutace má potenciál vyvolat již v časném věku těžkou obezitu, tzv. monogenně podmíněnou, tedy bez významného přispění dalších genů či faktorů zevního prostředí. Jedná se o geny kódující hormony, resp. neuropeptidy a jejich receptory, které se uplatňují v centrálním nervovém systému (CNS) buď v neuronální diferenciaci nucleus paraventricularis, či v leptino-melanokortinové signální cestě. Přestože jsou mutace těchto genů velmi vzácné, jejich identifikace nám umožňuje objasnit dosud neznámé patofyziologické procesy ovlivňující energetickou bilanci. Monogenně podmíněná obezita je nejčastěji vyvolána mutacemi genu pro melanokortinový receptor 4. typu (MC4R). Obezita podmíněná mutacemi ostatních genů je spíše vzácnější – jedná se o geny pro leptin (LEP), leptinový receptor (LEPR), proopiomelanokortin (POMC), prohormon konvertázu 1 (PC1), transkripční faktor single-minded homolog 1 (SIM1), brain-derived neurotrophic factor (BDNF) a receptor neurotrofické tyrosinové kinázy 2. typu (NTRK2). Pro úplnost a snazší orientaci v problematice si dovolujeme nastínit začátky výzkumu a též samotnou regulaci energetické bilance v lidském organismu.
REGULACE ENERGETICKÉ BILANCE NA ÚROVNI CENTRÁLNÍHO NERVOVÉHO SYSTÉMU
Rychle postupující genetický výzkum přinesl řadu objevů, které mají zásadní význam pro pochopení regulace energetické bilance a jídelního chování. Nejprve se jednalo o studie na zvířecích modelech, později i u lidských jedinců. Známý myší ob/ob model, popsaný již v padesátých letech 20. století (1, 2), je charakteristický excesivní tělesnou hmotností, nadbytkem tělesné tukové tkáně a porušenou reprodukcí (3). Příčina tohoto fenotypu nebyla známa dlouhá léta. V roce 1994 se podařilo identifikovat nejen LEP, ale i jeho produkt, hormon leptin (4). Další myší model, tzv. db/db, který se rovněž vyznačuje závažnou obezitou, umožnil objev LEPR (5). Jak u hlodavců, tak i u lidí, se leptin řadí mezi klíčové faktory v regulaci energetické bilance a tělesné hmotnosti.
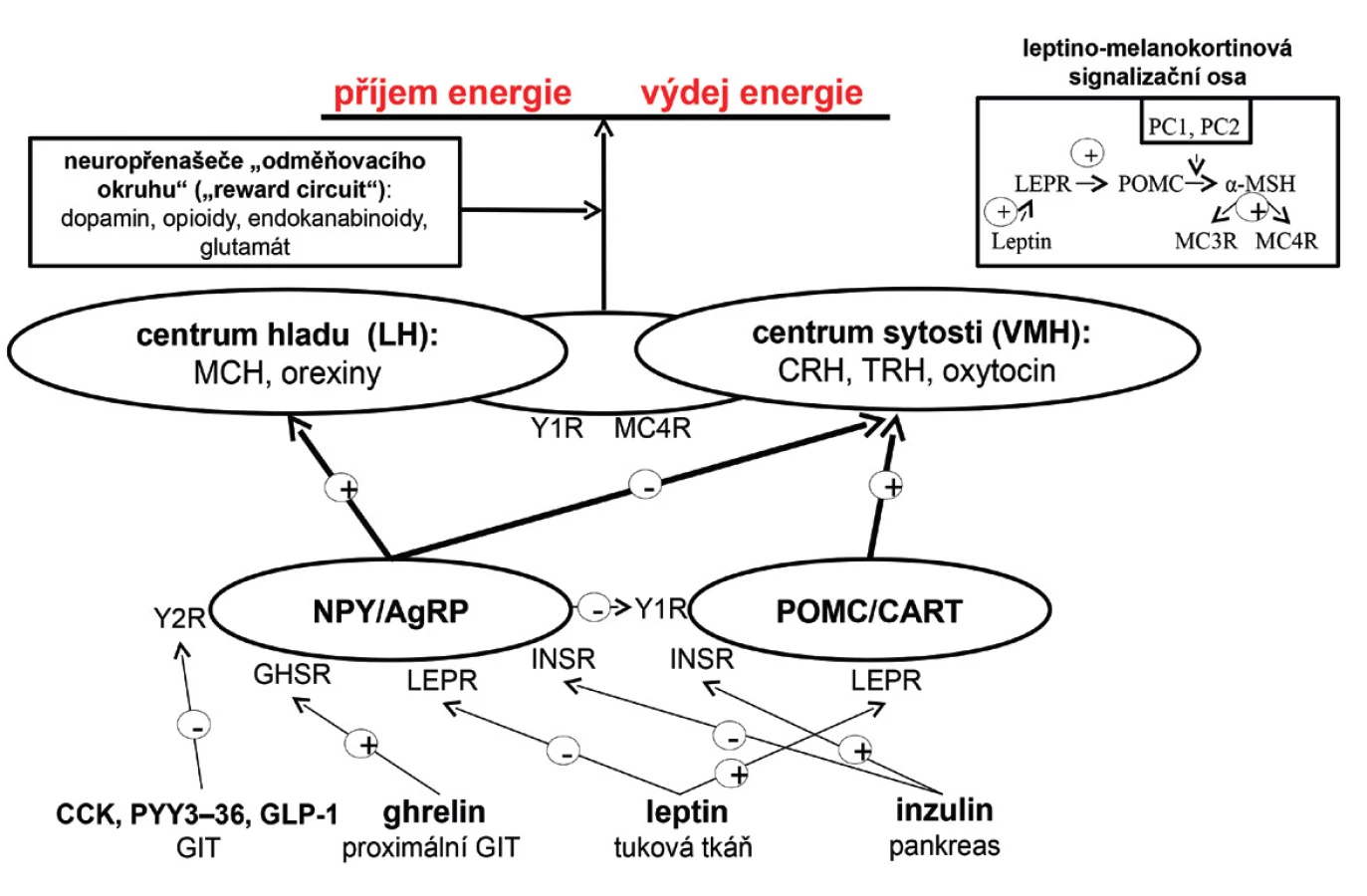
Regulace energetické bilance, pocitu hladu a sytosti, je dána interakcí periferních signálů s CNS, zejména s oblastí hypothalamu. V jeho ventromediální části se nachází centrum sytosti, zatímco v laterální části centrum hladu. Hypothalamus disponuje dvěma druhy neuronů exprimujících orexigenní a anorexigenní neuropeptidy, které společně s adrenergními, dopaminergními, serotoninergními a endokanabinoidními neuropřenašeči regulují v CNS energetickou bilanci. Mezi orexigenní neuropetidy se řadí neuropeptid Y a agouti-related peptide. K anorexigenním neuropeptidům patří POMC a cocaine-amphetamine related transcript. Orexigenní neuropeptidy aktivují centrum hladu, a tím stimulují příjem potravy a snižují energetický výdej. Oproti tomu anorexigenní neuropeptidy aktivují centrum sytosti, a tudíž příjem potravy potlačují. Hypothalamické neuropeptidy reagují na periferní signalizaci zprostředkovanou řadou působků – leptin a inzulin aktivují anorexigenní neurony a zároveň inhibují orexigenní. Ghrelin, hormon gastrointestinálního traktu, orexigenní neurony aktivuje prostřednictvím svého receptoru. Naopak peptid YY3–36, cholecystokinin a glucagon-like peptide-1 je tlumí přes Y2 receptory. Orexigenní neurony potlačují funkci anorexigenních neuronů prostřednictvím Y1 receptorů. Signály obou typů neuronů aktivací či inhibicí MC4R a Y1 receptorů ovlivňují neurony v hypothalamu (obr. 1).
Leptin
Od roku 1997, kdy byli identifikováni první homozygotní nositelé mutací LEP, bylo popsáno několik málo desítek případů těchto mutací na světě. Většinou se jednalo o nekavkazskou rasu, nejčastěji pakistánského či tureckého původu. Nedávná studie provedená na náhodně vybraných obézních pakistánských dětech rodičů z příbuzenského sňatku odhalila, že 16,1 % jedinců byli nositelé LEP mutace (6). Nositelé mutace se vyznačují těžkou časně nastupující obezitou spojenou s hyperfagií, hypogonadotropním hypogonadismem, zvýšeným výskytem infekcí souvisejících s porušenou T-buněčnou imunitou a poruchami sympatického nervového systému (7, 8). V laboratorním nálezu dominují velmi nízké až nulové hladiny leptinu (9). U některých jedinců byla popsána hyperinzulinémie, rozvoj cukrovky 2. typu a dyslipidémie.
Saeed et al. hodnotili změny periferních hormonů ovlivňující jídelní chování u homozygotních a heterozygotních nositelů a nenositelů LEP mutace. Na rozdíl od heterozygotů či kontrolní skupiny zůstávaly hladiny ghrelinu i po konzumaci stravy u homozygotních nositelů bez změny. Dále u homozygotů nebyl pozorován postprandiální vzestup peptidu YY (10). Ukázalo se, že i řada polymorfismů LEP je spojena s obezitou u lidí (11).
Děti i dospělí s mutací LEP jsou dosud jediní, u kterých je možné účinně terapeuticky zasáhnout. Každodenním subkutánním podáváním rekombinantního methionyl leptinu ve večerní dávce 0,02–0,04 mg/kg lze zcela zvrátit nepříznivý fenotyp doprovázený mutací tohoto genu. Dochází k výraznému snížení až normalizaci tělesné hmotnosti, snížení množství tělesného tuku, snížení energetického příjmu s vymizením hyperfagie, zvýšení pohybové aktivity bez snížení bazálního metabolického výdeje, zlepšení lipidového profilu, inzulinové senzitivity, imunitního systému (zvýšení počtu cirkulujících CD4+ T-lymfocytů, zlepšení porušené proliferace T-lymfocytů), vzestupu gonadotropních hormonů a navození puberty (12 až 15). V rámci jedné studie byla u dospívající dívky provedena funkční magnetická rezonance po 1 roce a po 2 letech terapie leptinem. Tímto vyšetřením docházelo k vizuální stimulaci, a to tak, že dívce byly předkládány obrázky vysoce a nízkokalorických potravin a obrázky neznázorňující potraviny. Cílem studie bylo sledovat změny v oblasti hypothalamu a v oblastech mozku souvisejících s odměňováním (striatum, orbitofrontální kůra, substantia nigra/ventrální tegmentální oblast, amygdala). Studie demonstrovala, že dlouhodobá terapie leptinem vede k pozitivním změnám jak v centrech pro homeostázu a ve frontálních oblastech mozku, tak v hedonických odpovědích (16). Po zavedení terapie dochází u pacientů k vývoji sekundárních pohlavních znaků, u ženského pohlaví navození ovulace a menstruačního cyklu. Kazuistiky jednotlivých pacientů ukazují, že terapie leptinem vede poměrně rychle k výraznému zlepšení jaterní steatózy (17). Vynechání léčby leptinem po dobu 6 týdnů vedlo k signifikantnímu hmotnostnímu nárůstu.
Leptinový receptor
Jedinci s deficitem LEPR vykazují obdobné fenotypové projevy jako jedinci s deficitem leptinu a též se řadí mezi velmi vzácné případy. Mutace LEPR byly identifikovány zejména u určitých specifických etnických skupin (např. Alžírsko, Turecko, Egypt), u kterých jsou příbuzenské sňatky poměrně běžné (18, 19). U homozygotních či složených heterozygotních nositelů dochází k porušení signalizace tohoto receptoru, což vede k hyperfagii a ke značnému nárůstu tělesné hmotnosti a tělesného tuku od prvních měsíců věku. Dále je pro nositele mutace charakteristická přítomnost endokrinních abnormalit, jako jsou hypogonadotropní hypogonadismus, porušená sekrece růstového hormonu a thyreotropinu, snížená tělesná výška v dospělosti a těžké infekce v důsledku porušené T-buněčné imunity (20). Sérové hladiny leptinu jsou zvýšené úměrně nadměrnému množství tukové tkáně. Polymorfismy LEPR též souvisí s obezitou u kavkazské populace (21).
Heterozygotní nositelé mutace LEPR mají tělesnou hmotnost normální či jen lehce zvýšenou. Toto bylo potvrzeno i metaanalýzou, která zkoumala v různých populacích tři jednonukleotidové polymorfismy LEPR (Lys109Arg, Gln223Arg a Lys656Asn) a nenalezla žádný vztah s BMI či obvodem pasu (22).
Účinná terapie není u těchto pacientů doposud známá. Terapie leptinem je na rozdíl od pacientů s mutací LEP neúčinná. Nicméně nedávno byla publikována kazuistika homozygotního nositele LEPR mutace způsobené uniparentální disomií chromozomu 1, který podstoupil vertikální gastroplastiku v období dospívání. Došlo ke snížení tělesné hmotnosti o 40 kg a jeho udržení do dospělosti (23).
Prohormon konvertáza 1
Deficit PC1 je autozomálně recesivně dědičné onemocnění způsobené vzácnými mutacemi genu pro proprotein konvertázu subtilisin/kexin typu 1 (PCSK1). PC1 je enzym zodpovědný za proteolytické štěpení řady prohormonů (např. proinzulinu) a proneuropeptidů (např. POMC) (24). Mutace PCSK1 jsou velmi vzácné. První kazuistika byla popsána v roce 1997 (25). Vrozený deficit PC1 vede k těžké hyperproinzulinémii, malabsorpci spojené s průjmy, hypogonadotropnímu hypogonadismu, částečným centrálním poruchám adrenální a tyroidální osy a těžké obezitě (25–27). Částečně lze tento klinický obraz vysvětlit poruchami fyziologické role PC1, tedy proteolytického štěpení mj. proinzulinu, POMC, proglukagonu, pro-gonadotropin uvolňujícího hormonu a pro-tyreotropin uvolňující hormonu (28). Nedávná studie zkoumala klinický obraz 13 dětí s deficitem PC1 (29). Demonstrovala, že vývoj endokrinopatií souvisí s věkem. Některé mutace způsobovaly závažné biochemické změny již in vitro. Pro novorozenecké období bylo typické neprospívání, těžké malabsorpční průjmy a vysoká mortalita. Ostatní endokrinopatie se postupně objevovaly s progresí onemocnění, včetně deficitu růstového hormonu, primárního hypogonadismu, adrenální insuficience, diabetu insipidu a hypotyreózy. I přes časnou poruchu růstu je toto onemocnění většinou doprovázeno těžkým stupněm obezity spojenou s polyfagií (29). Běžné varianty PCSK1 se též asociují s vyšším rizikem rozvoje obezity (30).
Proopiomelanokortin
POMC je polypeptid, který je exprimován nejen v nucleus arcuatus v hypothalamu, ale i v placentě a v pankreatu (31). Exprese POMC je stimulována navázáním leptinu na LEPR. POMC se následně štěpí na několik menších peptidů – adrenokortikotropní hormon (ACTH), α-, β - a γ-melanostimulující hormon (MSH), β-endorfin a β - a γ-lipotropin, z nichž některé jsou důležitými ligandy pro skupinu melanokortinových receptorů (32). Navázáním ligandů na tyto receptory je stimulována pigmentace (vazbou α-MSH na melanokortinový receptor 1. typu), sekrece kortizolu (vazbou ACTH na melanokortinový receptor 2. typu) a ovlivňována energetická bilance na úrovni CNS (vazbou α-MSH na melanokortinový receptor 3. typu (MC3R) a MC4R). Proto jsou typickými znaky nositelů homozygotních mutací POMC adrenální nedostatečnost, těžká obezita doprovázená hyperfagií, rusovlasost a bledá pleť. Byl popsán i případ těžce obézní nositelky mutace POMC, která se neprezentovala charakteristickou rusovlasostí a bledou pletí, avšak byla u ní prokázána nedostatečnost v řadě hormonů hypofýzy (33). Heterozygotní nositelé mají oproti homozygotům normální či jen mírné zvýšenou tělesnou hmotnost (34). Kompletní deficit POMC je u člověka velmi vzácný a ve světě bylo popsáno jen několik případů (34–36).
Přestože léčba obezity způsobené deficitem POMC zůstává nadále neznámá, literatura uvádí několik pokusů o ovlivnění tělesné hmotnosti u takto postižených jedinců (37). Nízkokalorická dvanáctiměsíční dieta vedla u tří dětí – heterozygotních nositelů mutace POMC – k normalizaci tělesné hmotnosti, množství tukové tkáně i inzulinové rezistence (38). Předchozí studie je velmi kontroverzní, jelikož se později ukázalo, že heterozygotní nositelství mutací POMC nemá vliv na tělesnou hmotnost. Intranazální podávání ACTH4-10, jež je obdobou α-MSH, dvěma pacientům s deficitem POMC ani ve své maximální dávce (5 mg/den) nijak neovlivnilo tělesnou hmotnost, tělesné složení či bazální energetický výdej (34). Jedním z možných vysvětlení je nižší afinita ACTH4–10 k MC4R. U pacientů s mírnou centrální hypotyreózou nevedla roční léčba hormony štítné žlázy k signifikantnímu úbytku tělesné hmotnosti (34).
Melanokortinový receptor 4. typu
MC4R, protein o 332 aminokyselinách kódovaný jediným exonem na chromozomu 18q22 (39), je jedním z klíčových faktorů hypothalamické regulace energetické bilance. Myši s inaktivní genovou variantou (nebo-li Mc4r knock-out myši) jsou velmi obézní, projevují se hyperfagií s patologickým nevnímáním pocitu sytosti, hyperinzulinémií, hyperglykémií, větším lineárním růstem, ale normální reprodukční schopností.
Mutace MC4R představují nejčastější monogenně podmíněnou formu obezity u lidí. Vyskytují se v rozmezí 2–6 % u extrémně obézních dětí a dospívajících a 1–2 % u obézních dospělých. Studie provedená na cca 300 českých dětech s těžkou obezitou vzniklou v raném dětství odhalila 2,4% prevalenci mutací MC4R (40).
Do současné doby bylo identifikováno více než 130 funkčních mutací, které většinou vedou k intracelulární retenci. Mutace se většinou dědí autozomálně dominantně s různým stupněm penetrance a expresivity – variabilní je věk nástupu obezity, stejně jako její závažnost, a to i pro stej-nou mutaci v rámci jedné rodiny. Podobně jako na zvířecích modelech byl prokázán i u lidských jedinců s deficitem MC4R vyšší lineární růst v dětství, vyšší finální výška v dospělosti a závažnější hyperinzulinémie než u stejně obézních jedinců bez mutace. Na základě nálezu zachování sekrece růstového hormonu v pulzech u nositelů MC4R mutace lze předpokládat, že MC4R hraje roli v regulaci růstu v oblasti hypothalamu (41). Pro úplnost uvádíme fakt, že u dvou poměrně častých polymorfismů MC4R byl naopak zjištěn malý protektivní účinek na vývoj obezity (42).
Vzhledem k tomu, že nositelství mutací MC4R je ze všech monogenně podmíněných forem obezity nejčastější, řada experimentálních studií se snaží najít terapeutické řešení. Studie s melanokortinovými agonisty ukázala, že mutovaný lidský MC4R s porušenou funkční odpovědí na endogenní ligandy může být aktivován některými z těchto agonistů a může představovat terapeutický cíl (43). Rovněž obnovení povrchové buněčné exprese a funkce mutovaných MC4R pomocí farmakologických chaperonů, má potenciál pro vývoj cílené terapie vhodné pro velkou část obézních pacientů s deficitem MC4R (44). Publikovány byly i studie, které sledovaly hmotnostní změny nositelů, resp. nenositelů mutací v odpověď na změnu životního stylu. U nositelů mutace byly zaznamenány obdobné hmotnostní úbytky jako u nenositelů jak po šestitýdenním léčebném pobytu (40), tak po roční ambulantní intervenci. (45). Nicméně, ve druhém případě nebyli nositelé mutace úspěšní v následném udržení tělesné hmotnosti.
Dále byl u obézních dětí jak s MC4R mutacemi, tak se syndromem Pradera-Williho zkoumán účinek sibutraminu, tj. inhibitoru zpětného vychytávání serotoninu, noradrenalinu a dopaminu. Výsledky ukázaly, že redukce hmotnosti po léčbě sibutraminem byla dosažena v obou skupinách, ale v menší míře u jedinců s MC4R mutacemi (46). Léčbu sibutra-minem podstoupil i náš pacient – homozygotní nositel mutace Gly181Asp (40, 47). V 18 letech dosáhl tělesné hmotnosti 174,4 kg a BMI 55 kg/m2. Po roční léčbě, která byla chlapcem dobře tolerována, jsme zaznamenali výrazně menší přibývání na váze než před léčbou (1,4 kg) (47). Dále došlo ke zlepšení dyslipidémie, zvýšení inzulinové senzitivity a snížení hladin jaterních enzymů, kyseliny močové, pocitu hladu a Beckova skóre deprese. Ovšem rok po ukončení farmakologické léčby se tělesná hmotnost pacienta zvýšila o 10,8 kg (47).
U několika nositelů MC4R mutace byly provedeny bariatrické zákroky. Laparoskopická bandáž žaludku vedla u nositelů mutace k menšímu snížení hmotnosti a menšímu zlepšení metabolického syndromu než u nenositelů mutace (48). Také byl zaznamenán vyšší výskyt pooperačních komplikací u nositelů oproti nenositelům MC4R mutací (48). Minimální hmotnostní pokles byl též popsán u adolescenta s kompletním nedostatkem MC4R po provedené laparoskopické adjustabilní bandáži žaludku a trunkální vagotomii (49). V roce 2011 publikovaná studie sledující výsledky Roux-en-Y žaludečního bypassu (RYGB) u heterozygotních nositelů MC4R mutací zjistila, že po ročním sledování byl pokles tělesné hmotnosti u nositelů srovnatelný s nenositeli mutace (50). O rok později výsledky s RYGB výkonem na myších modelech a na heterozygotních nositelích MC4R mutací potvrdily, že přítomnost jedné nemutované alely je dostatečná pro dosažení dobré odpovědi na tento výkon (51). Léčebné možnosti případů mutací MC4R představují slibné pole výzkumu.
PORUCHY SIGNALIZACE V KROCÍCH NÍŽE OD MC4R
Single Minded Homologue 1
SIM1 je transkripční faktor, který hraje významnou roli při diferenciaci buněk CNS, zejména paraventrikulární oblasti hypothalamu. U myší vedou heterozygotní mutace Sim1 k časně vzniklé obezitě, k urychlenému růstu, hyperinzulinémii a hyperleptinémii (52). De novo balancovaná translokace mezi chromozomy 1p.22.1. a 6q16.2, která vede k poruše SIM1, či missense mutace SIM1 způsobují těžkou časně nastupující obezitu spojenou s hyperfagií a vývojové opoždění různé tíže (53, 54). Nedávná studie zjistila, že delece 6q oblasti, která obsahuje SIM1, způsobuje fenotyp podobný syndromu Pradera-Williho včetně přítomnosti hypopituitarismu. Genotypizace SIM1 u obézních dětí a adolescentů a u normostenických dospělých vedla k objevení raritních variant tohoto genu jak u obézních, tak i normostenických jedinců. Následné funkční studie prokázaly, že varianty Thr481Lys a Ala517Val nalezené u obézních jedinců vedou ke snížené transkripční aktivitě SIM1 (55). Ve studii Ramachandrappa et al. bylo identifikováno devět různých heterozygotních variant SIM1 se sníženou aktivitou (56). Nositelé těchto variant se vyznačovali těžkou obezitou, zvýšeným ad libitum energetickým příjmem, normálním bazálním metabolickým výdejem a poruchou autonomního nervového systému. Větší část pacientů též vykazovala neurobehaviorální obtíže. Autoři spekulují o tom, zda obdobný klinický projev mezi nositeli mutací pro MC4R a SIM1 není v obou případech dán porušenou melanokortinovou signalizací. Další výzkum skutečně prokázal, že oblasti mozku, ve kterých se exprimují Sim1 neurony, jsou zásadními místy působení melanokortinové osy. Xu et al. zjistili, že glutamát je klíčovým neurotransmiterem, který zprostředkovává působení MC4R na Sim1 neuronech, a tím přispívá k regulaci tělesné hmotnosti (57). Lze očekávat, že tato místa budou předmětem studia cílené terapie.
Brain-derived neurotrophic factor a jeho receptor
BDNF a jeho receptor, receptor neurotrofické tyrosinové kinázy B (TrkB), jsou kódovány NTRK2 a ovlivňují jídelní chování a energetickou bilanci. U knock-out Bdnf myší se vyvine obezita a hyperfagie (58). Infuze BDNF snižuje příjem potravy u modelů s deficitem MC4R, což nasvědčuje tomu, že BDNF/TrkB signalizace se nejspíše vyskytuje distálně od MC4R (58). De novo missense mutace způsobující poruchu funkce TrkB vedla u 8leté dívky k časně vzniklé těžké obezitě s hyperfagií, mentální retardaci, poruše krátkodobé paměti, opoždění vývoje řeči, stereotypnímu chování a poruše nocicepce (59). Dále byl identifikován pacient s de novo chromozomální inverzí, 46,XX,inv(11)(p13p15.3), která zahrnuje BDNF lokus, a ruší tím jeho expresi (60). Han et al. zjistili, že pacienti s WAGR syndromem (Wilmsův tumor, aniridie, anomálie urogenitálu a mentální retardace), kteří měli několik delecí včetně chybění BDNF lokusu, vykazovali časně vzniklou obezitu (61). Funkční analýzy identifikovaných variant NTRK2 ve studii Gray et al. shledali, že pouze varianta Tyr722Cys NTRK2 způsobuje poruchu signalizace a vede k porušené neurogenezi hypothalamu. Nositel této varianty vykazoval kromě těžké obezity též poruchu paměti (62).
Diskutabilní melanokortinový receptor 3. typu
Role mutací MC3R je v regulaci energetické homeostázi stále předmětem diskuzí. Zvířecí modely prokázaly, že MC3R je společně s MC4R klíčovým receptorem v rámci leptino-melanokortinové signalizační kaskády. Myši s chybějícím Mc3r se prezentují normální tělesnou hmotností, zvýšeným tělesným tukem a mírnou hypofagií ve srovnání s kontrolní skupinou. Řada mutací byla nalezena u štíhlých lidí a ne všechny mutace vedly k poruše tohoto receptoru. Zároveň ale byli některé mutace MC3R častěji nalezeny u obézních jedinců (63). Nedávno publikovaná studie ukázala obdobnou prevalenci MC3R mutací u belgických obézních a štíhlých dětí a adolescentů (1,0 vs. 1,02 %) (64). Tyto výsledky poukazují na nezbytnost provedení funkčních studií u každé z identifikovaných variant.
Polymorfismy Thr6Lys a Val81Ile MC3R byly asociovány s dětskou obezitou, vyšším z-skóre BMI a větším množstvím tělesného tuku než u nenositelů. U obézních dětí, přítomnost obou těchto polymorfismů MC3R asociovala s větším množstvím tělesného tuku, vyššími hladinami leptinu a inzulinu a vyšší inzulinovou rezistencí ve srovnání s heterozygotními nositeli či nenositeli (65). In vitro studie prokázaly, že tyto polymorfismy vedou k porušené signalizaci v rámci MC3R. Několik studií sledovalo vztah mezi přítomností těchto polymorfismů MC3R a odpovědí obézních jedinců na redukční režim. Vyšší prevalence nositelů MC3R polymorfismů byla identifikována ve skupině dětí s nižšími hmotnostními úbytky po 6 a 12 měsících sledování (66). Výsledky v rámci NUGENOB studie naproti tomu neprokázaly vliv variant MC3R na účinek desetitýdenní intervence (67).
Mutace MC3R na rozdíl od mutací MC4R nejspíše nezpůsobují autozomálně dědičné formy monogenní obezity, jelikož studie na rodinách ve většině případů neprokázaly jasnou kosegraci mutace s daným fenotypem. Na druhou stranu výsledky dosavadních studií ukazují, že varianty tohoto genu přispívají k navýšení tělesného tuku (68). Rutinní screening MC3R u obézních jedinců zůstává tudíž stále diskutabilní.
POKROKY V MOLEKULÁRNĚ-GENETICKÉ LABORATORNÍ ANALÝZE
Výzkum i diagnostika genetických příčin monogenně podmíněné obezity byla donedávna omezena malým výkonem kapilárních sekvenátorů, technickou náročností metod a vysokými finančními náklady na stanovení. Před 10 lety došlo k technologickému zlomu v sekvenování deoxyribonukleové kyseliny (DNA). První světové laboratoře použily sekvenátory nové generace (next-generation sequencing, NGS), u nichž již není nutná separace a identifikace jednotlivých bází (čtení genetického kódu) pomocí elektroforézy. V nových přístrojích se paralelně ve stejný moment sekvenují miliony fragmentů DNA – sekvenování probíhá v reálném čase, čímž se o několik řádů zvýšila kapacita, rychlost a přesnost čtení genetické informace. Zjištěné sekvence DNA se porovnávají s referenčními pomocí bioinformatických algoritmů. Následně se nalezené odlišnosti v pořadí bází DNA ověřují s klinickými záznamy v databázích (např. ClinVar, COSMIC, 1000 Genomes, NHLBI Exome Variant Server, Variant Effect Predictor, PubMed). Hlavní část celého pracovního postupu se dnes přesouvá k analýze velkých objemů dat, vyhledávání subsekvencí v datech, srovnávání podobných DNA/ribonukleových kyselin (RNA) či proteinových sekvencí. Zatímco dříve byla časově nejnáročnější laboratorní část, dnes je to bioinformatické vyhodnocení a klinická interpretace výsledků. Zároveň se intenzivně vyvíjejí laboratorní postupy (69) jak z kompletní DNA konkrétního pacienta vybrat pouze geny požadované k analýze, tzv. příprava DNA knihoven. V současnosti je na trhu dostupných několik typů sekvenátorů upravených přímo pro diagnostiku, které začínají být využívány i rutinními molekulárně-genetickými laboratořemi. Endokrinologický ústav používá pro přípravu DNA knihoven genů spojených s monogenní obezitou reagenční soupravy HaloPlex firmy Agilent a sekvenátor MiSeq od firmy Illumina.
Je třeba si ale uvědomit, že většinu nově nalezených variant nedokážeme aktuálně zhodnotit z hlediska jejich klinického významu pro pacienta. Správná klinicko-genetická interpretace vyžaduje i rozsáhlé genetické/biochemické analýzy v rodině pacienta, funkční studie in vitro nebo na tkáňových kulturách. Nástup NGS je ale nyní tak masivní, že lze předpokládat rychlý nárůst dostupných geneticko-klinických záznamů v databázích z laboratoří celého světa, což tuto interpretaci u monogenní obezity usnadní a zpřesní.
ZÁVĚR
Dosud bylo identifikováno několik genů kódujících hormony, resp. neuropeptidy, a jejich receptory zapojené do regulace energetické bilance, jejichž mutace vedou k monogenně podmíněné obezitě. Nicméně v budoucnosti lze očekávat objevení dalších genů a chromozomálních lokusů zodpovědných za rozvoj obezity. Příkladem může být nedávno popsaná delece chromozomální oblasti 16p11.2, která obsahuje Src homology 2 B adapter protein 1 (SH2B1). Ten se podílí na signalizaci leptinu a inzulinu (70, 71). SH2B1 zřejmě hraje významnou roli nejen v regulaci jídelního chování a tělesné hmotnosti, ale i v sociálních aspektech jedince. V objevení nových faktorů genetického pozadí obezity určitě nadále sehraje svoji roli technický pokrok. Na druhou stranu nelze opomenout důležitost důkladného anamnestického a klinického vyšetření, které může napomoci stanovit diagnózu a následně určit další diagnostický a terapeutický postup. Lze očekávat, že u pacientů s monogenními formami obezity budou využívány nové lékové formy zapojené do regulace CNS. V budoucnosti se též u takto postižených jedinců dá očekávat i potenciální využití genového inženýrství či jasně cílených přístupů týkajících se nejen terapie, ale i prevence geneticky predisponovaných jedinců.
Seznam použitých zkratek
Zkratky v textu uvedené kurzívou označují gen, oproti tomu název jejich proteinového produktu je psán bez kurzívy.
ACTH adrenokortikotropní hormon
AgRP agouti-related peptide
BMI body mass index
BDNF brain-derived neurotrophic factor
CART cocaine-amphetamine related transcript
CCK cholecystokinin
DNA deoxyribonukleová kyselina
GHSR growth hormone secretagogue receptor
GIT gastrointestinální trakt
GLP-1 glucagon-like peptide-1
INSR inzulinový receptor
LEP leptin
LEPR leptinový receptor
LH laterální hypothalamus
MCH melanin koncentrující hormon
MC3R melanokortinový receptor 3. typu
MC4R melanokortinový receptor 4. typu
MSH melanostimulující hormon
NGS sekvenování nové generace (next-generation sequencing)
NTRK2 receptor neurotrofické tyrosinové kinázy 2. typu
PC1 prohormon konvertáza 1
PCSK1 proprotein konvertáza subtilisin/kexin typu 1
POMC proopiomelanokortin
PYY3–36 peptid YY3–36
RNA ribonukleová kyselina
RYGB Roux-en-Y žaludeční bypass
SH2B1 Src homology 2 B adapter protein 1
SIM1 single-minded homolog 1
TrkB neurotrofická tyrosinová kináza B
VMH ventromediální hypothalamus
Y1R Y1 receptor
Y2R Y2 receptor
Podpořeno granty IGA MZ ČR NT/13792-4/2012, NT12342-5/2011 a MZ ČR – RVO (Endokrinologický ústav – EÚ, 00023761).
ADRESA PRO KORESPONDENCI:
MUDr. Irena Aldhoon-Hainerová, Ph.D.
Klinika dětí a dorostu 3. LF UK a FNKV
Šrobárova 50, 100 34 Praha 10
e-mail: ihainer@hotmail.com
Sources
1. Ingalls AM, et al. Obese, a new mutation in the house mouse. Obes Res 1996; 4 : 101.
2. Ingalls AM, et al. Obese, a new mutation in the house mouse. J Hered 1950; 41 : 317–318.
3. Houseknecht KL, et al. The biology of leptin: a review. J Anim Sci 1998; 76 : 1405–1420.
4. Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372 : 425–432.
5. Chua SC, et al. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 1996; 271 : 994–996.
6. Saeed S, et al. High prevalence of leptin and melanocortin-4 receptor gene mutations in children with severe obesity from Pakistani consanguineous families. Mol Genet Metab 2012; 106(1): 121–126.
7. Montague CT, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997; 387 : 903–908.
8. Echwald SM, et al. Identification of two novel missense mutations in the human OB gene. Int J Obes Relat Metab Disord 1997; 21 : 321–326.
9. Karvonen MK, et al. Identification of new sequence variants in the leptin gene. J Clin Endocrinol Metab 1998; 83(9): 3239–3242.
10. Saeed S, et al. Changes in levels of peripheral hormones controlling appetite are inconsistent with hyperphagia in leptin-deficient subjects. Endocrine 2014; 45(3): 401–408.
11. Oksanen L, et al. Novel polymorphism of the human ob gene promoter in lean and morbidly obese subjects. Int J Obes Relat Metab Disord 1997; 21 : 489–494.
12. Farooqi IS, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 1999; 341(12): 879–884.
13. Farooqi IS, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 2002; 110(8): 1093–1103.
14. Galgani JE, et al. Leptin replacement prevents weight loss-induced metabolic adaptation in congenital leptin-deficient patients. J Clin Endocrinol Metab 2010; 95 : 851–855.
15. Andreev VP, et al. Deconvolution of insulin secretion, insulin hepatic extraction post-hepatic delivery rates and sensitivity during 24-hour standardized meals: time course of glucose homeostasis in leptin replacement treatment. Horm Metab Res 2009; 41 : 142–151.
16. Frank S, et al. Long-term stabilization effects of leptin on brain functions in a leptin-deficient patient. PLoS One 2013; 14: e65893.
17. von Schnurbein J, et al. Rapid improvement of hepatic steatosis after initiation of leptin substitution in a leptin-deficient girl. Horm Res Paediatr 2013; 79(5): 310–317.
18. Mazen I, et al. Homozygosity for a novel missense mutation in the leptin receptor gene (P316T) in two Egyptian cousins with severe early onset obesity. Mol Genet Metab 2011; 102(4): 461–464.
19. Andiran N, et al. Homozygosity for two missense mutations in the leptin receptor gene (P316:W646C) in a Turkmenian girl with severe early-onset obesity. J Pediatr Endocrinol Metab 2011; 24(11–12): 1043–1045.
20. Clement K, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998; 392 : 398–401.
21. Masuo K, et al. Leptin-receptor polymorphisms relate to obesity through blunted leptin-mediated sympathetic nerve activation in a Caucasian male population. Hypertens Res 2008; 31 : 1093–1100.
22. Heo M, et al. Pooling analysis of genetic data: the association of leptin receptor (LEPR) polymorphisms with variables related to human adiposity. Genetics 2001; 159(3): 1163–1178.
23. Le Beyec J, et al. Homozygous leptin receptor mutation due to uniparental disomy of chromosome 1: response to bariatric surgery. J Clin Endocrinol Metab 2013; 98(2): E397–402.
24. Zhou A, et al. The prohormone convertases PC1 and PC2 mediate distinct endoproteolytic cleavages in a strict temporal order during proopiomelanocortin biosynthesis processing. J Biol Chem 1993; 268(3): 1763–1769.
25. Jackson RS, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1. Na. Genet 1997; 16 : 303–306.
26. Jackson RS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest 2003; 112 : 1550–1560.
27. Farooqi IS, et al. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab 2007; 92 : 3369–3373.
28. Seidah NG. The proprotein convertases, 20 years later. Methods Mol Biol 2011; 768 : 23–57.
29. Martín MG, et al. Congenital proprotein convertase 1/3 deficiency causes malabsorptive diarrhea and other endocrinopathies in a pediatric cohort. Gastroenterology 2013; 145(1): 138–148.
30. Benzinou M, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nat Genet 2008; 40 : 943–945.
31. O’Donohue TL, Dorsa DM. The opiomelanotropinergic neuronal and endocrine system. Peptides 1982; 3(3): 353–395.
32. Whitfeld PL, et al. The human pro-opiomelanocortin gene. DNA 1982; 143(1): 133–143.
33. Clément K, et al. Unexpected endocrine features and normal pigmentation in a young adult patient carrying a novel homozygous mutation in the POMC gene. Clin Endocrinol Metab 2008; 93(12): 4955–4962.
34. Krude H, et al. Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4-10. J Clin Endocrinol Metab 2003; 88(10): 4633–4640.
35. Krude H, et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998; 19(2): 155–157.
36. Krude H, Gruters A. Implications of proopiomelanocortin (POMC) mutations in humans: the POMC deficiency syndrome. Trends Endocrinol Metab 2000; 11(1): 15–22.
37. Aldhoon-Hainerová I, Lebl J. Treatment options for children with monogenic forms of obesity. World Rev Nutr Diet 2013; 106 : 105–112.
38. Santoro N, et al. Weight loss in obese children carrying the proopiomelanocortin R236G variant. J Endocrinol Invest 2006; 29 : 226–230.
39. Gantz I, et al. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem 1993; 15(20): 15174–15179.
40. Hainerová I, et al. Melanocortin 4 receptor mutations in obese Czech children: studies of prevalence, phenotype development, weight reduction response, and functional analysis. J Clin Endocrinol Metab 2007; 92 : 3689–3696.
41. Martinelli CE, et al. Obesity due to melanocortin 4 receptor (MC4R) deficiency is associated with increased linear growth and final height, fasting hyperinsulinemia, and incompletely suppressed growth hormone secretion. J Clin Endocrinol Metab 2011; 96(1): E181–E188.
42. Hinney A, et al. Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population-based sample and obese adults from Germany. J Clin Endocrinol Metab 2006; 91 : 1761–1769.
43. Roubert P, et al. Novel pharmacological MC4R agonists can efficiently activate mutated MC4R from obese patient with impaired endogenous agonist response. J Endocrinol 2010; 207 : 177–1783.
44. René P, et al. Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J Pharmacol Exp Ther 2010; 335 : 520–532.
45. Reinehr T, et al. Lifestyle intervention in obese children with variations in the melanocortin 4 receptor gene. Obesity (Silver Spring) 2009; 17 : 382–389.
46. Danielsson P, et al. Impact sibutramine therapy in children with hypothalamic obesity or obesity with aggravating syndromes. J Clin Endocrinol Metab 2007; 92 : 4101–4106.
47. Aldhoon Hainerová I, et al. Hypogonadotropic hypogonadism in a homozygous MC4R mutation carrier and the effect of sibutramine treatment on body weight and obesity-related health risks. Obes Facts 2011; 4(4): 324–328.
48. Potoczna N, et al. Gene variants and binge eating as predictors of comorbidity and outcome of treatment in severe obesity. J Gastrointest Surg 2004; 8 : 971–981.
49. Aslan IR, et al. Bariatric surgery in a patient with complete MC4R deficiency. Int J Obes (Lond) 2011; 35 : 457–461.
50. Aslan IR, et al. Weight loss after Roux-en-Y gastric bypass in obese patients heterozygous for MC4R mutations. Obes Surg 2011; 21 : 930–934.
51. Hatoum IJ, et al. Melanocortin-4 receptor signaling is required for weight loss after gastric bypass surgery. J Clin Endocrinol Metab 2012; 97(6): E1023–E1031.
52. Michaud JL, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet 2001; 10 : 1465–1473.
53. Holder JL, et al. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet 2000; 9 : 101–108.
54. Hung CC, et al. Studies of the SIM1 gene in relation to humanobesity and obesity-related traits. Int J Obes (Lond) 2007; 31(3): 429–434.
55. Zegers D, et al. Mutation screen of the SIM1 gene in pediatric patients with early-onset obesity. Int J Obes (Lond) 2013; Epub ahead of print.
56. Ramachandrappa S, et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest 2013; 123(7): 3042–3050.
57. Xu Y, et al. Glutamate mediates the function of melanocortin receptor 4 on Sim1 neurons in body weight regulation. Cell Metab 2013; 18(6): 860–870.
58. Xu B, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci 2003; 6 : 736–742.
59. Yeo GS, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 2004; 7 : 1187–1189.
60. Gray J, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 2006; 55(12): 3366–3371.
61. Han JC, et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med 2008; 359(9): 918–927.
62. Gray J, et al. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int J Obes (Lond) 2007; 31(2): 359–364.
63. Lee YS, et al. The role of melanocortin 3 receptor gene in childhood obesity. Diabetes 2007; 56(10): 2622–2630.
64. Zegers D, et al. Prevalence of rare MC3R variants in obese cases and lean controls. Endocrine 2013; 44(2): 386–390.
65. Feng N, et al. Co-occurrence of two partially inactivating polymorphisms of MC3R is associated with pediatric-onset obesity. Diabetes 2005; 54(9): 2663–2667.
66. Santoro N, et al. Effect of the melanocortin-3 receptor C17A and G241A variants on weight loss in childhood obesity. Am J Clin Nutr 2007; 85(4): 950–953.
67. Santos JL, et al. Consortium Allelic variants of melanocortin 3 receptor gene (MC3R) and weight loss in obesity: a randomised trial of hypo-energetic high - versus low-fat diets. PLoS One 2011; 6(6): e19934.
68. Lee YS. Melanocortin 3 receptor gene and melanocortin 4 receptor gene mutations: the Asian Perspective. Diabetes Metab Res Rev 2012; 28 : 26–31.
69. Bonnefond A, et al. Highly sensitive diagnosis of 43 monogenic forms of diabetes or obesity through one-step PCR-based enrichment in combination with next-generation sequencing. Diabetes Care 2014; 37(2): 460–467.
70. Bochukova EG, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 2010; 463 : 666–670.
71. Doche ME, et al. Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J Clin Invest 2012; 122(12): 4732–4736.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Monogenně podmíněná obezita – současný stav molekulárně genetického výzkumu a význam v klinické praxi*
- Genetické pozadí běžných forem obezity – od studií identických dvojčat po studium kandidátních genů obezity*
- MikroRNA a ledviny