
Nadměrný růst u dětí a dospělých: nový klinický pohled, nové geny, nové fenotypy
Overgrowth in children and in adults: novel clinical view, novel genes, novel phenotypes
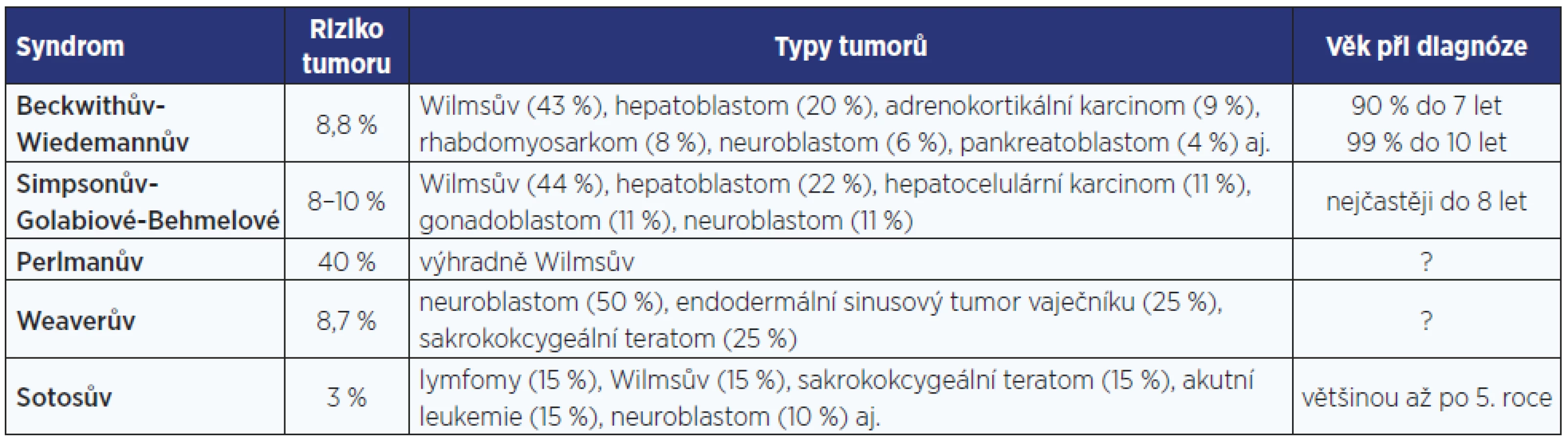
Novel genetic findings allow to more reliably elucidate the aetiology and pathogenesis of overgrowth syndromes in children and in adults. The relatively prevalent overgrowth syndromes in foetuses and neonates include Beckwith-Wiedemann (BWS) and Sotos syndromes; in addition, several rare conditions may occur e.g. Simpson-Golabi-Behmel and Weaver syndromes. These syndromes are not connected with overproduction of growth hormone. Their carriers are at risk of hypoglycaemia (in BWS), of congenital malformations and of childhood tumours. Targeted oncologic screening may improve the outcomes.
Despite rapid growth even postnatally, the final height is mostly normal. In childhood and adolescence, the increased growth velocity results from hormonal overproduction – of precocious production of sexual hormones, hyperthyroidism, or of growth hormone overproduction due to pituitary adenoma that may lead to gigantism or acrogigantism and may be familiar (familiar isolated pituitary adenoma; FIPA). In 15–25 % of affected families, FIPA is caused by autosomal dominantly inherited mutations of AIP gene encoding a tumour suppressor protein named AIP (aryl hydrocarbon receptor-interacting protein). X-linked acrogigantism (X-LAG) is due to GPR101 gene mutations or microduplications of Xq26 chromosomal region. GPR101 encodes G-protein coupled receptor with unknown ligand. X-LAG is associated with recurrent and highly-penetrant pituitary macroadenomas. Mutations of additional at least 10 genes may lead to pituitary tumour with growth hormone overproduction. Gigantism in adults results from untreated or insufficiently treated pituitary adenoma in childhood. Some of the well-known current or past giants were found to carry pathogenic genetic variants of GPR101 or AIP.
Keywords:
overgrowth, overgrowth syndromes, Beckwith-Wiedemann syndrome, Sotos syndrome, Simpson-Golabi-Behmel syndrome, gigantism, acrogigantism, GPR101, AIP
Authors:
Jan Lebl; Lukáš Plachý; Květa Bláhová; Lenka Elblová; Filip Fencl; Stanislava Koloušková; Štěpánka Průhová
Authors‘ workplace:
Pediatrická klinika 2. LF UK a FN Motol, Praha
Published in:
Čas. Lék. čes. 2017; 156: 233-240
Category:
Review Article
Overview
Nové genetické poznatky pomáhají přesněji porozumět etiopatogenezi syndromů s nadměrným růstem u dětí i dospělých. Mezi syndromy nadměrného růstu u fétu a novorozence patří dva častější – Beckwithův-Wiedemannův (BWS) a Sotosův – a několik vzácných, např. Simpsonův-Golabiové-Behmelové (SGBS) a Weaverův. Nejsou spojeny s nadprodukcí růstového hormonu. Jejich nositelé mohou být ohroženi hypoglykemií (zejména u BWS), vrozenými vývojovými vadami a mají vysoké onkologické riziko v dětství. Cílený onkologický screening významně zlepší prognózu.
Rychlý růst pokračuje i postnatálně, ale dospělá výška bývá normální. Příčinou syndromů nadměrného růstu v dětství a adolescenci bývá hormonální nadprodukce. Vedle předčasné produkce pohlavních hormonů a hypertyreózy se může rozvinout gigantismus či akrogigantismus při nadprodukci růstového hormonu z pituitárního adenomu, často familiárního typu (FIPA – familiární izolovaný pituitární adenom). U 15–25 % rodin jsou příčinou FIPA autosomálně dominantně dědičné mutace genu AIP, který kóduje nádorový supresor AIP (aryl hydrocarbon receptor-interacting protein). X-vázaný akrogigantismus (X-LAG) je způsoben mutacemi genu GPR101 nebo mikroduplikacemi oblasti Xq26. GPR101 kóduje receptor spojený s G-proteinem s neznámým ligandem. Pro X-LAG jsou typické torpidní makroadenomy hypofýzy s vysokou penetrancí. Pituitární tumor s nadprodukcí růstového hormonu může být důsledkem mutací nejméně 10 dalších genů. Gigantismus v dospělosti je důsledkem neléčeného či neúspěšně léčeného hypofyzárního adenomu v dětství. U některých mediálně známých gigantů současnosti i minulosti byly prokázány patogenní varianty genů GPR101 nebo AIP.
Klíčová slova:
nadměrný růst, syndromy nadměrného růstu, Beckwithův-Wiedemannův syndrom, Sotosův syndrom, Simpsonův-Golabiové-Behmelové syndrom, gigantismus, akrogigantismus, GPR101, AIP
ÚVOD
Nadměrný růst bývá definován jako tělesná výška nad 97. percentilem a/nebo růstová rychlost nad 75. percentilem pro daný věk a pohlaví.
Nadměrný růst je v pediatrické klinické praxi důvodem k návštěvě lékaře vzácněji než růst nedostatečný, protože fyziologická varianta vyšší postavy (tall-normal) je s výjimkou extrémních případů vnímána jako společensky výhodná. Ostatně ve Spojených státech amerických je tělesná výška jedním z prediktorů volebního úspěchu: Ve 26 amerických prezidentských volbách během posledních 100 let (1916–2016) pouze sedmkrát zvítězil menší z obou kandidátů, přičemž vítěz byl v průměru o 1 palec (cca o 0,4 SD tělesné výšky) vyšší než poražený kandidát (1).
Předpokládáme, že i na tzv. fyziologických variantách vysoké postavy se podílejí určité genové varianty – vyvolávají však podstatně menší výzkumný zájem než varianty postavy malé (2). V některých případech je však vysoký vzrůst zjevně abnormální a zasluhuje podrobnou diagnostiku a adekvátní terapii. Z etiopatogenetického i z didaktického hlediska rozdělíme tyto situace podle věkového kontextu na 3 skupiny:
- syndromy nadměrného růstu u fétu a novorozence
- syndromy nadměrného růstu v dětství a adolescenci
- paralely syndromů nadměrného růstu v dospělosti
SYNDROMY NADMĚRNÉHO RŮSTU U FÉTU A NOVOROZENCE
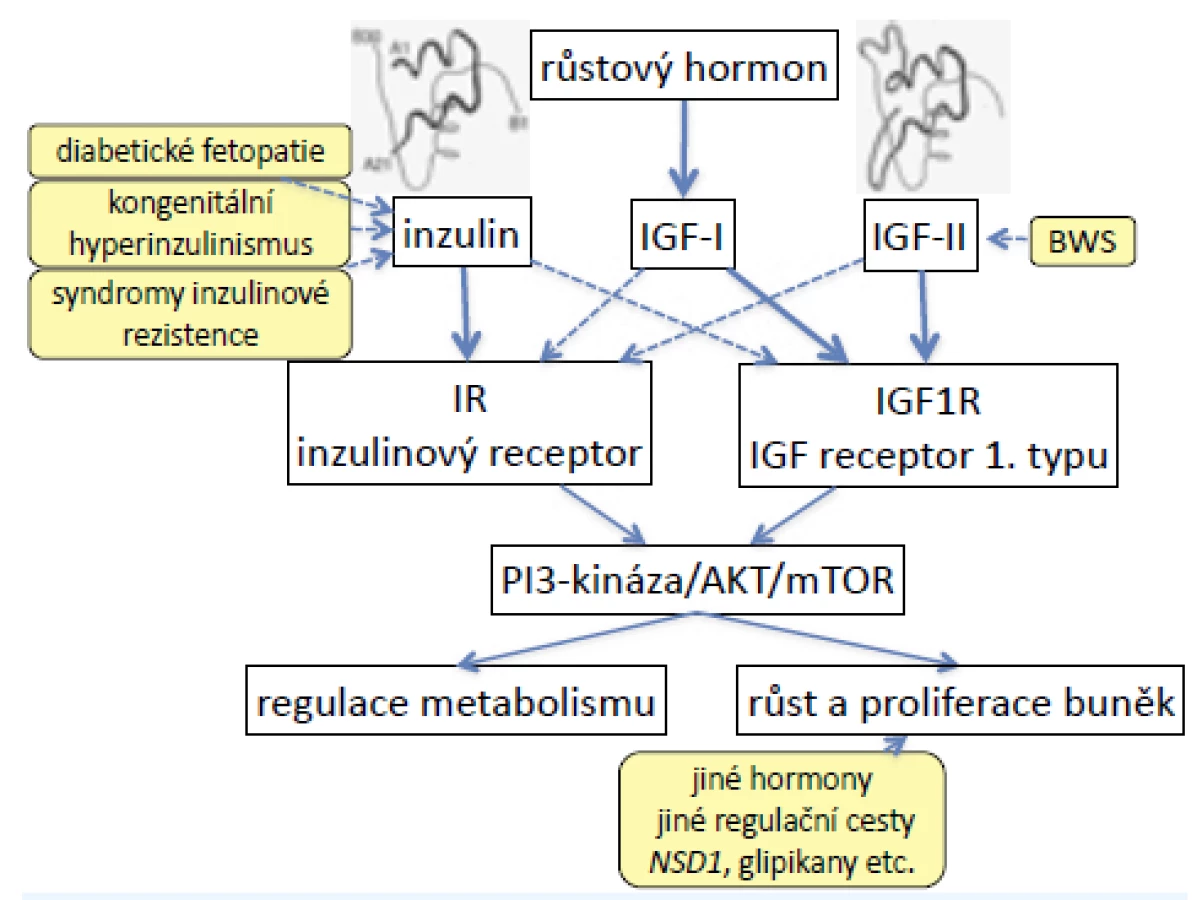
Stavy s prenatálním začátkem nadměrného růstu jsou poměrně vzácné (incidence nejčastějších z nich, Sotosova a Beckwithova-Wiedemannova syndromu, činí 1 : 10–15 000), ale ve svých důsledcích závažné. Některé ohrožují své nositele hypoglykemií s rizikem poškození mozku, jiné zvyšují onkologické riziko (3–5). Genetické mechanismy syndromů nadměrného růstu umožňují lépe porozumět vzájemnému prolínání účinků inzulinu podobných růstových faktorů (IGFs – insulin-like growth factors) a inzulinu, ale také regulaci buněčného růstu, buněčné proliferace, apoptózy a onkogeneze.
Inzulinový receptor a 1. typ receptoru IGF jsou heterotetramery se dvěma podjednotkami α a dvěma podjednotkami β (2). Jsou strukturně podobné a částečně homologní, proto mají částečnou zkříženou afinitu ke svým ligandům (obr. 1).
Vysoké hladiny inzulinu in utero (diabetická fetopatie, kongenitální hyperinzulinismus) proto vedou nejen k nadměrným přírůstkům hmotnosti plodu, ale vazbou inzulinu na receptor IGF narůstá také fetální délka. U některých syndromů inzulinové rezistence (pseudoakromegalie) jsou zablokované metabolické účinky inzulinu, vysoké hladiny inzulinu ovšem vedou k nadměrnému růstu do délky a rozvoji akromegaloidních rysů (6).
Naopak nadměrná exprese genu IGF2 (kóduje fetální růstový promotor IGF-II) u Beckwithova-Wiedemannova syndromu (BWS) způsobí nejen nadměrný růst, ale vazbou na inzulinový receptor vyvolá také hypoglykemie. Současně se u BWS nadměrně exprimuje také INS (gen pro inzulin), takže účinky obou hormonů se prolínají a lze je obtížně odlišit (7).
Beckwithův-Wiedemannův syndrom (BWS)
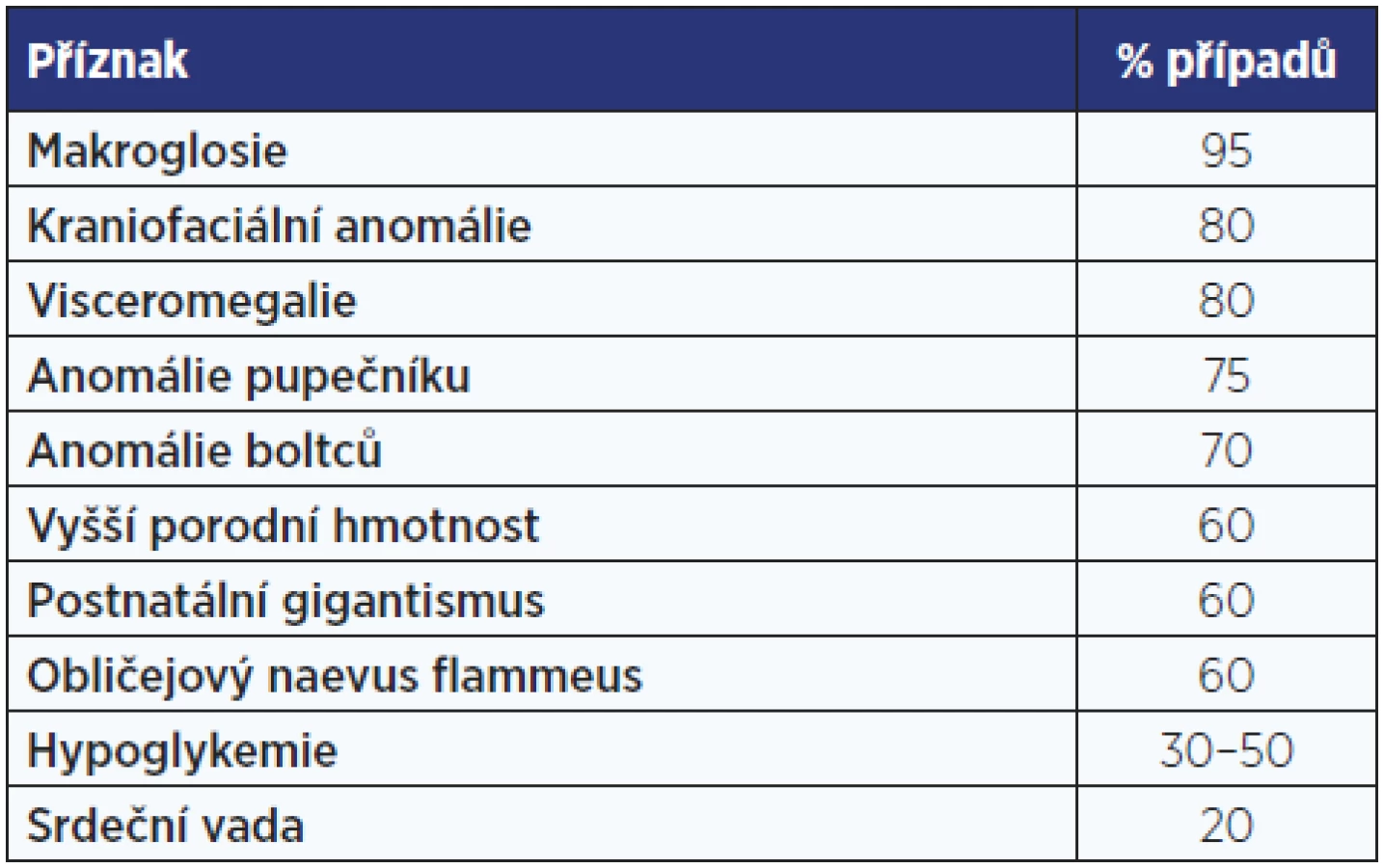
Beckwithův-Wiedemannův syndrom má příznačný fenotyp s umbilikální kýlou, makroglosií a gigantismem, který se projevuje již in utero (tab. 1, obr. 2). Příčinou jsou genetické, ale častěji epigenetické změny (imprinting) v oblasti 11p15.5 – v úseku chromosomu, kde jsou geny pro růstové faktory. V této oblasti může být trisomie s duplikaturou od otce nebo paternální disomie se ztrátou mateřského chromosomu.


V kritické oblasti se nadměrně exprimují geny INS (kóduje inzulin) a IGF2. Je také narušena exprese tumor-supresorových genů CDKN1C a H19 (3, 4).
Zásadním problémem dětí s BWS jsou hyperinzulinemické hypoglykemie, vrozené srdeční vady a vysoká prevalence embryonálních nádorů v útlém věku (tab. 2, obr. 3). I po narození pokračuje rychlý, avšak nikoliv extrémní růst.

Mortalita dětí s BWS během prvního roku života je vysoká – dosahuje 20–25 %. Příčinou časného úmrtí může být nedonošenost, srdeční vady, hypoglykemie nebo nádory. Toto riziko může poněkud omezit důsledně prováděný onkologický screening (tab. 3). Intelektový vývoj závisí zejména na poškození hypoglykemiemi. Po překonání kojeneckého období již vývoj bývá poměrně příznivý, hypoglykemie ustupují. Dospělá výška je zpravidla normální (3–5).

Pseudoakromegalie
Pseudoakromegalie je stav s akromegaloidním fenotypem, inzulinovou rezistencí, střevní polypózou a suprimovanými hladinami IGF-I (6). Příčinou je postreceptorový defekt inzulinové signalizace, který narušuje metabolickou signalizační dráhu cestou inzulinového receptoru, ale zachovává prorůstovou (mitotickou) signalizaci cestou receptoru IGF 1. typu (srovn. obr. 1).
Sotosův syndrom
Nositelé Sotosova syndromu mají dlouhý úzký obličej, vysoké čelo, začervenalé tváře a malou bradu. Po nadměrném růstu ve fetálním období pokračuje růstová akcelerace i v dětství, dospělá výška je ale normální. Postupně se rozvíjí makrocefalie (obr. 4). Bývá přítomna hypotonie s mírným vývojovým opožděním, lehký až střední intelektový deficit, porucha vývoje řeči a poruchy chování – časté jsou ADHD (attention deficit and hyperactivity disorder), fobie, obsese, nutkavé a impulzivní chování. U 25 % dětí se rozvine epilepsie. K dalším klinickým problémům patří skolióza, křeče, poruchy sluchu a zraku, některé vrozené vývojové odchylky. Mírně zvýšené je onkologické riziko (tab. 2).
Příčinou Sotosova syndromu jsou mutace genu NSD1 (nuclear receptor binding SET domain protein 1), který kóduje transkripční faktor přímo zodpovědný za genovou expresi, včetně genů zúčastněných v regulaci růstu a vývoje (8, 9).



Simpsonův-Golabiové-Behmelové syndrom (SGBS)
Simpsonův-Golabiové-Behmelové syndrom je vázaný na chromosom X. Postižení chlapci se často rodí s makrosomií a jejich rychlý růst a hmotnostní přírůstky mohou pokračovat i po narození. Fenotyp je variabilní – závažné formy jsou letální, nositelé mírnějších forem s různou tíží klinického postižení žijí do dospělosti. Typické faciální rysy zahrnují hypertelorismus, makrostomii (velká široká ústa), makroglosii, široký antevertovaný nos a poruchy uzávěru patra. V pozdějším věku má obličej hrubé rysy a bývá označován jako „buldočí tvář“. Na hrudníku mohou být nadpočetné prsní bradavky, na břiše diastáza přímých břišních svalů a umbilikální hernie. Někdy se chlapci rodí s brániční kýlou, vrozenou srdeční vadou, nefro-, hepato - a splenomegalií (obr. 5). Intelektový vývoj je variabilní (10–13).
Tyto děti mají významně zvýšené onkologické riziko, u 8–10 % se objeví některý z embryonálních tumorů, především nefroblastom a hepatoblastom (tab. 2 a 3) (14).
Příčinou jsou mutace nebo delece genu GPC3, který kóduje glypikan 3, vzácně genu GPC4 kódujícího glypikan 4. Glypikany jsou heparansulfátové proteiny uchycené na buněčné membráně, které pomáhají akumulovat ligandy pro určité membránové receptory. Tím se významnou měrou podílejí na regulaci buněčného růstu, proliferace a apoptózy.




Weaverův syndrom
Weaverův syndrom se projevuje makrosomií, někdy ve spojení s makrocefalií a mírným intelektovým deficitem. Pro facies jsou charakteristické široké čelo, hypertelorismus, velké nízko posazené boltce a mikrognacie. Může docházet ke svalovým kontrakturám s deformitami kloubů, zvláště prstů rukou i nohou, které omezují hybnost, a ke kyfoskolióze. Svalový tonus může být snížený i zvýšený, může být přítomná umbilikální hernie. Také nositelé Waeverova syndromu mají v útlém dětství zvýšené onkologické riziko (tab. 2 a 3).
Příčinou Weaverova syndromu jsou mutace genu EZH2, který kóduje enzym histon-methyltransferázu. Její methylační aktivita usnadňuje tvorbu heterochromatinu a tím potlačuje expresi genů. Histon-methyltransferáza také zajišťuje remodelaci chromosomového heterochromatinu nutnou pro buněčné dělení (9, 15).
SYNDROMY NADMĚRNÉHO RŮSTU V DĚTSTVÍ A V ADOLESCENCI
Nadměrný růst s vysokou růstovou rychlostí v dětství a dospívání je zpravidla důsledkem hormonální nadprodukce. Příčinou může být předčasná produkce sexuálních steroidů v dětství (pubertas praecox nebo pseudopubertas praecox) – v těchto případech zpravidla upoutá dříve pozornost předčasný pubertální vývoj než nadměrný růst. K růstovému urychlení vede také nadprodukce hormonů štítné žlázy (Gravesova-Basedowova tyreotoxikóza, autonomní hyperfunkční uzel štítné žlázy nebo ojediněle ektopická nadprodukce tyreoidálních hormonů – tzv. struma ovarii). I v těchto případech příznaky hypertyreózy upoutají pozornost jako první, před rozvojem nadměrného růstu (16).
Klasikou příčinou nadměrného růstu je nadprodukce růstového hormonu s klinickým obrazem gigantismu. Ten může být provázen prvky akromegalie – akromegalogigantismus (akrogigantismus), které se zvýrazňují s věkem nebo objevují v průběhu dospívání a později. Příčinou nadměrné výroby růstového hormonu je zpravidla hypofyzární adenom – sporadický nebo familiární (FIPA – familiární izolovaný pituitární adenom) (17).
FIPA – familiární izolovaný pituitární adenom (autosomálně dominantní)
Geneticky podmíněné familiární formy hypofyzárních adenomů se dědí autosomálně dominantně. Jejich genová podstata je v současné době předmětem intenzivního výzkumu. U 15–25 % rodin s FIPA jsou příčinou patogenní varianty genu AIP. U ostatních autosomálně dominantních forem zatím genetická příčina objasněna nebyla.
Gen AIP kóduje bílkovinu reagující s aryl-hydrokarbonovým receptorem (aryl hydrocarbon receptor-interacting protein). AIP protein působí jako nádorový supresor – brání nadměrné proliferaci buněk adenohypofýzy. Mutace genu AIP naruší schopnost proteinu AIP regulovat buněčný růst a proliferaci, což vede ke zmnožení buněk a ke vzniku adenomu (18).
Adenomy se při nosičství mutací AIP projeví zpravidla v mladém věku, často již v dětství, a jsou obecně větší než tumory bez přítomnosti mutací AIP. V 90 % případů produkují růstový hormon, někdy společně s prolaktinem. 9 % z nich produkuje pouze prolaktin, 1 % má jinou hormonální produkci (17, 18).
Obr. 6a–b znázorňují nadměrný růst a morfologii pituitárního makroadenomu u dívky staré 7,5 roku s patogenní mutací v genu AIP. Tumor byl endoskopicky exstirpován transnazální cestou pod MRI kontrolou (obr. 6c – stav po exstirpaci). Dívka měla již při diagnóze makroadenomu centrální hypotyreózu, po výkonu se rozvinul panhypopituitarismus.
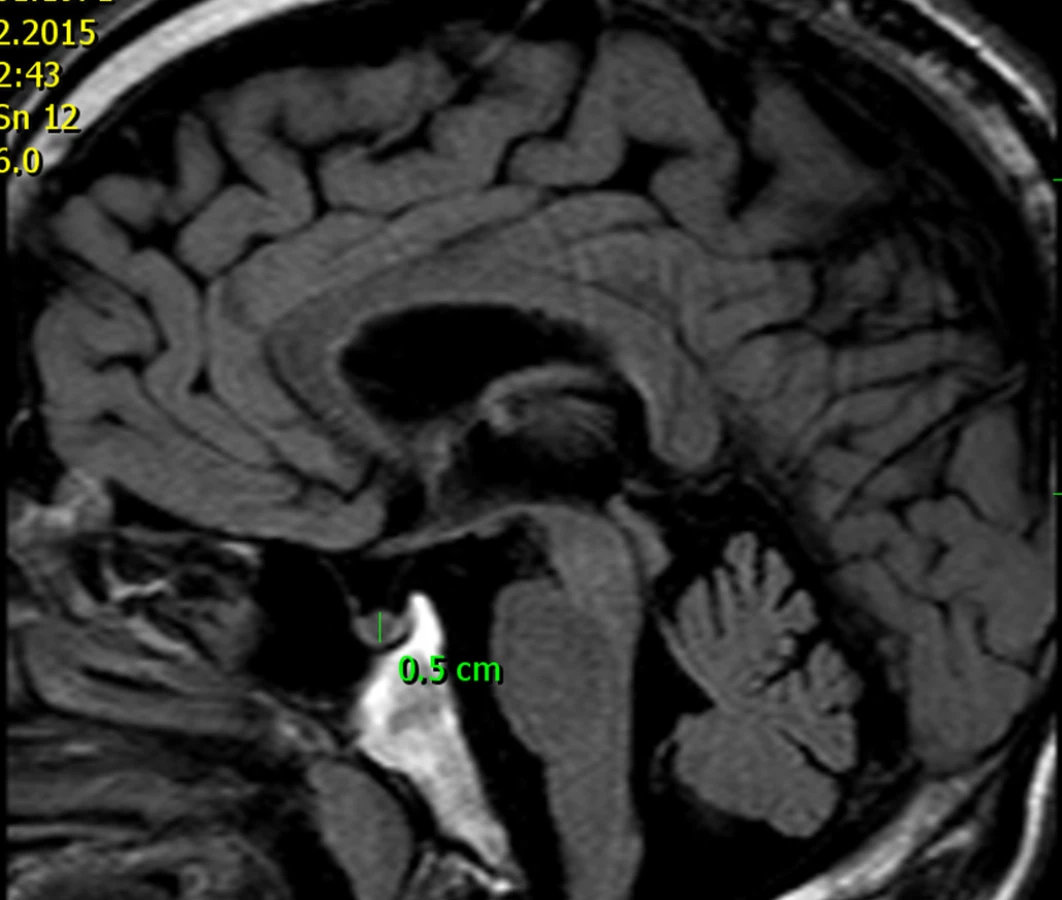
U tatínka této dívky jsme prokázali shodnou patogenní mutaci genu AIP. Ve věku 46 let měřil 177 cm a při vyšetření magnetickou rezonancí (obr. 7) jsme nalezli pituitární mikroadenom velikosti 5 mm, endokrinně němý. I u jiných rodin bylo zjištěno, že nosičství patogenní mutace AIP se může projevit také hormonálně a klinicky němým mikroadenomem.



X-vázaný akrogigantismus
Mutace genu GPR101, který je lokalizován na chromosomu X v oblasti Xq26, a mikroduplikace v této oblasti způsobují gigantismus s akromegalií. Gen GPR101 kóduje receptor spojený s G-proteinem, jehož ligand je zatím neznámý (19).
X-vázaný akrogigantismus (X-LAG – X-linked acrogigantism) je charakterizován normální porodní hmotností, ale poté navazuje časná růstová akcelerace (často již kolem 1 roku, nejpozději do 5 let věku) v důsledku nadměrné produkce růstového hormonu, jež bývá spojena s nadprodukcí prolaktinu. Příčinou je makroadenom hypofýzy s vysokou penetrancí, který je rezistentní k běžné terapii, brzy recidivuje a vyžaduje opakované neurochirurgické intervence ve spojení s medikamentózní léčbou kabergolinem nebo pegvisomantem, případně s radioterapií.
Právě vysoká penetrance těchto adenomů může způsobovat klinicky extrémní formy akrogigantismu (20).
Další geny spojené s hypofyzárními tumory
Nejméně 10 dalších genů je spojeno s rizikem hypofyzárního tumoru. Patří mezi ně MEN1, PRKAR1A, PRKACB, CDKN1B, DICER1 a geny ze skupiny SDH (A, B, C, D a AF2). Sukcinátdehydrogenáza (SDH) je enzymový komplex složený ze 4 hlavních podjednotek A, B, C a D. Je lokalizovaný na vnitřní mitochondriální membráně a je součástí citrátového cyklu i dýchacího řetězce. Germinální mutace obou alel kódujících genů vedou k syndromu familiárního paragangliomu, v případě heterozygotní mutace způsobí somatická inaktivace druhé alely deficit kódované podjednotky, destabilizaci celého komplexu a ztrátu jeho enzymatické funkce, což zásadním způsobem ovlivňuje riziko onkogeneze.
Kromě genů ze skupiny SDH se u mutací všech dalších uvedených genů může hypofyzární tumor projevit již v dětském věku a dospívání. Patogenní varianty PRKAR1A způsobují Carneyho komplex, patogenní varianty DICER1 vedou ke vzniku hypofyzárního blastomu s Cushingovou nemocí ve věku do 2 let. K dalším možným projevům patří mimo jiné pleuropulmonální blastom.
Adenom hypofýzy s nadprodukcí růstového hormonu se také může objevit u některých dětí s McCuneovým-Albrightovým syndromem v důsledku buněčného chimérismu s mutací GNAS (21).
Jiné případy nadměrného růstu v dětském věku a dospívání
Nadměrný vzrůst s narušenou proporcionalitou může být důsledkem primární poruchy metabolismu pojiva nebo důsledkem hypogonadismu.
Mezi poruchy spojené s disproporcionálním nadměrným vzrůstem (tj. s dlouhými končetinami) patří Marfanův syndrom, homocystinurie, Klinefelterův syndrom nebo jiné formy hypogonadismu (16).
PARALELY SYNDROMŮ NADMĚRNÉHO RŮSTU V DOSPĚLOSTI
Před érou moderní medicíny vedla neléčená nadprodukce růstového hormonu v dětství ke gigantismu v dospělosti. V některých částech světa k tomu dochází i dnes. Takoví lidé potom zaujímají pomyslné první příčky mezi „nejvyššími muži světa“ v Guinessově knize rekordů. Nejvyšší muž současnosti s výškou 251 cm je nositelem mutace v genu GPR101, která způsobuje urputnou a obtížně léčitelnou formu gigantismu (20). Spekuluje se o tom, že nositeli GPR101 mutací mohli být další slavní giganti minulosti.
„Irský obr“ Charles Byrne, známý pod uměleckým jménem O’Brien, se narodil v roce 1761 v Littlebridge v Severním Irsku. Proslavil se v 80. letech 18. století v Londýně, kde se předváděl jako kuriozita. Zemřel ve 22 letech v důsledku problémů s alkoholem. Údajně měřil více než 8 stop (244 cm). Podle kostry je však zřejmé, že měřil 7 stop a 7 palců (231 cm). Po smrti získal Byrneovo tělo chirurg John Hunter. Kostru vystavuje Hunterovo muzeum v londýnském sídle Royal College of Surgeons (obr. 8).

Prof. Marta Korbonitsová si vypůjčila Byrneův zub a extrahovala z něj DNA. Zjistila, že Byrne měl mutaci genu AIP R304* (22). Stejnou mutaci mají současní pacienti s gigantismem a akromegalií v Ulsteru v Severním Irsku. Jejich předkové pocházejí z několika blízkých vesnic, ve kterých byl historicky zaznamenán nápadně častý výskyt gigantismu. V jedné z těchto vesnic trávil jedno léto spisovatel Jonathan Swift a našel tam zřejmě inspiraci ke svému příběhu o Gulliverovi mezi Liliputy. Profesorka Korbonitsová se svým týmem nakonec analýzou haplotypů prokázala, že ancestrální mutace R304* pochází od společného předka, který žil přibližně před 66 generacemi (23).
Matematickým výpočtem bylo zjištěno, že tuto mutaci může nést 200–300 žijících osob, z nichž většina o ní neví. Včasná genetická diagnóza by u nich umožnila odhalit a řešit hormonální nadprodukci ještě před klinickými projevy akromegalie či gigantismu. Rozsáhlá osvětová akce v Ulsteru, při které byl zájemcům nabídnut odběr krve v několika krytých parkovištích supermarketů, vedla k identifikaci dalších 153 současných nositelů „Byrneovy“ mutace (24).
ZÁVĚR
Nadměrný růst dětí se nyní dostává do popředí zájmu dětských endokrinologů. Správná klinická diagnóza spojená s genetickou diagnostikou přináší nové léčebné možnosti a umožňuje cílený screening zaměřený na včasnou detekci nádorů u fetálních a novorozeneckých syndromů nadměrného růstu, případně na rozpoznání presymptomatických nosičů genové mutace v rodinách s FIPA. Dokáže tak zlepšit kvalitu života nebo dokonce zachránit život při včasné diagnostice onkologického onemocnění.
Problematiku genetické podmíněnosti růstu autoři řeší s podporou grantu Agentury pro zdravotnický výzkum (AZV) Ministerstva zdravotnictví ČR č. 16-31211A.
Adresa pro korespondenci:
prof. MUDr. Jan Lebl, CSc.
Pediatrická klinika 2. LF UK a FN Motol
V Úvalu 84
150 06 Praha 5
Tel.: 224 432 000, 224 432 001
e-mail: jan.lebl@lfmotol.cuni.cz
Sources
1. Heights of presidents and presidential candidates of the United States. Dostupné na: https://en.wikipedia.org/wiki/Heights_of_presidents_and_presidential_candidates_of_the_United_States
2. Kiess W, Kratzsch J, Kruis T et al. Genetics of human stature: insight from single gene disorders. Horm Res Paediatr 2011; 76(Suppl. 3): 11–13.
3. Edmondson AC, Kalish MK. Overgrowth syndromes. J Pediatr Genet 2015; 4 : 136–143.
4. Visser R, Kant SG, Wit JM, Breuning MH. Overgrowth syndromes: from classical to new. Pediatr Endocrinol Rev 2009; 6 : 375–394.
5. Lapunzina P. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet 2005; 137 : 53–71.
6. Flier JS, Moller DE, Moses AC et al. Insulin-mediated pseudoacromegaly: clinical and biochemical characterization of a syndrome of selective insulin resistance. J Clin Endocrinol Metab 1993; 76 : 1533–1541.
7. Tan TY, Amor DJ. Tumour surveillance in Beckwith-Wiedemann syndrome and hemihyperplasia: a critical review of the evidence and suggested guidelines for local practice. J Pediatr Child Health 2006; 42 : 486–490.
8. Sotos JF. Sotos syndrome 1 and 2. Pediatr Endocrinol Rev 2014; 12 : 2–16.
9. Opitz JM, Weaver DW, Reynolds JF jr. The syndromes of Sotos and Weaver: reports and review. Am J Med Genet 1998; 79 : 294–304.
10. Weichert J, Schrorer A, Amari F et al. A 1Mb-sized microdeletion Xq26.2 encompassing the GPC3 gene in a fetus with Simpson-Golabi-Behmel syndrome Report, antenatal findings and review. Eur J Med Genet 2011; 54 : 343–347.
11. Waterson J, Stockley TL, Segal S, Golabi M. Novel duplication in glipican-4 as an apparent cause of Simpson-Golabi-Behmel syndrome. Am J Med Genet 2010; 152A: 3179–3181.
12. Cottereau E, Mortemousque I, Moizard MP et al. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet 2013; 163C: 92–105.
13. Tenorio J, Arias P, Martínez-Glez V et al. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis 2014; 9 : 138–145.
14. DeBraun MR, Ess J, Saunders S. Simpson-Golabi-Behmel syndrome: progress toward understanding the molecular basis for overgrowth, malformation, and cancer predisposition. Mol Genet Metab 2001; 72 : 279–286.
15. Tatton-Brown K, Murray A, Hanks S et al. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet 2013; 161A: 2972–2980.
16. Albuquerque EV, Scalco RC, Jorge AA. Management of endocrine disease: diagnostic and therapeutic approach of tall stature. Eur J Endocrinol 2017; 176: R339–R353.
17. Hernández-Ramírez LC, Gabrovska P, Dénes J et al. Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J Clin Endocrinol Metab 2015; 100: E1242–E1254.
18. Hernández-Ramírez LC, Trivellin G, Stratakis CA. Role of phosphodiesterases on the function of aryl hydrocarbon receptor-interacting protein (AIP) in the pituitary gland and on the evaluation of AIP gene variants. Horm Metab Res 2017; 49 : 286–295.
19. Naves LA, Daly AF, Dias LA et al. Aggressive tumor growth and clinical evolution in a patient with X-linked acro-gigantism syndrome. Endocrine 2016; 51 : 236–244.
20. Beckers A, Lodish MB, Trivellin G et al. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer 2015; 22 : 353–367.
21. Rostomyan L, Daly AF, Petrossians P et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocr Relat Cancer 2015; 22 : 745–757.
22. Chahal HS, Stals K, Unterländer M et al. AIP mutation in pituitary adenomas in the 18th century and today. N Engl J Med 2011; 364 : 43–50.
23. Salvatori R, Radian S, Diekmann Y et al. In-frame seven amino-acid duplication in AIP arose over the last 3000 years, disrupts protein interaction and stability and is associated with gigantism. Eur J Endocrinol 2017; 177 : 257–266.
24. Radian S, Diekmann Y, Gabrovska P et al. Increased population risk of AIP-related acromegaly and gigantism in Ireland. Hum Mutat 2017; 38 : 78–85.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Hypogonadismus u mužů a jeho léčba
- Nadměrný růst u dětí a dospělých: nový klinický pohled, nové geny, nové fenotypy
- Adrenokortikální insuficience
- Diagnostika a další péče o pacienty s tyreoidálními uzly: doporučení American Thyroid Association 2015 modifikované pro podmínky České republiky