
Mitochondrie – od vzniku po dnešní terapeutické možnosti
Mitochondria – from origin to current therapies
Mitochondria are part of almost all eukaryotic cells. Their function is to produce and release energy for the needs of the cell, provide beta-oxidation, participate in the synthesis of steroids, serve for heat production through non-shaking thermoregulation or for calcium ions storage. They are also involved in the cell apoptosis and membrane potential regulation.
Mitochondrial energy production affects cell proliferation, changes in gene expression, and the formation of reactive oxygen species (ROS).
Mitochondrial DNA (mtDNA) is located in the matrix of mitochondria and is inherited exclusively maternally. Hence it is a suitable tool for studying the evolution of the human population, for elucidating evolutionary relationships, and for mapping the migration throughout history.
Gene mutations in nuclear DNA or mtDNA negatively impact the mitochondrial activity. As a result, various mitochondrial diseases arise, which are characterized by a specific type of heredity and various clinical manifestations. Their origin has most often been explained by the theory of mitochondrial aging.
The quality of mitochondria is negatively affected, among other, by environmental effects, mainly radiation. Most of all they benefit from healthy lifestyle including diets rich in vitamins, phytonutrients, and antioxidants.
Keywords:
Mutation – Mitochondrial DNA – Transplantation – Mitochondrial diseases – Regeneration – organelle – cellular energy – ATP – haplotype
Authors:
Vít Smejkal 1; Ilona Hromadníková 2
![]() ; Anežka Palmová 1; Jana Šimková 1; Radek Klubal 1
; Anežka Palmová 1; Jana Šimková 1; Radek Klubal 1
Authors‘ workplace:
Medicínské centrum Praha, s. r. o.
1; Oddělení molekulární biologie a patologie buňky 3. LF UK v Praze
2
Published in:
Čas. Lék. čes. 2021; 160: 332-339
Category:
Review Article
Overview
Mitochondrie jsou součástí téměř všech eukaryotických buněk. Jejich funkcí je produkce a uvolňování energie k potřebám buňky, zajišťují beta oxidaci, podílejí se na syntéze steroidů, slouží k produkci tepla netřesovou termoregulací či ke skladování vápníkových iontů. Účastní se také apoptózy buňky a regulace membránového potenciálu.
Energetická produkce mitochondrií ovlivňuje proliferaci buněk, změny genové exprese a tvorbu reaktivních forem kyslíku (ROS).
Mitochondriální DNA (mtDNA) je uložená v matrix této organely a dědí se výhradně maternálně, proto je vhodným nástrojem pro zkoumání evoluce lidské populace, objasnění evolučních vztahů a také pro mapování migrace v průběhu historie.
Genové mutace v jaderné DNA nebo mtDNA negativně ovlivňují mitochondriální aktivitu. V jejich důsledku vznikají různá mitochondriální onemocnění, která se vyznačují specifickým typem dědičnosti a různorodými klinickými projevy. Jejich vznik se nejčastěji vysvětluje teorií mitochondriálního stárnutí.
Na kvalitu mitochondrií negativně působí mimo jiné některé vlivy prostředí, zejména záření. Příznivý vliv na mitochondrie má především zdravý životní styl, včetně stravy bohaté na vitaminy, fytonutrienty a antioxidanty.
Klíčová slova:
mutace – transplantace – mitochondriální onemocnění – regenerace – organela – buněčná energie – ATP – mitochondriální DNA – haplotyp
ÚVOD
Mitochondrie jsou přítomny v cytoplazmě většiny eukaryotických buněk a představují vysoce dynamické organely uspořádané do souvislé sítě, která se může měnit v závislosti na stavu energetického metabolismu buňky.
Jedním z nejdůležitějších metabolických pochodů odehrávajících se v mitochondriích je proces oxidativní fosforylace, pokrývající až 90 % energetických nároků organismu savců (1). Mitochondrie slouží tedy především jako zdroj energie, ale plní i další významné funkce nezbytné pro život.
Díky pokročilému mikromolekulárnímu výzkumu je stále více patrné, jak fungování mitochondrií ovlivňuje vitalitu, nemocnost i rychlost stárnutí. Problematika mitochondrií, která se ukazuje být jednou z klíčových pro lidské zdraví a dlouhověkost, je tak v popředí zájmu nejen výzkumných a lékařských pracovišť, ale i veřejnosti.
ENDOSYMBIOTICKÁ TEORIE
Původ mitochondrií není doposud zcela uspokojivě vysvětlen a na toto téma existuje řada hypotéz. Mezi nimi je zatím nejuznávanější endosymbiotická teorie o formování eukaryotické buňky, podle které jsou mitochondrie považovány za endosymbionty původních amitochondriálních prokaryotických hostitelských buněk.
Podle této hypotézy došlo k tomu, že amitochondriální prokaryotická hostitelská buňka pohltila jinou jednobuněčnou bakterii (alfaproteobakterii). Z pohlcené bakterie zůstala jen část (organela se dvěma membránami) (2). Změny, které nastaly po ustanovení vztahu mezi těmito bakteriemi (alfaproteobakterií a hostitelskou buňkou), vyústily ve ztrátu postradatelných genů a přenos většiny genů do buněčného jádra. Tak došlo k přerozdělení genomu na jaderný a mitochondriální a vznikla nefotosyntetizující protoeukaryotická buňka (1).
VÝSKYT V ZÁVISLOSTI NA TYPU BUNĚK
Mitochondrie jsou až na výjimky součástí eukaryotických buněk všech živočichů. Nevyskytují se například u prvoků žijících v anaerobním prostředí (diplomonády, archaméby či mikrosporidie), kteří mají vlastní způsob výroby energie (3).
V lidských buňkách mitochondrie chybí pouze v oční čočce, zrohovatělých buňkách a v červených krvinkách (pasivní pohyb erytrocytů krevním řečištěm nevyžaduje žádnou energii, absence mitochondrií dává více prostoru hemoglobinu) (4, 5).
Nejčastěji se vyskytují v cytoplazmě buněk společně s dalšími organelami. Tvar, velikost (0,75–3 μm) a počet mitochondrií závisí na typu a funkci buněk, kterých jsou součástí. Počet se v různých buňkách liší. V lidské kosterní svalovině nebo v játrech se vyskytují ve stovkách až tisících na jednu buňku.
Mitochondrie lze pozorovat fluorescenční konfokální mikroskopií. Většinou zaujímají určité pozice, ale mohou se v buňce přesunout na místa potřeby. Mohou mít formu rozvětvených tubulárních organel (ve svalových vláknech, kde tvoří síť okolo sarkomer) nebo sférické částice (např. v lymfocytech) (6).
STAVBA MITOCHONDRIÍ
Mitochondrie jsou obaleny dvěma membránami s odlišnými vlastnostmi. Vnější membrána je hladká, velmi permeabilní, obsahuje větší množství lipidů a nacházejí se v ní poriny (translokátorové integrální membránové proteiny).
Vnitřní membrána je zvrásněná (má tak větší plochu), a to buď do podoby záhybů (krist), nebo trubiček (tubulů). Obsahuje méně lipidů a více proteinů. Je málo prostupná pro ionty v důsledku vysokého obsahu kardiolipinu.
Vnitřní prostor vyplňuje matrix, mitochondriální cytoplazma, v níž se nacházejí elementární tělíska (ATP-ozómy, na kterých probíhá syntéza adenosintrifosfátu [ATP]), cirkulární mitochondriální deoxyribonukleová kyselina (mtDNA), enzymy a proteosyntetický aparát (ribozomy). Stavba mitochondrie je znázorněna na obr. 1.

FUNKCE MITOCHONDRIÍ
Hlavní funkcí těchto organel je produkce a uvolňování energie k potřebám buňky. Slouží také k regulaci buněčného metabolismu – zajišťují beta oxidaci (aerobní odbourávání mastných kyselin) a podílejí se na syntéze steroidů, dále k produkci tepla tzv. netřesovou termoregulací či ke skladování vápníkových iontů. Účastní se rovněž apoptózy buňky a regulace membránového potenciálu.
Proces produkce energie v aerobních buňkách spočívá ve spřažení exergonních reakcí a syntézy ATP z adenosindifosfátu (ADP).
MITOCHONDRIÁLNÍ VS. JADERNÁ DNA
MtDNA je uložena v matrix mitochondrie. Mitochondriální genom je tvořen dvouřetězcovou cirkulární DNA. Ta se u člověka skládá z 16 569 párů nukleových bází. MtDNA obsahuje 37 genů, z nichž 24 kódují části proteosyntetického aparátu mitochondrie (7).
Většina mitochondriálních proteinů je však kódována jadernou DNA. V mtDNA se nevyskytují introny, což vylučuje možnost vzniku více různých produktů z jednoho genu mechanismem alternativního sestřihu, a také se neuplatňuje introny zprostředkovaná regulace exprese genů. Zároveň chybí nekódující regulační region, tzv. D-loop (displacement loop) (8). MtDNA se liší od jaderné DNA i stopkodony, jež jsou zde zastoupeny: pro mtDNA jsou typické stopkodony AGA (adenin-guanin-adenin) a AGG (adenin-guanin-guanin) (7).
Nemá takové množství opravných mechanismů jako jaderná DNA. Při dělení buňky se nedělí jen jedna mitochondrie, nýbrž všechny, které jsou v buňce přítomné, čímž v porovnání s dělením jádra stoupá pravděpodobnost vzniku chyb (mutací) (9). Frekvence vzniku mutací mtDNA je tak 10–20× větší než u jaderné DNA (1).
MtDNA se dědí výhradně maternálně, a tak u mutací přímo v její sekvenci dochází ke genetickému přenosu na potomky pouze z matčiny strany (8). Oproti tomu mutace způsobené poruchou mitochondriální replikace, jež je regulována jadernými geny, se dědí od obou rodičů (1).
MITOCHONDRIÁLNÍ HAPLOSKUPINY
MtDNA je vhodným nástrojem pro zkoumání evoluce lidské populace, objasnění evolučních vztahů a také pro mapování migrace v průběhu historie. Nízký počet mitochondriálních rekombinací u lidí společně s maternální dědičností napomáhá mapování genealogických vztahů (10). Data získaná z mtDNA však můžeme sledovat pouze v omezeném období, zhruba posledních 200 tisíc let. Vedou nás ke společnému předkovi (tzv. mitochondriální Evě) žijící tehdy na africkém kontinentu (11).
Na základě mutací v mtDNA v dnešní době rozlišujeme 23–38 hlavních mitochondriálních haplotypů (např. L, G, H…) a až 4000 vedlejších (např. H2a2a, G2a2…) (11). Z genetické analýzy mtDNA a podle mitochondriálního haplotypu je možné určit predispozici jedince k různým onemocněním a také jeho genealogii.
Evoluce mitochondriálních haploskupin
Makrohaploskupina L, která vznikla jako první, je specifická pro subsaharskou Afriku. Nejstarší haploskupiny vznikly z ní – L0, L1, L2 a L3; nejběžněji se vyskytují ve východní a saharské Africe. L3 je předchůdkyní haploskupin M a N, z nichž se vyvinula většina neafrických mitochondriálních haploskupin. Populace lidí s haploskupinami M a N následně kolonizovaly Evropu a Asii, a daly tak vzniknout novým haploskupinám: z N vznikly H, V, J, T, U, K, I, W, X a z M a N se vytvořily A, B, C, D, G, X, Y, Z (12). Na obr. 2 je znázorněna migrace lidí a vznik nových mitochondriálních haploskupin.

Předpokládá se, že geografická distribuce různých mitochondriálních haploskupin (různých variant mtDNA) je výsledkem adaptační klimatické a nutriční selekce. Zdá se, že určité populace se dokázaly adaptovat na studené klimatické podmínky funkčními změnami v mtDNA zvyšujícími oxidační fosforylaci, a tím zvětšily produkci ATP. To umožnilo lidem s původně africkou haploskupinou L přežít na severní polokouli (12).
ENERGETICKÁ PRODUKCE MITOCHONDRIÍ A NOVÁ BIOMEDICÍNSKÁ PARADIGMATA
Energetická produkce mitochondrií se většinou hodnotí na základě efektivity oxidační fosforylace (tvorby ATP), která následně ovlivňuje tepelnou bilanci jedince. Je spojena s proliferací buněk, změnou genové exprese a tvorbou reaktivních forem kyslíku (ROS), které dokážou snadno reagovat s dalšími molekulami v buňce. Ovlivňuje i pufrování vápníku (proces stabilizace koncentrace volných iontů vápníku v buňkách) a regulaci apoptózy skrze přechodový pór mitochondriální permeability (13, 14). Se sníženou produkcí ATP souvisejí i menší produkce ROS, pomalejší proces stárnutí a oddálení rozvoje degenerativních, metabolických a nádorových onemocnění (13).
Energetická produkce mitochondrií ovlivňuje téměř každou činnost buňky v organismu (15). Prohlubováním chápání mitochondriální biologie vznikají i nová biomedicínská paradigmata:
- První z nich zní, že na propuknutí onemocnění se podílí celá řada biochemických a bioenergetických procesů v buňce. Proto musejí být bioenergetické změny vzniklé v buňce analyzovány prostřednictvím systémové biologie (15). Důsledkem tohoto poznání je také odklon od tradičního konceptu „jeden gen à jeden polypeptid à jedno onemocnění“.
- Druhým paradigmatem je maternální dědičnost mtDNA, jež je příkladem nemendelovské dědičnosti. Maternálně jsou děděny mitochondriální geny, mutace v mitochondriální DNA a regulace exprese mitochondriálních genů (15).
- Třetí paradigma postuluje nestejné energetické nároky různých tkání a orgánů. V případě lokálního deficitu energie se začnou objevovat klinické symptomy specifické pro daný orgán (tkáň) (15).
GENOVÉ MUTACE A MITOCHONDRIÁLNÍ ONEMOCNĚNÍ
Genové mutace v jaderné DNA nebo mtDNA negativně ovlivňují mitochondriální aktivitu. V jejich důsledku vznikají různá mitochondriální onemocnění, která se vyznačují specifickým typem dědičnosti a různorodými klinickými projevy.
Od procentuálního zastoupení mitochondrií s mutovanou DNA se odvíjí tzv. prahová hypotéza, podle které určitá výše tohoto podílu způsobí daný defekt. Prahová hranice pro vznik defektu např. u mozku je dána 60% účinností mitochondrií (16) a u srdce 40% účinností (17). Mitochondriální onemocnění se mohou projevit kdykoliv během života (energetická zásoba a životnost mitochondrií se v závislosti na věku stále snižuje) a klinicky se projevují onemocněním zdánlivě nesouvisejících orgánů. Jsou-li mutacemi postiženy všechny molekuly mtDNA, mluvíme o tzv. homoplastické poruše, pokud mutaci vykazují jen některé molekuly mtDNA, jedná se o tzv. heteroplastii (18).
TEORIE STÁRNUTÍ
MtDNA se replikuje nezávisle na dělení buňky či replikaci jaderné DNA a častěji než jaderný genom. Tento fenomén se nazývá „relaxovaná replikace“ (19). Mitochondrie plně odrážejí proces stárnutí. Stárnutí vede k nahromadění poškození v mtDNA, která nemohou být efektivně opravena. V důsledku těchto akumulujících se změn buňka nakonec není schopna vykonávat svoje funkce a přechází do fáze programované buněčné smrti – apoptózy. V mtDNA se mutace hromadí rychleji než v jaderném genomu. Je však nutné podotknout, že čistá kauzalita mezi produkcí ROS a stárnutím není zatím stoprocentně potvrzena. Fyziologické koncentrace ROS působí příznivě na dlouhověkost mitochondrií a chovají se jako signální molekuly (hormeze, hypotéza o příznivém působení mírné zátěže na živý organismus) (8, 19, 20).
V mtDNA dochází během replikace v průběhu celého života jedince (od embryogeneze po celé prenatální a postnatální období) k mutacím, zejména bodovým (SNP) a delecím, které jsou přítomny u každého jedince v různém množství. Vzniká tzv. heteroplastie (mitochondrie obsahuje směs mutovaných a nepoškozených molekul mtDNA), která po překonání prahového efektu vede k propuknutí celé řady onemocnění, a to kdykoli v průběhu života jedince. V důsledku četné replikace mtDNA a také pod vlivem vnitřních a vnějších podnětů vyvolávajících tvorbu ROS dochází k postupné akumulaci delecí a SNP v mtDNA v jednotlivých tkáních (8, 19, 20). Oba mechanismy ukazuje obr. 3.

(A) Replikační chyby (bílé a černé tečky) vznikají již v raném období a v průběhu celého života. Tyto mutace jsou po celý život
přenášeny a množeny mitotickou segregací a replikací.
(B) Mutace v mtDNA se objevují během života v důsledku oxidačního stresu. Oxidační stres narůstá s věkem, a tím také přibývá
mutací v mtDNA. U starých lidí se vyskytuje vysoký podíl mutované mtDNA s nízkým poměrem individuální heteroplazmie,
přičemž heteroplazmie > 80 % všech molekul mtDNA je již závažná. Dominance mutované mtDNA nad zdravou mtDNA způsobuje
mitochondriální nedostatečnost a apoptózu buňky u starších jedinců.
Teorie kalorické restrikce hovoří o tom, že délka života se prodlužuje u lidí s kalorickým deficitem. Množstvím stravy lze podle ní nejen ovlivňovat choroby časté ve stáří, ale i prodloužit maximální délku života (21).
Jiná teorie stárnutí koreluje délku života s rychlostí metabolismu (rate of living theory of aging). Jedná se o předpoklad, že pomalý metabolismus je asociován s pomalým stárnutím (prodlužuje se životnost buněk). Tím se vysvětluje, proč živočichové s rychlým metabolismem mívají zpravidla život kratší a živočichové s pomalým metabolismem zpravidla žijí déle (22).
Další teorie stárnutí je založena na tom, že cvičením a otužováním dochází k biogenezi mitochondrií. Dochází k tzv. aktivaci mechanismů přežití. V důsledku sníženého množství pohybu a sedavého způsobu života vznikají mutace v mtDNA, nedochází k dostatečné reparaci mtDNA a biogenezi nových mitochondrií. Tímto způsobem člověk ztrácí značné množství funkčních mitochondrií (23).
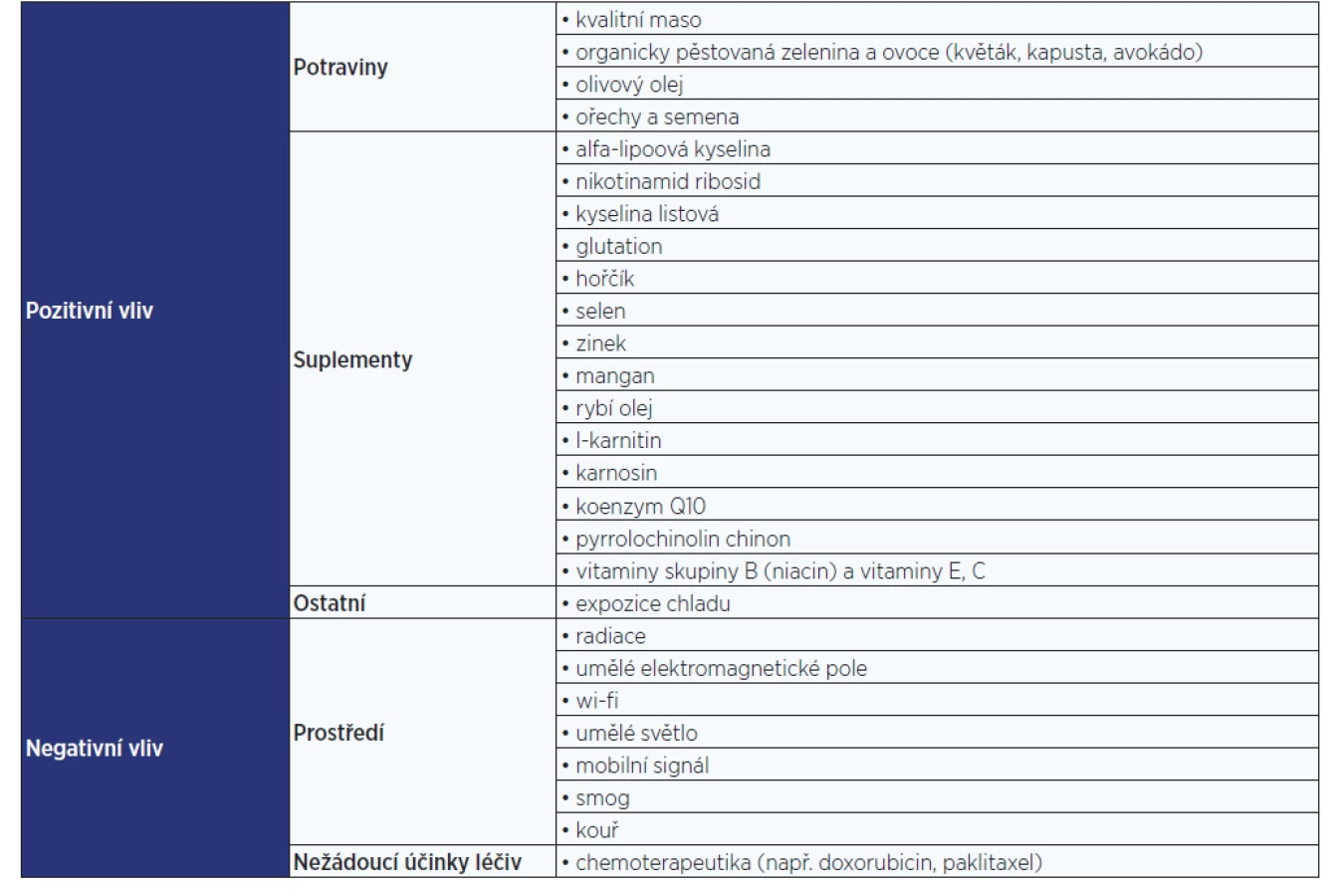
CO MITOCHONDRIÍM ŠKODÍ A CO PROSPÍVÁ
Negativní vlivy
Mitochondrie podléhají mnoha vnějším i vnitřním vlivům. Mezi negativní vnější patří vliv prostředí. Například radiace způsobuje rychlé množení buněk a následné přibývání mutací v mtDNA. Umělé elektromagnetické pole způsobuje strukturální poškození mitochondrií (kavitace, zlomené a neuspořádané kristy) následované degenerací mitochondrií. Mitochondriím také neprospívá expozice signálům mobilních telefonů, wi-fi, inteligentním měřičům nebo umělému světlu (24). Bylo zdokumentováno, že k redukci počtu mitochondrií v buňce dochází pod vlivem chemoterapeutik (např. doxorubicinu či paklitaxelu) (25, 26). K negativním vlivům prostředí patří i kouř a smog. Vůči nim jsou mitochondrie jedny z nejcitlivějších organel. Nejvíce škodí oxid dusný (27).
Pozitivní vlivy
Zvýšit efektivitu a regeneraci mitochondrií můžeme například stravou bohatou na vitaminy, fytonutrienty a antioxidanty (vysoce kvalitní maso, organicky pěstovaná zelenina [např. květák, růžičková kapusta] a ovoce [např. avokádo]). Pomáhají i olivový olej a ořechy (28).
Ke správné funkci mitochondrií dopomáhají i některé suplementy, jako jsou nikotinamid ribosid, alfa-lipoová kyselina, kyselina listová, glutation, vitaminy skupiny B (zvláště niacin), hořčík, selen, zinek, mangan, rybí olej, l-karnitin, karnosin, a koenzym Q10. Za velice účinný stimulant mitochondrií je považován také pyrrolochinolin chinon (28, 29). Zajímavý způsob, jak podpořit činnost mitochondrií, je vystavení se chladnému prostředí, kdy se tělo dostane do tzv. módu přežití a dojde ke zvýšení produkce adrenalinu a následné aktivaci buněk (a mitochondrií). Tento efekt vykazují i stále populárnější ledové koupele, které dokážou „nabudit“ mitochondrie, čímž je následně docíleno zvýšení jejich tvorby i účinnosti (30).
MOŽNOSTI REGENERACE MITOCHONDRIÍ
V průběhu posledních třiceti let značně pokročila identifikace mechanismů vzniku mitochondriálních poruch, které stojí za klinickou manifestací mnoha onemocnění a stárnutím organismu. Vznikly různé techniky obnovy a reparace mitochondrií, například:
- Aktivování mitochondriální biogenetiky pomocí PGC1α (peroxisome proliferator-activated receptor gamma coactivator 1), kdy se očekává zvýšení syntézy ATP. PGC1α dokáže aktivovat transkripční faktory, jako jsou nukleární respirační faktor 1 (NRF1), receptory aktivované proliferátory peroxisomů (PPAR) či geny oxidační fosforylace (20, 31).
- Ovlivnění dynamiky a tvaru mitochondrií (autofagie – apoptóza): Mitochondrie jsou vysoce dynamické organely, jejichž tvar a velikost úzce souvisí s aktivitou a fúzí proteinů, jako jsou (mitofuzín 1 (MFN1) a mitofuzín 2 (MFN2), protein atrofie optiku (OPA1) a dynaminu příbuzný protein 1 (DRP1). Kompletní delece některého z těchto genů vede k závažné klinické symptomatologii, jako jsou srdeční selhání (delece DRP1), dominantní optická atrofie (delece OPA1) či infertilita u žen (delece MFN1) (32, 33, 34). Vysoká exprese OPA1 může napomáhat při stabilizacích defektivních komplexů oxidační fosforylace (I–IV) u encefalopatií, myopatií, poškození jater a ischemického poškození mozku (35, 36).
- Náhrada poškozených mitochondrií formou transplantace mitochondrií – cílí na výměnu mitochondrií s mutovanou mtDNA za zdravé mitochondrie s nepoškozenou mtDNA z embryí v raném stadiu (mitochondrie z embryonálních kmenových buněk) či maternálních oocytů (37).
Mezi nové regenerační postupy patří transplantace/přenos mitochondrií samotných, či v pluripotentních kmenových buňkách, ať se jedná o hematopoetické (HSC) či mezenchymové (MSC), s cílem obnovy mitochondrií nebo nahrazení daných kmenových buněk buňkami novými. Pro izolaci mitochondrií z kmenových buněk se uplatňují jako zdroj zejména MSC. Naopak HSC se k tomuto účelu tak často nevyužívají, neboť při jejich transplantacích dochází v experimentálních studiích na zvířecích modelech k vysoké mortalitě (20, 38). Transplantace mitochondrií se dá využít v případě potřeby k dodání krátkodobé energie do orgánu, kdy se na omezenou dobu vytvoří orgán s vyšším množstvím energie, než sám dokáže vyprodukovat, tzv. supercharged orgán (39).
TRANSPLANTACE MITOCHONDRIÍ V LÉČBĚ ORGÁNOVÝCH POŠKOZENÍ
Existuje několik zajímavých studií zabývajících se mitochondriální transplantací u srdečních onemocnění.
Nejprve byly provedeny in vitro experimenty přenosu alogenních mitochondrií izolovaných diferenciální centrifugací z buněčné linie L6 (buňky kosterní svaloviny potkanů) do buněčné linie H9c2 (kardiomyocyty potkanů). Následně byly mitochondrie obarveny. Mitochondrie byly pohlcovány kardiomyocyty postupně a dokázaly se samy umístit na potřebné místo. Byly změřeny míra spotřeby kyslíku (OCR) a hodnoty extracelulární acidifikace. Přenos mitochondrií doprovázela zvýšená bioenergetika v cílových buňkách, a to již 2 dny po provedení transplantace. V porovnání s kontrolními buňkami hodnoty bazální respirace a produkce ATP v buněčné linii H9e2 vzrostly. Zvýšena byla i hodnota maximální respirace. Bylo také prokázáno, že přenos mitochondrií nemá z hlediska dlouhodobého udržení energie v cílových buňkách žádný efekt. V rámci této studie byl také testován přenos mitochondrií z buněčné linie L6 do lidské buněčné linie ARPE-19 (epiteliální buňky sítnice). Ukázalo se, že při stejném postupu bylo docíleno podobných výsledků jako u buněčné linie H9c2 (39).
První experimentální klinická transplantace mitochondrií byla provedena v roce 2016. Do studie bylo vybráno 5 dětí s myokardiální ischémií. V této studii byly aplikovány autologní mitochondrie izolované z přímého břišního svalu přímo do hypokinetických oblastí myokardu. Všem pacientům se po aplikaci výrazně zlepšila systolická funkce srdečního svalu. Čtyři z pěti nemocných mohli být následně odpojeni od mimotělní podpory (extrakorporální membránové oxygenace), a to již druhý den po výkonu. Po provedení mitochondriální transplantace nebyly u pacientů zaznamenány porucha srdečního rytmu, intramyokardiální hematomy ani jizvy myokardu (40).
Předpokládá se, že mitochondriální transfer by se mohl využívat i u vaskulopatie štěpu. Jak bylo zjištěno, pacienti s touto poruchou mají nižší hodnoty produkce ATP než zdraví lidé. Snížená mitochondriální respirační funkce je asociována se špatnou funkcí myokardu (41).
Mitochondriální transplantace byla využita i k obnově funkce nervové soustavy. V testech na experimentálních zvířecích modelech byl proveden přenos exogenních mitochondrií z jaterní tkáně potkanů do očního sklivce (intravitreální injekční aplikace) jiným potkanům (pod účinkem sedativ). Mitochondrie byly izolovány z jater, neboť hepatocyty obsahují mnoho mitochondrií (okolo 20–25 mg na 1 g). Transplantace mitochondrií do retiny dokázala obnovit mitochondriální biogenezi, a tím zvýšit i produkci ATP potřebnou pro správnou funkci očních nervů. Neurologické poškození bohužel většinou nelze vyléčit a samotný transfer mitochondrií pravděpodobně tento problém zcela nevyřeší. Popsaný úspěch však poukazuje na slibné výsledky transplantace mitochondrií při léčbě poškozené centrální nervové soustavy (CNS) (42).
Bylo již také provedeno přes padesát experimentálních klinických studií u pacientů po cévní mozkové příhodě (CMP). Jako zdroj mitochondrií se nejčastěji využívají autologní MSC a mononukleární buňky (MNC) izolované z kostní dřeně pacienta, MSC izolované z pupečníku a neurální kmenové buňky. MSC a MNC izolované z kostní dřeně se podávají intravenózně, intracerebrálně a intraarteriálně. MSC izolované z pupečníku se aplikují intravenózně a neurální kmenové buňky intracerebrálně (43).
Mitochondrie se následně dokážou přesunout z jedné buňky do buňky druhé za pomoci tunelujících nanotrubic (TNT), tj. útvary zajišťujícími komunikaci s okolními buňkami (obdoba rostlinných plazmidů). Díky této obnově mitochondrií dochází k redukci neurologické deficience ATP. Terapie CMP pomocí kmenových buněk jako zdroje mitochondrií je stále na počátku výzkumu (zatím nejsou kompletně prozkoumány mechanismy účinku), podle studií provedených na experimentálních zvířecích modelech se však tato cesta zdá být správná a do budoucna by se mohlo jednat o velice slibnou metodu léčby poškození CNS (43).
Mitochondriální transplantace byla rovněž využita k obnově funkce dýchací soustavy. Ukázalo se, že u myší (C57BL/6J) transplantace zdravých autologních mitochondrií získaných z trojhlavého svalu lýtkového v dávce 1–3 × 108 mitochondrií dokáže zmírnit průběh syndromu akutní dechové tísně (ARDS). Transplantace mitochondrií se provádí jak intravaskulárně (do plicní tepny), tak intratracheálně (endotracheální kanylou, lze provést i injekčně přes kůži přímo do průdušnice); v obou případech se dostávají do alveolárního epitelu. Po aplikaci mitochondrií dochází k zmírnění projevů ARDS do 24 hodin od podání. Tento způsob terapie ARDS má však jen velice obtížné uplatnění v humánní medicíně (44).
S věkem klesající funkčnost mitochondrií se projevuje také sníženou schopností reprodukce v důsledku stárnutí ovarií. Regenerace mitochondrií ve vaječnících by mohla vést k zlepšení kvality oocytů a následně prodloužení reprodukční schopnosti a zvýšení fertility (45).
ZÁVĚR
Mitochondrie jsou předmětem intenzivního zájmu. Výsledky výzkumu poodhalují zajímavou historii jejich vzniku, přinášejí informace o možnostech regenerace a posílení funkce mitochondrií či prevence mitochondriálního poškozování. Očekává se vývoj progresivních léčebných postupů závažných onemocnění asociovaných s mitochondriální dysfunkci.
Čestné prohlášení
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou ani jinou firmou.
Seznam zkratek
ARDS syndrom akutní dechové tísně
ATP adenosintrifosfát
DNA deoxyribonukleová kyselina
DRP1 dynaminu příbuzný protein 1
HSC hematopoetické kmenové buňky
MNC mononukleární buňky
MSC mezenchymové kmenové buňky
mtDNA mitochondriální deoxyribonukleová kyselina
OPA1 protein atrofie optiku
PGC1α koaktivátor 1-alfa peroxisomového proliferátoru aktivovaného receptoru gamma
ROS reaktivní formy kyslíku
SNP bodové mutace
Adresa pro korespondenci:
Ing. Vít Smejkal
Medicínské centrum Praha, s. r. o.
Mezi vodami 205/29, 143 00 Praha 4
Tel.: 720 597 510
e-mail: vit.smejkal@mc-praha.cz
Sources
1. Ješina P. Mitochondriální nemoci způsobené genetickými poruchami F1Fo-ATP syntázy. Habilitační práce 2017. Dostupné na: dspace.cuni.cz/bitstream/handle/20.500.11956/111557/Jesina_habilitacniprace_2017.pdf?sequence=1&isAllowed=y
2. Martin WF, Neukirchen S, Zimorski V et al. Energy for two: New archaeal lineages and the origin of mitochondria. Bioessays 2016; 38(9): 850–856.
3. Karnkowska A, Vacek V, Zubáčová Z et al. A eukaryote without a mitochondrial organelle. Curr Biol 2016; 26(10): 1274–1284.
4. Zhang Z-W, Cheng J, Xu F et al. Red blood cell extrudes nucleus and mitochondria against oxidative stress. IUBMB Life 2011; 63(7): 560–565.
5. Costello MJ, Brennan LA, Basu S et al. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res 2013; 116 : 141–150.
6. Picard M, Taivassalo T, Gouspillou G, Hepple RT. Mitochondria: Isolation, structure and function. J Physiol 2011; 589(18): 4413–4421.
7. Šeda O, Liška F, Šedová L. Aktuální genetika – multimediální učebnice lékařské biologie, genetiky a genomiky. Praha: Ústav biologie a lékařské genetiky 1. LF UK a VFN, 2005–2006.
8. Holt IJ, Spinazzola A. Mechanisms of onset and accumulation of mtDNA mutations. In: Gasparre G, Porcelli AM (eds.): The Human Mitochondrial Genome. Cambridge: Academic Press 2020 : 195–219.
9. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012; 337(6098): 1062–1065.
10. Fritsch ES, Chabbert CD, Klaus B, Steinmetz LM. A genome-wide map of mitochondrial DNA recombination in yeast. Genetics 2014; 198(2): 755–771.
11. Kivisild T. Maternal ancestry and population history from whole mitochondrial genomes. Investig Genet 2015; 6 : 3.
12. Tranah GJ, Manini TM, Lohman KK et al. Mitochondrial DNA variation in human metabolic rate and energy expenditure. Mitochondrion 2011; 11(6): 855–861.
13. Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010; 5 : 297–348.
14. Kenney MC, Chwa M, Atilano SR et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: Implications for age-related macular degeneration. PLoS One 2013; 8(1): e54339.
15. Wallace DC, Lott M, Procaccio V. Mitochondrial medicine: The mitochondrial biology and genetics of metabolic and degenerative diseases, cancer, and aging. In: Rimoin D, Pyeritz R, Korf B (eds.): Emery and Rimoin’s Principles and Practice of Medical Genetics. Cambridge: Academic Press 2013 : 1–153.
16. Area-Gomez E, Guardia-Laguarta C, Schon EA, Przedborski S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J Clin Invest 2019; 129(1): 34–45.
17. Tsuda M, Fukushima A, Matsumoto J et al. Protein acetylation in skeletal muscle mitochondria is involved in impaired fatty acid oxidation and exercise intolerance in heart failure. J Cachexia Sarcopenia Muscle 2018; 9(5): 844–859.
18. Stefano GB, Bjenning C, Wang F et al. Mitochondrial heteroplasmy. Adv Exp Med Biol 2017; 982 : 577–594.
19. Szczepanowska K, Trifunovic A. Mitochondrial DNA mutations and aging. In: Gasparre G, Porcelli AM (eds.): The Human Mitochondrial Genome. Cambridge: Academic Press 2020 : 221–242.
20. Quadalti C, Garone C. MtDNA maintenance: Disease and therapy. In: Gasparre G, Porcelli AM (eds.): The human mitochondrial genome. Cambridge: Academic Press 2020 : 411–442.
21. Lanza IR, Nair KS. Mitochondrial function as a determinant of life span. Pflugers Arch 2010; 459(2): 277–289.
22. Hwang AB, Jeong D-E, Lee S-J. Mitochondria and organismal longevity. Curr Genomics 2012; 13(7): 519–532.
23. Bo H, Zhang Y, Ji LL. Redefining the role of mitochondria in exercise: A dynamic remodeling. Ann N Y Acad Sci 2010; 1201(1): 121–128.
24. Santini SJ, Cordone V, Falone S et al. Role of mitochondria in the oxidative stress induced by electromagnetic fields: Focus on reproductive systems. Oxid Med Cell Longev 2018; 2018 : 5076271.
25. Gorini S, De Angelis A, Berrino L et al. Chemotherapeutic drugs and mitochondrial dysfunction: focus on doxorubicin, trastuzumab, and sunitinib. Oxid Med Cell Longev 2018; 2018 : 7582730.
26. Kober KM, Olshen A, Conley YP et al. Expression of mitochondrial dysfunction-related genes and pathways in paclitaxel-induced peripheral neuropathy in breast cancer survivors. Mol Pain 2018; 14 : 1744806918816462.
27. Fetterman JL, Sammy MJ, Ballinger SW. Mitochondrial toxicity of tobacco smoke and air pollution. Toxicology 2017; 391 : 18–33.
28. Dai D-F, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res 2012; 110(8): 1109–1124.
29. Niyazov DM, Kahler SG, Frye RE. Primary mitochondrial disease and secondary mitochondrial dysfunction: Importance of distinction for diagnosis and treatment. Mol Syndromol 2016; 7(3): 122–137.
30. Chung N, Park J, Lim K. The effects of exercise and cold exposure on mitochondrial biogenesis in skeletal muscle and white adipose tissue. J Exerc Nutrition Biochem 2017; 21(2): 39–47.
31. Viscomi C, Zeviani M. Strategies for fighting mitochondrial diseases. J Intern Med 2020; 287(6): 665–684.
32. Dorn GW. Gone fission: diverse consequences of cardiac Drp1 deficiency. Circ Res 2015; 116(2): 225–228.
33. Marchbank NJ, Craig JE, Leek JP et al. Deletion of the OPA1 gene in a dominant optic atrophy family: Evidence that haploinsufficiency is the cause of disease. J Med Genet 2002; 39(8): 47–47.
34. Zhang M, Bener MB, Jiang Z et al. Mitofusin 1 is required for female fertility and to maintain ovarian follicular reserve. Cell Death Discov 2019; 10(8): 1–15.
35. Varanita T, Soriano ME, Romanello V et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 2015; 21(6): 834–844.
36. Civiletto G, Varanita T, Cerutti R et al. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab 2015; 21(6): 845–854.
37. Hyslop LA, Blakeley P, Craven L et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016; 534(7607): 383–386.
38. Roushandeh AM, Kuwahara Y, Roudkenar MH. Mitochondrial transplantation as a potential and novel master key for treatment of various incurable diseases. Cytotechnology 2019; 71(2): 647–663.
39. Ali Pour P, Kenney MC, Kheradvar A. Bioenergetics consequences of mitochondrial transplantation in cardiomyocytes. J Am Heart Assoc 2020; 9(7): e014501.
40. Shin B, Cowan DB, Emani SM et al. Mitochondrial transplantation in myocardial ischemia and reperfusion injury. Adv Exp Med Biol 2017; 982 : 595–619.
41. Lichscheidt ED, Jespersen NR, Nielsen BR et al. Mitochondrial function is impaired in heart transplant patients with cardiac allograft vasculopathy. J Heart Lung Transplant 2020; 39(4): S89.
42. Nascimento-dos-Santos G, de-Souza-Ferreira E, Lani R et al. Neuroprotection from optic nerve injury and modulation of oxidative metabolism by transplantation of active mitochondria to the retina. Biochim Biophys Acta Mol Basis Dis 2020; 1866(5): 165686.
43. Sarmah D, Kaur H, Saraf J et al. Getting closer to an effective intervention of ischemic stroke: The big promise of stem cell. Transl Stroke Res 2018; 9(4): 356–374.
44. Hepokoski M. Mitochondrial transplantation: Respiration rescue in respiratory failure. Am J Physiol Lung Cell Mol Physiol 2020; 318(1): L76–L77.
45. Chiang JL, Shukla P, Pagidas K et al. Mitochondria in ovarian aging and reproductive longevity. Ageing Res Rev 2020; 63 : 101168.
46. Benton MC. Mitochondrial genome variation and metabolic traits in a Maori community. 2009. Dostupné na: www.researchgate.net/publication/43193294_Mitochondrial_Genome_Variation_and_Metabolic_Traits_in_a_Maori_Community
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

2021 Issue 7–8
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- COVID-19, postcovid syndrome and postvaccination complications at neurology outpatient clinic
- Manuál k elektronickému zdravotnictví
- Mitochondria – from origin to current therapies
- Legal framework of telemedicine for provision of healthcare