
Srdeční amyloidóza v zobrazovacích metodách – pohled kardiologa
Cardiac amyloidosis in imaging methods – a view of cardiologist
Cardiac amyloidosis (CA) previously considered to be an untreatable disease is getting deserved attraction now, namely because of advances in imaging and recent approval of breakthrough therapies. Our review focuses on the two most frequent types of CA, immunoglobulin light chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis. It deals predominantly with the diagnostic imaging procedures and briefly describe the therapy with the future perspective. The recent findings and progress in imaging made it possible early diagnosis and differentiation from other different hypertrophic cardiomyopathies. Magnetic resonance imaging enabled deeper understanding of basic pathophysiological processes in CA most importantly thanks to its ability to characterize tissue properties. A wide use of bone scintigraphy reduced the need of myocardial biopsy and improved diagnostic certainty in ATTR. There will be a higher demand for imaging procedures to diagnose CA early, to monitor therapeutic response and to be able to change properly therapeutic strategy thanks to a rapid progress in new therapies.
Keywords:
cardiac amyloidosis – cardiac magnetic resonance – cardiomyopathy – immunoglobulin light chain – transthyretin – echocardiography – bone scintigraphy
Authors:
Adéla Morávková; Petr Povolný
Authors‘ workplace:
Cardiocentrum Kladno, ČR
Published in:
NuklMed 2020;9:76-84
Category:
Review Article
Overview
Srdeční amyloidóza (CA), kdysi považovaná za neléčitelné onemocnění, si nyní získává zaslouženou pozornost díky pokroku v zobrazování a nedávnému schválení cílených průlomových terapií. Náš přehled se zaměřuje na dva nejčastěji se vyskytující typy srdeční amyloidózy, monoklonální imunoglobulinovou amyloidózu lehkých řetězců (AL) a transthyretinový typ amyloidózy (ATTR). Zabývá se především jejich diagnostikou pomocí různých zobrazovacích technik, stručně nastiňuje i léčbu a prognózu s výhledem do budoucna. Nedávná zjištění a pokroky v zobrazování vedly k časnější diagnostice CA s vyšší schopností odlišit CA od jiných hypertrofických kardiomyopatií. Použití magnetické rezonance srdce (CMR) vedlo k hlubšímu pochopení základních patofyziologických procesů u CA, a to především díky její schopnosti charakterizovat tkáňové vlastnosti. Široké využití kostní scintigrafie snížilo potřebu srdeční biopsie a zlepšilo diagnostickou jistotu u ATTR. Vzhledem k rychlému rozvoji nové léčby CA bude větší potřeba zobrazovacích technik pro časnější diagnostiku tohoto onemocnění, monitoraci odpovědi na léčbu a možnosti měnit odpovídajícím způsobem léčebnou strategii.
Klíčová slova:
srdeční amyloidóza – zobrazování magnetickou rezonancí – kardiomyopatie – imunoglobulinový lehký řetězec – transthyretin – echokardiografie – scintigrafie kostí
ÚVOD
Systémové amyloidózy zahrnují heterogenní skupinu onemocnění. Podstatou onemocnění je agregace a depozice bílkovinných substancí ve formě nerozpustných amyloidových fibril v antiparalelním β-uspořádání uložených extracelulárně v podobě homogenních amorfních hmot, tj. amyloidu. Když je akumulace dostatečná, dochází k narušení struktury a integrity postižených orgánů. Každý typ amyloidu má svůj specifický fibrilární prekurzorový protein, od něhož se odvíjí i jeho název. Pro specifické znázornění amyloidu v histologickém preparátu je nejčastěji používáno barvení konžskou červení, kdy depozita amyloidu vykazují oranžové až červené zbarvení. Při vyšetření polarizačním mikroskopem vzniká při použití konžské červeně ve vazbě na amyloid tzv. dvojlom („birefrigerence“) a dichroismus, vyznačující se typickou metachromázií, tj. výskytem žlutozeleného „apple-green“ zbarvení.
Srdeční amyloidóza (CA) vzniká ukládáním amyloidních fibril do extracelulárního prostoru myokardu, čímž je způsobeno narušení kontraktilních elementů myokardu, zvýšená tuhost komor a systolická a diastolická dysfunkce. Ačkoli je amyloidóza často multiorgánové onemocnění, srdeční selhání je hlavní příčinou nemocnosti a úmrtnosti. 1,2 Většina případů CA je zastoupena dvěma typy prekurzorových proteinů. Prvním je monoklonální imunoglobulin, jehož prekurzorem jsou lehké řetězce (AL) a který je produkován při monoklonální proliferaci plazmocytů v kostní dření („plasmocelulární dyskrazie“). Druhým je protein transthyretin (ATTR), který pochází z jater a je normálně zodpovědný za transport tyroxinu a retinolu. 3,4 Pokroky v zobrazovacích metodách jako je scintigrafie kostí (BS) a zobrazování magnetickou rezonancí srdce (CMR) pomohly rozšířit povědomí o CA jako o nedostatečně diagnostikované restriktivní kardiomyopatii, umožnily dřívější diagnostiku, lepší pochopení základních chorobných procesů a schopnost sledovat reakci na léčbu u tohoto onemocnění. V následujícím přehledovém článku jsou shrnuty klíčové rysy těchto dvou nejčastějších typů srdeční amyloidózy se zaměřením na klinickou aplikaci a užitečnost zobrazovacích metod při diagnostice CA a vlivu zobrazovacích metod na algoritmy léčby.
Patofyziologie
Srdeční amyloidóza vede ke zvýšené tloušťce stěn obou komor a zvýšené tuhosti stěn, což jsou charakteristické rysy této restriktivní kardiomyopatie. Infiltrace svaloviny předsíní amyloidovým proteinem pravděpodobně přispívá k vysoké prevalenci síňové fibrilace a zvyšuje riziko tvorby trombů síní a tromboembolie, a to i při sinusovém rytmu. 5–7 Dále dochází k častému výskytu supraventrikulárních arytmií a AV blokád různého stupně. Kromě toho se u AL onemocnění může amyloid ukládat uvnitř a/nebo kolem malých arteriol v srdci a může vést ke klinickému syndromu anginy pectoris nebo v některých případech i k infarktu myokardu. 8 Abnormality koronárního průtoku při vyšetření pozitronovou emisní tomografií (PET) byly pozorovány u pacientů s mikrovaskulární infiltrací amyloidem. 9 Kromě intersticiální infiltrace může také docházet k srdeční dysfunkci u AL amyloidózy z přímé toxicity lehkého řetězce. 10 Amyloidogenní volné lehké řetězce mohou podporovat lysozomální dysfunkci, která vede ke generování volných kyslíkových radikálů a následné buněčné smrti. 11
AL a ATTR srdeční amyloidóza
Systémová AL amyloidóza je považována za nejběžnější typ amyloidózy s odhadovanou prevalencí 8 až 12 případů na milion osob na rok. 12,13 Klinický fenotyp a symptomatologie jsou různorodé, což odráží potenciální infiltraci amyloidem ovlivňující více orgánů. Diagnostická prodleva často nastává kvůli nespecifické povaze příznaků zahrnující únavu, dušnost a úbytek hmotnosti. Specifičtější klinické příznaky, jako je makroglosie a periorbitální hematomy, jsou v zásadě patognomické, ale vyskytují se pouze v jedné třetině případů. 14 Srdeční postižení nacházíme až u 80 % pacientů s AL CA 15 a projevuje se symptomy srdečního selhání. Protože onemocnění postihuje obě srdeční komory, je obvykle přítomna dysfunkce obou komor, i když nejčastějším projevem je závažné pravostranné srdeční selhání. Navzdory nejlepší lékařské léčbě zůstává prognóza AL CA špatná. 16
ATTR CA je dělena podle typu transthyretinového proteinu na dědičnou formu (hATTR) nebo nedědičnou formu, která je známá jako divoký typ ATTR (wtATTR). 17 Diagnóza wtATTR v posledních letech značně vzrostla, odhady prevalence jsou 13–16 % u starších pacientů se srdečním selháním se zachovanou ejekční frakcí (EF). 18 Divoký typ ATTR CA (dříve známý jako senilní CA) má převahu v mužské populaci a projevuje se nejčastěji restriktivní kardiomyopatií, ale může být spojen i s bederní stenózou, syndromem karpálního tunelu a/nebo tendinopatií. 19–22 Naopak klinický fenotyp amyloidózy hATTR, který se vyskytuje v mladším věku, se vyznačuje proměnlivým klinickým projevem, který obvykle zahrnuje periferní neuropatii, autonomní neuropatii a/nebo kardiomyopatii. 23 Existuje více než 120 příčinných mutací TTR 24, nejběžnější je V122I, která je přítomna až u 3,4 % afroameričanů v USA a klinický obraz a nástup velmi napodobuje wtATTR. 25 Odhaduje se, že přibližně 2 miliony lidí v USA jsou nositeli této varianty a jsou vystaveni riziku rozvoje CA. U pacientů s postižením nervového systému často dochází k neurologickým projevům, avšak podobně jako u AL amyloidózy má srdeční postižení u ATTR nejdůležitější dopad na prognózu s mediánem přežití 4–5 let. 26
Diagnóza srdeční amyloidózy: neinvazivní zobrazování
Elektrokardiografie
Klasickým rysem srdeční amyloidózy je nízká voltáž EKG způsobená zvýšenou tloušťkou stěn levé komory patrnou při echokardiografickém vyšetření. 27 Pseudoinfarktové změny se mohou vyskytovat přibližně u 50 % pacientů s AL srdeční amyloidózou. 28 Atrioventrikulární blok může být pozorován až u 22 % pacientů se srdeční amyloidózou. Prodloužené nitrokomorové vedení a blokády větví Hissova svazku jsou také běžnými příznaky na EKG u srdeční amyloidózy a jsou častěji pozorovány u ATTR typu. 29 EKG známky hypertrofie levé komory lze pozorovat také u přibližně 10–15 % pacientů s AL amyloidózou, ale to častěji odráží již existující hypertenzní onemocnění srdce s nasedající amyloidózou. 28
Echokardiografie
Echokardiografie je nejdostupnějším zobrazovacím nástrojem první linie k hodnocení pacientů s jakoukoliv kardiomyopatií. Pro amyloidózu je jedním z charakteristických rysů zesílení stěn obou komor s malými, nedilatovanými komorami a zesílením stěn levé komory (LV) obvykle větší než 12 mm. U AL CA existuje tendence k symetrickému nárůstu tloušťky stěny LV, zatímco u ATTR CA se často vyskytuje asymetrická hypertrofie. 30,31 Tloušťka stěny je obecně výraznější u pacientů s ATTR CA v době diagnózy, protože pacienti s AL amyloidózou jsou symptomatičtější v dřívějším stadiu onemocnění. Přestože pacienti s ATTR CA mají při diagnóze obvykle vyšší hmotnost levé a pravé komory než pacienti s AL CA 32, izolované stanovení objemu levé komory (LV mass) není vhodné k rozlišení mezi jednotlivými typy CA. Dobře popsané, ale nespecifické nálezy CA zahrnují zesílený a „jiskřivý“ vzhled chlopní a interatriálního septa stejně jako „skvrnitý“ vzhled myokardu. (Obr. 1 a 2) Perikardiální a pleurální výpotky jsou také poměrně častým nálezem, zejména u AL amyloidózy. Infiltrace amyloidu v extracelulárním prostoru vede k tuhnutí komor, zhoršené relaxaci a diastolické dysfunkci obou komor, která v kombinaci s přímou infiltrací předsíní amyloidem vede k jejich dilataci, stagnaci krve a vyššímu riziku tvorby trombů. 33–36 Ačkoli je CA tradičně označována jako příčina „srdečního selhání se zachovanou EF“, toto onemocnění je charakterizováno postižením jak systolické, tak diastolické funkce. Diastolická funkce pacientů s CA je téměř vždy postižena a projevuje se od zhoršené relaxace po restriktivní typ plnění. Ejekční frakce není spolehlivým indikátorem systolické funkce u CA, protože EF reflektuje radiální kontrakci, která je často zachována až do konečného stadia onemocnění. Longitudinální kontrakce je typicky postižena dříve než radiální kontrakce a indexy longitudinální funkce lze použít jako markery rané nemoci. Toto bylo zpočátku prokázáno měřením systolických exkurzí mitrálního anulu pomocí tkáňového Dopplerova zobrazování (TDI), přičemž TDI mitrálního anulu je často nižší než 6 cm/s nebo ze systolické amplitudy pohybu mitrálního anulu (MAPSE) pomocí M-modu 38,39 a později pomocí metody hodnocení strainu (metoda speckle tracking). Měření longitudinálního strainu (LS) pomocí TDI a sledování EKG se ukázalo být cenným nástrojem při diagnostice CA, stejně jako při odlišení CA od ostatních typů hypertrofických kardiomyopatií. 1 Metoda speckle tracking demonstruje nejen zmenšení longitudinální kontrakce, ale také redukci LS, které postihují převážně bazální segmenty a šetří apikální segmenty. Disociace mezi zachováním EF levé komory a redukcí globálního longitudinálního strainu (GLS), vyjádřená jako poměr EF/GLS, byla uváděna jako reprodukovatelný a přesný prostředek k odlišení srdeční amyloidózy a jiných příčin zesílení LV. GLS je navíc nezávislý prediktor přežití u pacientů s AL srdeční amyloidózou.
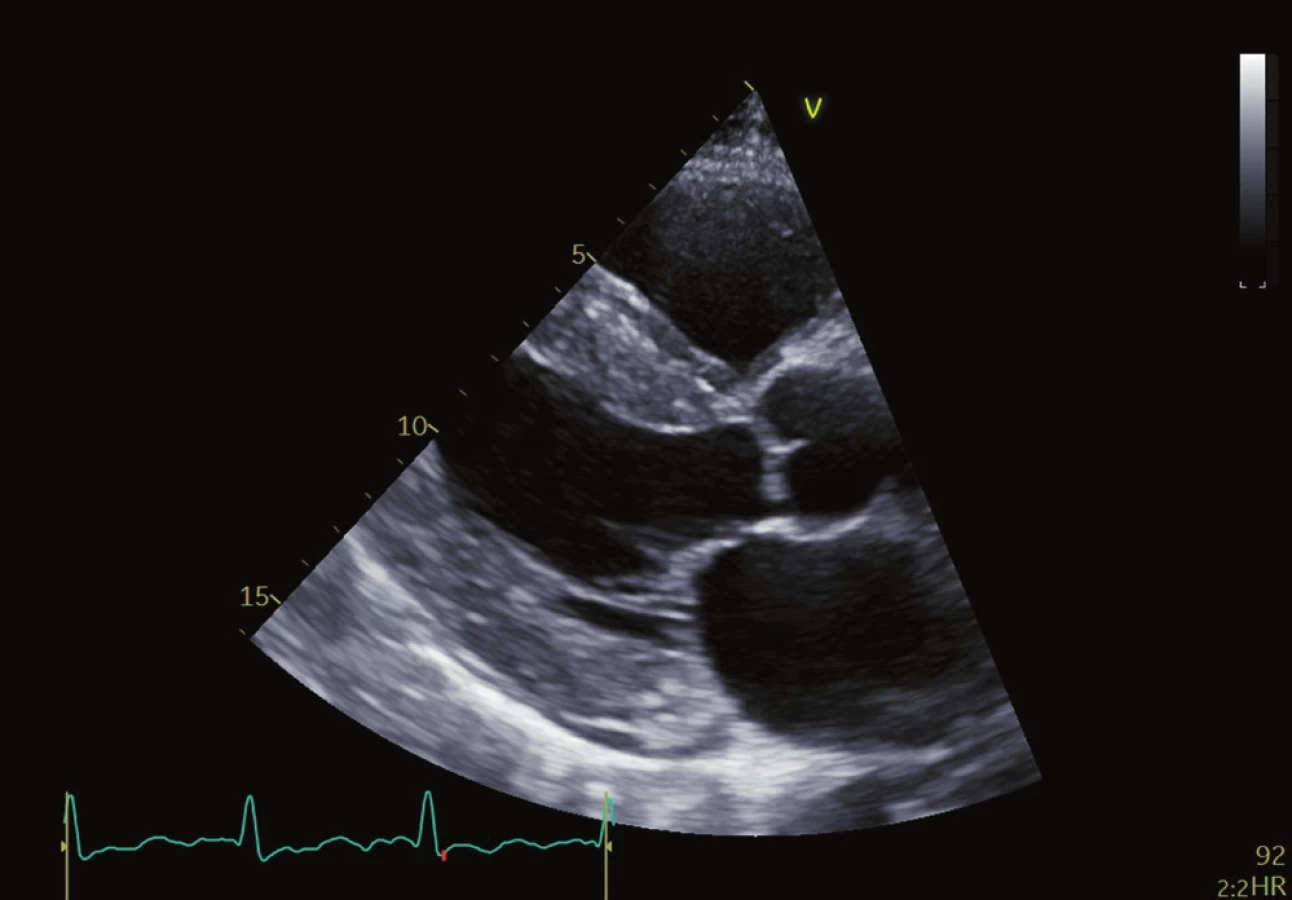
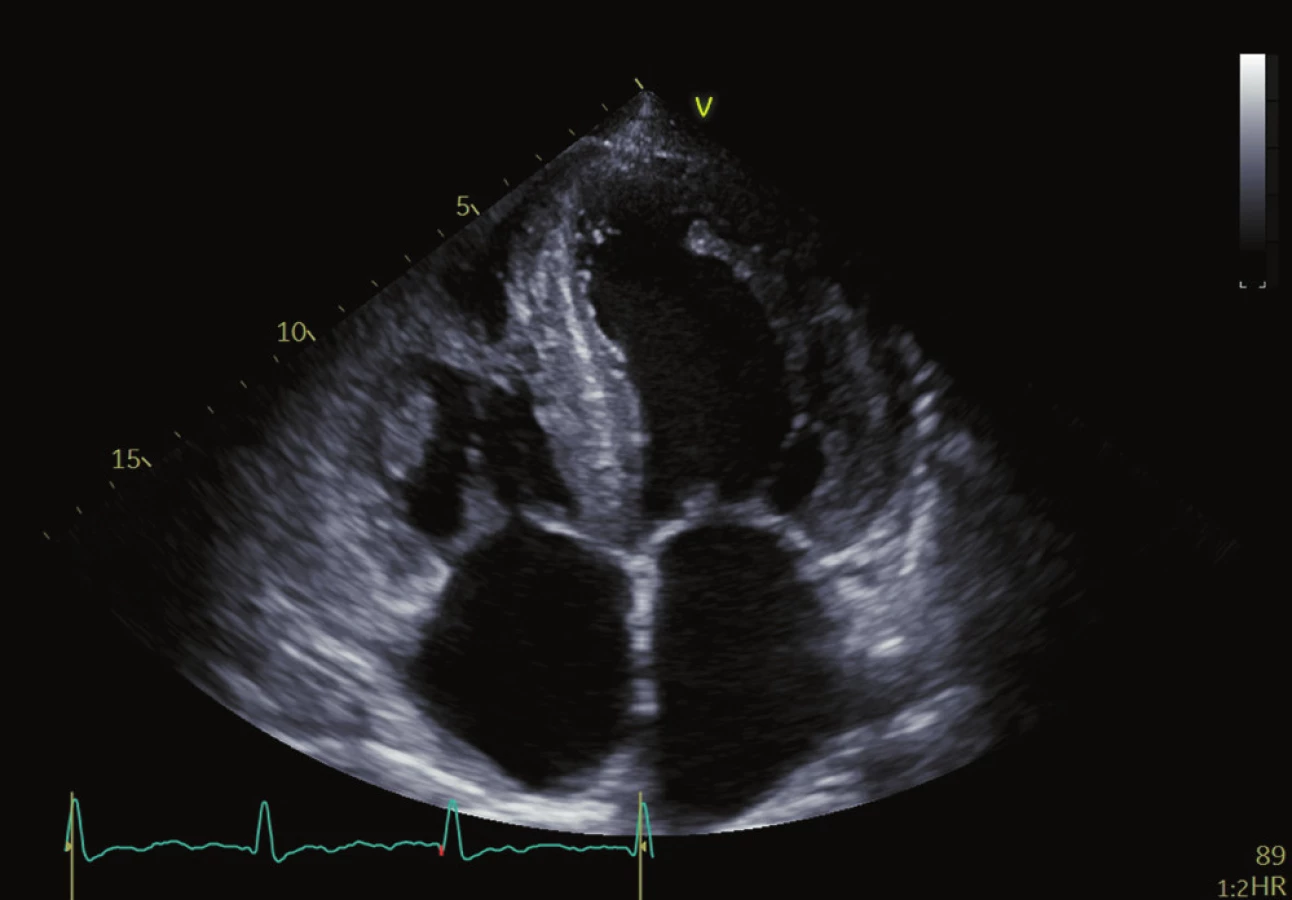
Magnetická rezonance srdce
Magnetická rezonance srdce poskytuje jedinečnou přesnost v hodnocení morfologie srdce a informuje o složení tkáně prostřednictvím své schopnosti definovat tkáňovou charakteristiku. Depozice amyloidových fibril v extracelulárním myokardiálním prostoru vede k expanzi extracelulárního objemu, což je dobře vizualizováno po podání kontrastních látek na bázi gadolinia a je označováno jako „late gadolinium enhancement “(LGE). (Obr. 3) Gadolinium se pasivně akumuluje v prostorech mezi buňkami myokardu a vede u srdeční amyloidózy ke vzniku difuzního subendokardiálního nebo transmurálního LGE v přítomnosti abnormálních charakteristik gadolinia v myokardu a v krvi. Tento jev byl rozpoznán před více než 10 lety. 40 LGE odlišuje normální a abnormální myokard na základě předpokladu, že existují oblasti normálního myokardu vzdálené od myokardu postiženého. Tento rozdíl však nemusí být pozorovatelný u difuzně infiltračních onemocnění, jako je CA. To může způsobovat chybu v hodnocení a postižená oblast může být chybně označena za normální myokard, což s sebou nese riziko „falešně negativních“ výsledků nebo riziko „zrcadlového obrazu“ skutečného vzoru. 41 Technika LGE již dospěla a tím došlo k širokému přijetí spolehlivější techniky „fázově citlivé rekonstrukce obrazů “ (PSIR). S přístupem PSIR LGE byly stanoveny 3 vzorce LGE; žádný, subendokardiální a transmurální. Toto rozdělení vykazuje dobrou korelaci se stupněm infiltrace myokardu. 41 Důležitým nedostatkem LGE je skutečnost, že kontrastní látky na bázi gadolinia (GBCA) byly spojeny se vznikem nefrogenní systémové fibrózy (NSF). Jedná se o závažný a potenciálně smrtelný stav. I když riziko vzniku NSF silně souvisí se vstupní renální funkcí (je nejvyšší, když eGFR < 30 ml/min), při stanovení rizika hraje důležitou roli také základní chemická struktura kontrastní látky. Nedávné pokyny z American College of Radiology doporučují preferenční použití přípravků skupiny II u pacientů s rizikem NSF a zdůrazňují posouzení rizik podávání GBCA. 42 Zatímco původní představa byla, že gadoliniový iont zůstal po intravenózním podání v chelátovaném stavu, několik studií prokázalo retenci ve tkáních, a to i u pacientů s normální funkcí ledvin 43, včetně zpráv o retenci v nervové tkáni (nucleus dentatum, thalamus, pons a globus pallidus) 44–46 a kostní tkáni, 47 jejichž klinické důsledky nejsou zcela známy. Další omezení LGE spočívá v tom, že jej nelze použít ke sledování změn stavu nemoci v průběhu času kvůli jeho nekvantitativní povaze. Tato omezení lze překonat pomocí mapování T1, které přímo měří vnitřní signál z myokardu, podélný relaxační čas v pixelech. Nativní (předkontrastní) myokardiální T1 sleduje infiltraci amyloidu v srdci, markery systolické a diastolické dysfunkce a závažnost onemocnění. 48 Důležitými výhodami nativního T1 zobrazení myokardu je jeho diagnostická přesnost pro detekci CA u AL i ATTR typů a jeho role jako časného markeru onemocnění, který je často zjištěn před nástupem známek onemocnění, jako je hypertrofie levé komory nebo LGE. 48,49 Nativní T1 je složený signál z extra i intracelulárního prostoru. Po podání kontrastních látek s gadoliniem, z poměru T1, T1 po podání kontrastu a hematokritu, lze měřit signál z extracelulárního prostoru a stanovit extracelulární objem (ECV). ECV je první neinvazivní metoda pro kvantifikaci srdeční zátěže amyloidem a několik studií prokázalo souvislost se závažností u obou typů CA. 30,50 ECV je globálně zvýšen, u ATTR o více než 40 % ve srovnání s AL CA. Důležité výhody měření ECV u CA zahrnují jeho jedinečnou schopnost měřit kontinuum infiltrace amyloidem, sledovat ukazatele aktivity onemocnění a dokáže působit jako marker nemoci v časném stadiu a jedinečným způsobem sleduje změny v čase. 50 Například téměř polovina pacientů ve studované kohortě, kteří dosáhli dobré klonální odpovědi na chemoterapii u AL amyloidózy, měla regresi depozit amyloidu v srdci na ECV. 51 Ve spojení s detailním morfologickým a funkčním hodnocením poskytuje tkáňová charakteristika pomocí CMR úplné pochopení mnoha patologických procesů, které existují u CA, překračující koncept CA jako pouhé infiltrativní choroby. Relaxační čas T2 je časová konstanta představující útlum příčné magnetizace a detekuje edém u různých patologických stavů, mimo jiné včetně akutního infarktu myokardu, myokarditidy a Takotsubo kardiomyopatie. 52 V poslední době mapování T2 u CA významně přispělo k chápání CA jako heterogenního stavu, zahrnujícího více chorobných procesů, což ukazuje, že hladiny T2 byly vyšší v kohortě pacientů s neléčenou AL CA ve srovnání s léčenou AL a ATTR CA. To znamená, že edém má důležitou jak patofyziologickou, tak i prognostickou roli. 53
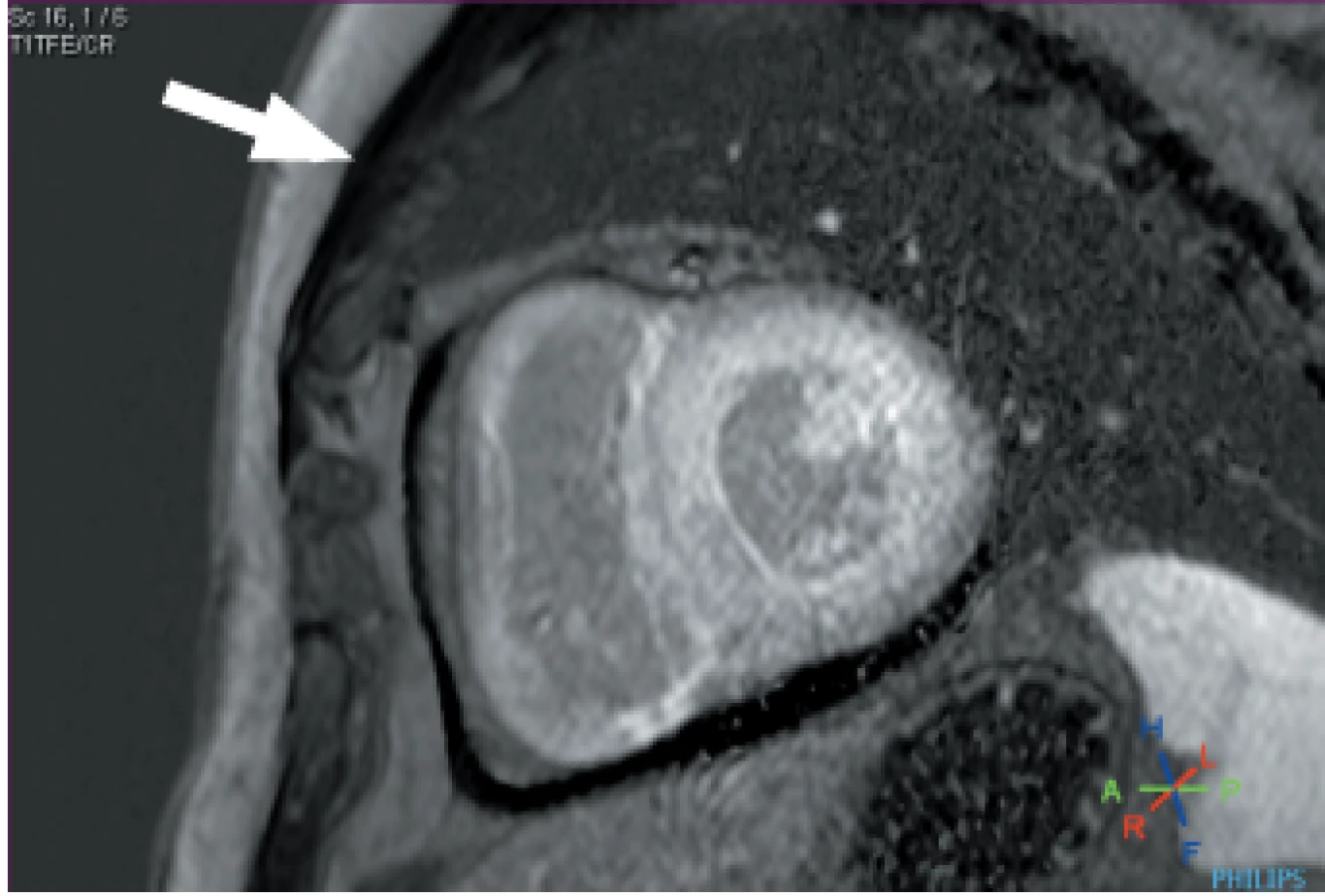
Kostní scintigrafie
Od 80. let 20. století bylo u pacientů postižených CA náhodně pozorováno, že docházelo k akumulaci určitého derivátu fosfátu značeného 99mTc a následně se začala využívat kostní scintigrafie při diagnostice CA. Při amyloidóze mají fibrilární depozita tři hlavní strukturální složky, prekurzorový protein, proteoglykan heparinsulfátu a P–složku závislou na vápníku, která váže dohromady fibrily. Cirkulující P–složka amyloidu, univerzální složka všech typů amyloidu, by se mohla vázat na amyloidní fibrily prostřednictvím mechanismu zprostředkovaného vápníkem, což vysvětluje akumulaci kostních radiofarmak značených 99mTc. Tato metoda je citlivá na diagnostiku ATTR CA zřejmě kvůli vysoké hladině vápníku v amyloidových ATTR depozitech. V roce 2005 prokázala malá, ale klíčová práce silný diagnostický potenciál 99mTc značeného 3,3-dikarboxypropan-2,1-difosfonátu (99mTc-DPD) při identifikaci ATTR CA. 54 Další studie potvrdily toto zjištění i schopnost dalších kostních radiofarmak detekovat ATTR CA. Tyto klíčové nálezy byly nedávno posíleny výsledky z velké multicentrické studie, která prokázala schopnost kostní scintigrafie diagnostikovat srdeční ATTR CA spolehlivě bez nutnosti histologie, a tento diagnostický algoritmus je v klinické praxi široce uznáván a přijímán. 55 Stručně shrnuto, u pacientů, u kterých v krvi a moči chybí volné lehké řetězce a je negativní scintigrafie s použitím 99mTc-PYP (99mTc značený pyrofosfát)/99mTc-DPD/99mTc-HMDP (99mTc značený hydroxymethylenedifosfonát), je diagnosa CA velmi nepravděpodobná. Pokud je srdeční sken s 99mTc-PYP/99mTc-DPD/99mTc-HMDP pozitivní pro stupeň 2 nebo 3 a nejsou přítomny volné řetězce v krvi a/nebo v moči, lze diagnostikovat ATTR CA bez endomyokardiální biopsie (specificita a pozitivní prediktivní hodnota > 98%). Zajímavým a dosud ne zcela prozkoumaným polem je potenciál kostních radiofarmak pro hodnocení extrakardiálního postižení při systémové amyloidóze. Typický model akumulace 99mTc-DPD ve svalech a měkkých tkáních byl popsán již dříve 56 a infiltrace amyloidní tkáně byla později prokázána biopsií měkkých tkání u většiny těchto pozitivních pacientů. 57 Akumulaci v plicích lze nalézt na 99mTc-HMDP scintigrafii 58 s vysokou selektivitou pro ATTR. Klinické důsledky těchto nálezů nejsou zcela objasněny.
Planární versus SPECT zobrazení
Většina zkušeností v zobrazovacích protokolech pro 99mTc-PYP, 99mTc-DPD, 99mTc-HMDP je se současným planárním a SPECT zobrazováním 56 nebo s planárním zobrazováním následovaným SPECT, pokud je planární pozitivní. 59–61 Samotné planární zobrazování je omezeno, vychytávání v myokardu nelze rozeznat od vychytávání v krvi, nadměrné vychytávání v oblasti žeber se může sumarizovat s oblastí srdce a korekce zeslabení není proveditelná. SPECT tyto obtíže překonává. Segmentové vyhodnocení a korekce zeslabení záření nabízejí potenciální výhody lepší kvantifikace a lépe zaznamenává změny.
Hodnocení
Vizuální metoda – Perugini a kolegové 62 popsali klasifikační schéma pro zobrazování po 3 hodinách, kde stupeň 0 = žádná akumulace v myokardu; stupeň 1 = akumulace v myokardu < akumulace v kostech; stupeň 2 = akumulace v myokardu se rovná akumulaci v kostech; stupeň 3 = akumulace v myokardu > akumulace v kostech (se sníženou akumulací v kostech na celotělových obrazech). Toto je velmi jednoduchá metoda a zahrnuje všechna omezení tříhodinového zobrazování. Použití radiofarmaka se liší podle jednotlivých pracovišť. U 99mTc-DPD/HMDP a zobrazování celého těla je pro stupeň 3 vyžadováno kritérium zeslabení akumulace v dlouhých kostech (Obr. 4), zatímco u 99mTc-PYP se většinou provádí pouze zobrazování hrudníku a jako stupeň 3 je hodnocena akumulace v myokardu větší než akumulace v žebrech.
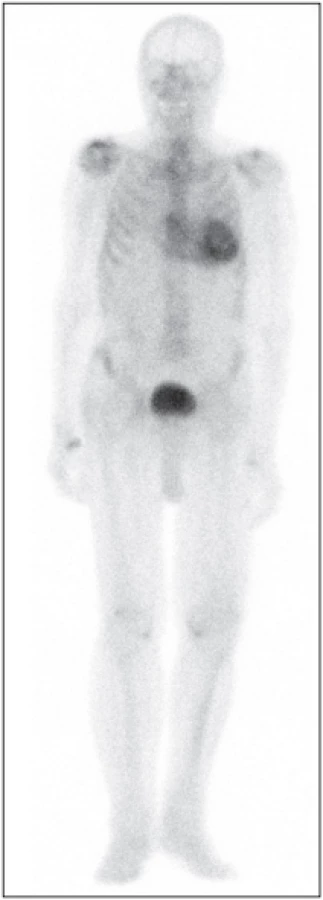
Semikvantitativní metoda – Rapezziho metoda 61 stanovuje poměr radiofarmaka akumulovaného v srdci a v celém těle pomocí časných snímků (po 5 minutách) a pozdních snímků (za 3 hodiny). Výsledek je nutné korigovat na rozpad (korekce na rychlost rozpadu, rychlost skenování a s odečtením aktivity v ledvinách a v močovém měchýři). Tato metoda má výhodu kvantifikace retence radiofarmaka, ale vyžaduje dlouhou dobu (dva skeny celého těla, 3 hodiny od sebe). Pro lepší kvantifikaci a objektivizaci jsou k dispozici nové programy pro výpočet SUV (standardized uptake value) i pro snímání technikou SPECT.
Bokhariho metoda 60 hodnotí poměr aktivity akumulované v srdci a v pravé plíci (poměr H/CL) na planárním zobrazení (za 1hodinu a za 3 hodiny po podání radiofarmaka) pro rozlišení ATTR od AL amyloidózy (≥ 1,5 za 1 hodinu a ≥ 1,3 za 3 hodiny). Optimální metoda je pravděpodobně kombinace výše uvedeného a závisí na použitém radiofarmaku, načasování skenování a použití planárních vs SPECT obrazů.
Pozitronová emisní tomografie
Zobrazování pozitronovou emisní tomografií (PET) nabízí vysoké prostorové rozlišení a může usnadnit absolutní kvantifikaci srdeční a extrakardiální amyloidní zátěže. 63 PET radiofarmaka s vazbou na amyloid, která byla studována u pacientů s AL a ATTR CA, zahrnují sloučeninu 11-C-Pittsburgh B (11CPiB) 64,65, 18F-florbetapir a 18F-florbetaben. Tyto sloučeniny se pravděpodobně vážou na beta skládanou komponentu amyloidových vláken, což vysvětluje jejich schopnost zobrazovat amyloidní depozity nezávisle na prekurzorovém proteinu. Radiofarmaka značená 18F mají další výhodu dlouhého poločasu přeměny (109,7 minut) a možnosti použití i na pracovištích bez dostupnosti cyklotronu. Pozoruhodné je, že PET radiofarmaka vázající amyloid jsou jedinými klinicky dostupnými radioaktivními stopovacími látkami pro adekvátní zobrazení AL amyloidové zátěže v srdci. Ačkoli tyto výsledky jsou povzbuzující, další hodnocení pomocí PET radiofarmak čeká na začlenění do klinické praxe.
Diagnostika
K nedostatečné diagnostice CA přispívá několik faktorů. Patří mezi ně fenotypová heterogenita, nízký index klinického podezření v případě překrývajících se příznaků u častěji se vyskytujících onemocnění (hypertenze, chronické selhání ledvin, hypertrofická kardiomyopatie, aortální stenóza), historický nedostatek neinvazivních diagnostických testů a omezené porozumění dostupným možnostem léčby. Současné neinvazivní diagnostické algoritmy se snaží o integrovaný a multimodální přístup k diagnostice CA. Mezi důležité faktory, které je třeba vzít v úvahu, patří přítomnost nebo výskyt dyskrazie plazmatických buněk, typické nálezy na echokardiografii, na zobrazování CMR a na scintigrafii skeletu. Hodnocení CA na pozadí známé systémové AL amyloidózy je omezeno AL postižením ledvin, kde nelze používat kontrastní látky na bázi gadolinia, a tím je diagnostika obtížnější. Echokardiografie je nejčastěji prováděnou zobrazovací metodou první linie u pacientů se známkami srdečního selhání. Většina echokardiografických nálezů u CA je nespecifických, mohou však být vysoce podnětné a mohou ovlivnit vysokou předtestovou pravděpodobnost.
Diagnostika srdeční amyloidózy u hypertrofických fenotypů
Pokud echokardiografie vzbudí podezření na CA, je nutné myslet v diferenciální diagnostice na jinou příčinu hypertrofie myokardu (hypertenze, hypertrofická kardiomyopatie, Anderson Fabry choroba). Po pozitivním CMR vyšetření by mělo být provedeno skenování 99mTc-PYP/99mTc-DPD/99mTc-HMDP v kombinaci s hodnocením volných lehkých řetězců v krvi a moči, aby bylo možné rozlišit mezi AL a ATTR amyloidózou.
Diagnostika srdečního postižení při známé systémové AL nebo ATTR amyloidóze
U pacientů se systémovou AL amyloidózou by měla být metodou volby CMR k potvrzení srdečního postižení nebo k časnému záchytu onemocnění. Ukázalo se, že CMR má vysokou senzitivitu a specificitu pro AL CA, která zachytí časná stadia onemocnění, i když není patrná dostatečná infiltrace srdce při echokardiografické diagnostice. U pacientů s nositeli polyneuropatie nebo mutací ATTR by měla být zvážena CMR nebo kostní scintigrafie, ale u těchto populací pacientů jsou zapotřebí další studie.
Diagnostika srdeční ATTR amyloidózy
Několik studií prováděných v jednom centru potvrdilo vysokou diagnostickou přesnost (citlivost a specificitu > 90 %) pro zobrazování pomocí 99mTc-PYP, -DPD 61,62 a -HMDP 66 srdeční ATTR amyloidózy. Specifičnost byla nižší v přítomnosti cirkulujícího monoklonálního proteinu při AL amyloidóze. Asi 25–50 % pacientů v těchto studiích prokázalo akumulaci 99mTc-PYP a 99mTc-DPD v myokardu obvykle nízkého stupně (stupeň 1 nebo 2) a byli to pacienti s AL srdeční amyloidózou. V nedávné velké mezinárodní studii provedené Gillmorem et al. 55 bylo sledováno 1217 pacientů, kteří byli posláni pro podezření na srdeční amyloidózu na vyšetření pomocí kostních radiofarmak (99mTc-PYP = 199, 99mTc-HMDP = 141 a 99mTc-DPD = 877). Autoři dospěli k závěru, že nálezy vychytávání radiofarmaka ≥ 2. stupně myokardem na scintigrafii kostí spolu s absencí monoklonální gamapatie při analýze séra a moči prokázaly specificitu a pozitivní prediktivní hodnotu 100 % pro srdeční amyloidózu ATTR.
Prognóza
Při stratifikaci prognózy AL a ATTR CA hrají primární roli biomarkery krve. Mayo klasifikace AL CA používá měření NT–proBNP a troponinu ke kategorizaci pacientů do stupně 0 (obě hodnoty pod prahovou hodnotou), stupně 1 (jedna hodnota nad prahovou hodnotou) a stupně 2 (obě hodnoty nad prahovou hodnotou), což poskytuje cenný prognostický nástroj jako doplněk k dalším vyšetřením. U ATTR CA byly vyvinuty dvě různé prognostické klasifikace, jedna na základě troponinu a NT–proBNP a druhá na základě klasifikace NT–proBNP a eGFR (stanovení glomerulární filtrace): stadium 1 (obě hodnoty pod prahovou hodnotou), stadium 2 (jedna hodnota nad prahovou hodnotou), a stadium 3 (obě hodnoty nad prahovou hodnotou). 67,68 Na echokardiografii a CMR je vidět několik strukturálních a funkčních parametrů, které korelují s prognózou v AL i ATTR amyloidózy, nicméně jako nejpřínosnější se jeví hodnocení systolické exkurze trikuspidálního anulu (TAPSE) a měření srdečního výdeje (SV). 69 Hlavním důvodem prognostického významu TAPSE u CA je pravděpodobně spíše přímá subendokardiální infiltrace než sekundární dysfunkce pravé komory při zhoršení funkce levé komory. Prognostická role stanovení srdečního výdeje u CA 69 je v souladu s očekávanými rysy restriktivní kardiomyopatie charakterizované nízkým objemem se zachovanou EF levé komory. Globální longitudinální strain je navíc nezávislý prediktor přežití u pacientů s AL srdeční amyloidózou. Parametry CMR, které mají prognostickou roli, zahrnují transmuralitu LGE, ECV, T1 a T2 u AL CA a ECV u ATTR CA. 30
Aktuální terapie a budoucnost
Léčba srdečního selhání u srdeční amyloidózy má za cíl zmírnit městnání při infiltrační kardiomyopatii. Na rozdíl od jiných terapeutických intervencí srdečního selhání neexistují žádné randomizované studie, na nichž by bylo možné založit rozhodnutí o léčbě; ta je řízena na základě zkušeností nebo údajů z malých kohortových studií. Diuretika jsou základem léčby, často se používají kličková diuretika v kombinaci s antagonisty mineralokortikoidních receptorů (jako je spironolakton). Standardní terapie srdečního selhání představující beta blokátory a inhibitory angiotensin konvertujícího enzymu nebo blokátory receptorů pro angiotensin často vede k hypotenzi a únavě pacientů se srdeční amyloidózou, což omezuje snášenlivost této léčby. Běžný scénář, který by měl zvýšit klinické podezření na srdeční amyloidózu, je vývoj hluboké hypotenze a únavy po zahájení terapie beta blokátory. Pacienti se srdeční amyloidózou jsou velmi závislí na srdeční frekvenci a kontraktilitě pro udržení srdečního výdeje a blokáda beta receptorů zasahuje do této adaptace. U některých pacientů mohou být betablokátory tolerovány, pokud jsou použity v nízkých dávkách pro kontrolu supraventrikulárních arytmií. Tolerance vysoké dávky beta blokády naznačuje, že srdeční amyloidóza není přítomna nebo není klinicky významná. ACE inhibitory a blokátory angiotensinových receptorů způsobují ortostatickou hypotenzi, která se často vyskytuje při amyloidóze kvůli autonomní dysfunkci.
Důležitou expanzi zažívá oblast léčby jak u AL, tak u ATTR CA. U AL amyloidózy jsou léčebné strategie zaměřeny na rychlé potlačení produkce amyloidogenních lehkých řetězců, z nichž ústřední metodou je cytotoxická chemoterapie. 14 Přestože je u pacientů časně hodnocena způsobilost k transplantaci kmenových buněk, většina je kontraindikována z důvodu věku, dysfunkce ledvin a pokročilého srdečního postižení. 70 Možnosti léčby jsou přizpůsobeny individuálnímu profilu pacienta na základě funkčního stavu a zkušeností, které vycházejí z léčby mnohočetného myelomu. Chemoterapeutické látky zahrnují kombinace bortezomibu, melfalanu, dexamethasonu, cyklofosfamidu, lenalidomidu a dalších látek. Daratumumab, který je protilátkou proti plazmatickým buňkám pro léčbu relapsu mnohočetného myelomu, vykazuje u pacientů s AL amyloidózou slibné výsledky. 70 Mezi další slibné oblasti vývoje patří léčba monoklonálními protilátkami, která je zaměřena na cílení stávajících depozit amyloidů. 71 Došlo k výraznému rozšíření farmakoterapie zaměřené na ATTR CA a přístup k léčbě zahrnuje snížení nebo eliminaci produkce transthyretinu, narušení již uložených amyloidových fibril nebo stabilizujícího proteinu. 72 Inotersen, 2′-O-methoxyethyl-modifikovaný oligonukleotid, který potlačuje jaterní produkci TTR, byl použit v randomizované kontrolované studii u pacientů s ATTR polyneuropatií a vedl ke zlepšení kvality života a úpravě neurologického postižení. 73 V další studii, kde byl testován tafamidis, stabilizátor TTR, bylo prokázáno, že u pacientů s ATTR CA dochází ke snížení mortality ze všech příčin a ke snížení hospitalizací souvisejících s kardiovaskulárním onemocněním. Ve srovnání s placebem byly také pozorovány výhody ve funkční kapacitě a kvalitě života. 74 V lékové studii s pataranem, látkou interferující s RNA, byla ve srovnání s placebem po osmnácti měsících zlepšena hypertrofie stěn na echokardiografii, globální podélný strain a NT–proBNP. 75
Závěr
Srdeční amyloidóza je onemocnění způsobené extracelulárním ukládáním nesprávně složeného proteinu odvozeného nejčastěji z monoklonálních lehkých řetězců v prostředí s dyskrazií plazmatických buněk (AL amyloidóza) nebo z akumulace divokého typu nebo mutantu transthyretinu produkovaného játry (ATTR amyloidóza). Postižení srdce při systémové amyloidóze je důležité diagnostikovat, protože je spojeno s významnou morbiditou a mortalitou. Zobrazovací techniky hrají klíčovou roli při vymezení a pochopení různých mechanismů onemocnění, které jsou součástí CA. Srdeční zobrazování kostními radiofarmaky přineslo revoluci ve schopnosti neinvazivně specificky diagnostikovat srdeční amyloidózu ATTR, čímž se odstranila potřeba provedení endomyokardiální biopsie. Kostní scintigrafie je vhodná zejména proto, že je široce dostupná a má vysokou senzitivitu. 76 Vzhledem k rychlému pokroku v léčebných strategiích je základním cílem zobrazovacích technik zaměření na časnější diagnostiku, léčbu a následné zlepšení kvality života pacientů a jejich přežití.
Sources
- Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis..Clin Med (Lond). 2018;18 : 30–35. https://doi.org/10.7861/clinmedicine.18-2-s30
- Fontana M, Banypersad SM, Treibel TA et al. Differential Myocyte Responses in Patients with Cardiac Transthyretin Amyloidosis and Light-Chain Amyloidosis: A Cardiac MR Imaging Study. Radiology. 2015;277 : 388–397. https://doi.org/10.1148/radiol.2015141744.
- Falk RH, Alexander KM, Liao R, et al. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016;68 : 1323–41. https://doi.org/10.1016/j.jacc. 2016.06.053.
- Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J Am Coll Cardiol. 2015;66 : 2451–66. https://doi.org/10.1016/j.jacc.2015.09.075.
- Stables RH, Ormerod OJ. Atrial thrombi occurring during sinus rhythm in cardiac amyloidosis: evidence for atrial electromechanical dissociation. Heart 1996;75 : 426.
- Feng D, Edwards WD, Oh JK, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116 : 2420–2426.
- Feng D, Syed IS, Martinez M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation. 2009;119 : 2490–2497.
- Tsai SB, Seldin DC, Wu H,et al. Myocardial infarction with “clean coronaries” caused by amyloid light-chain AL amyloidosis: a case report and literature review. Amyloid 2011;18 : 160–164.
- Dorbala S, Vangala D, Bruyere J Jr, et al. Coronary microvascular dysfunction is related to abnormalities in myocardial structure and function in cardiac amyloidosis. JACC Heart failure. 2014;2 : 358–367.
- Brenner DA, Jain M, Pimentel DR, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res 2004;94 : 1008–1010.
- Guan J, Mishra S, Qiu Y, et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO molecular medicine. 2014;6):1493–1507.
- Kyle RA, Linos A, BeardCM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992;79 : 1817–1822.
- Pinney JH, Smith CJ, Taube JB, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013;161 : 525–532. https://doi.org/10.1111/bjh.12286.
- Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387 : 2641–54. https://doi.org/10.1016/S0140-6736(15)01274-X.
- Aimo A, Buda G, Fontana M, et al. Therapies for cardiac light chain amyloidosis: An update. Int J Cardiol. 2018;271 : 152–160. https://doi.org/10.1016/j.ijcard.2018.05.018.
- Grogan M, Dispenzieri A, GertzMA. Light-chain cardiac amyloidosis: strategies to promote early diagnosis and cardiac response. Heart. 2017;103 : 1065–1072. https://doi.org/10.1136/heartjnl-2016-310704.
- Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016;23 : 209–213. https://doi.org/10.1080/13506129.2016.1257986.
- CastañoAND, Hamid N, Khalique OK, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38 : 2879–87.
- Pinney JH, Whelan CJ, Petrie A, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2: e000098. https://doi.org/10.1161/JAHA.113.000098.
- Carr AS, Pelayo-Negro AL, Evans MR, et al. A study of the neuropathy associated with transthyretin amyloidosis (ATTR) in the UK. J Neurol Neurosurg Psychiatry. 2016;87 : 620–627. https://doi.org/10.1136/jnnp-2015-310907.
- Yanagisawa A, Ueda M, Sueyoshi T, et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol. 2015;28 : 201–207. https://doi.org/10.1038/modpathol. 2014.102.
- Geller HI, Singh A, Alexander KM, et al. Association Between Ruptured Distal Biceps Tendon and Wild-Type Transthyretin Cardiac Amyloidosis. JAMA. 2017;318 : 962–963. https://doi.org/10.1001/jama.2017.9236.
- Coelho T, Maurer MS, Suhr OB. THAOS - The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29 : 63-76. doi: 10.1185/03007995.2012.754348.
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35:E2403–2412. https://doi.org/10.1002/humu.22619.
- Jacobson DR, Alexander AA, Tagoe C, et al. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333African-Americans. Amyloid 2015;22 : 171–174. https://doi.org/10.3109/13506129.2015.1051219.
- Castano A, Drachman BM, Judge D, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging diseasemodifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20 : 163–178. https://doi.org/10.1007/s10741-014-9462-7.
- Cyrille NB, Goldsmith J, Alvarez J, et al. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 2014;114 : 1089–1093.
- Murtagh B, Hammill SC, Gertz MA, et al. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2005; 95 : 535–537.
- Huang J, Zhao S, Chen Z, et al. Contribution of Electrocardiogram in the Differentiation of Cardiac Amyloidosis and Nonobstructive Hypertrophic Cardiomyopathy. Int Heart J 2015; 56 : 522–526.
- Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol. 2017;70 : 466–477. https://doi.org/10.1016/j.jacc.2017.05.053.
- Gonzalez-Lopez E, Gagliardi C, Dominguez F, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J. 2017;38 : 1895–1904. https://doi.org/10.1093/eurheartj/ehx043
- Siddiqi OK, Ruberg FL. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28 : 10–21. https://doi.org/10.1016/j.tcm.2017.07.004.
- Murphy L, Falk RH. Left atrial kinetic energy in AL amyloidosis: can it detect early dysfunction? Am J Cardiol 2000;86 : 244–246.
- Modesto KM, Dispenzieri A, Cauduro SA, et al. Left atrial myopathy in cardiac amyloidosis: implications of novel echocardiographic techniques. Eur Heart J 2005;26 : 173–179. https://doi.org/10.1093/eurheartj/ehi040.
- Feng D, Edwards WD, Oh JK, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116 : 2420–2426. https://doi.org/10.1161/CIRCULATIONAHA.107.697763.
- Martinez-Naharro A, Gonzalez-Lopez E, Corovic Aet al. High Prevalence of Intracardiac Thrombi in Cardiac Amyloidosis. J Am Coll Cardiol 2019;73 : 1733–1734. https://doi.org/10.1016/j.jacc.2019.01.035.
- Falk RH, Quarta CC. Echocardiography in cardiac amyloidosis.Heart Fail Rev. 2015;20 : 125–131. https://doi.org/10.1007/s10741-014-9466-3.
- Siepen FAD, Bauer R, Voss A, et al. Predictors of survival stratification in patients with wild-type cardiac amyloidosis. Clin Res Cardiol. 2018;107 : 158–169. https://doi. org/10.1007/s00392-017-1167-1.
- Riffel JH, Mereles D, EmamiM, et al. Prognostic significance of semiautomatic quantification of left ventricular long axis shortening in systemic light-chain amyloidosis. Amyloid. 2015;22 : 45–53. https://doi.org/10.3109/13506129.2014.992515.
- Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111 : 186–193. https://doi.org/10.1161/01.CIR.0000152819.97857.9D.
- Fontana M, Pica S, Reant P, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation. 2015;132 : 1570–1579. https://doi.org/10.1161/CIRCULATIONAHA.115.016567.
- ACR Manual On Contrast Media. 2020. American College of Radiology. Available from: https://www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf.
- McDonald RJ, Levine D, Weinreb J, et al. Gadolinium Retention: A Research Roadmap from the 2018 NIH/ACR/RSNA Workshop on Gadolinium Chelates. Radiology. 2018;289 : 517–534. https://doi.org/10.1148/radiol.2018181151.
- McDonald RJ, McDonald JS, Kallmes DF, et al. Intracranial Gadolinium Deposition after Contrast-enhanced MR Imaging. Radiology. 2015;275 : 772-782. https://doi.org/10.1148/radiol.15150025.
- Kanda T, Fukusato T, Matsuda M, et al. Gadolinium-based Contrast Agent Accumulates in the Brain Even in Subjects without Severe Renal Dysfunction: Evaluation of Autopsy Brain Specimens with Inductively Coupled Plasma Mass Spectroscopy. Radiology. 2015;276 : 228–232. https://doi.org/10.1148/radiol.2015142690.
- Stojanov DA, Aracki-Trenkic A, Vojinovic S, et al. Increasing signal intensity within the dentate nucleus and globus pallidus on unenhanced T1W magnetic resonance images in patients with relapsing-remitting multiple sclerosis: correlation with cumulative dose of a macrocyclic gadolinium based contrast agent, gadobutrol. Eur Radiol. 2016;26 : 807–815. https://doi.org/10.1007/s00330-015-3879-9.
- Murata N, Gonzalez-Cuyar LF, Murata K, et al. Macrocyclic and Other Non-Group 1 Gadolinium Contrast Agents Deposit Low Levels of Gadolinium in Brain and Bone Tissue: Preliminary Results From 9 Patients With Normal Renal Function. Investig Radiol. 2016;51 : 447–453. https://doi.org/10.1097/RLI.0000000000000252.
- Karamitsos TD, Piechnik SK, Banypersad SM, et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging. 2013;6 : 488–6497. https://doi.org/10.1016/j.jcmg.2012.11.013.
- Fontana M, Banypersad SM, Treibel TA, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2014;7 : 157–165. https://doi.org/10.1016/j.jcmg.2013.10.008.
- Banypersad SM, Sado DM, Flett AS, et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. 2013;6 : 34–39. https://doi.org/10.1161/CIRCIMAGING.112.978627.
- Martinez-Naharro A, Abdel-Gadir A, Treibel TA, et al. CMR-Verified Regression of Cardiac ALAmyloid After Chemotherapy. JACC Cardiovasc Imaging.2018;11 : 152–154. https://doi.org/10.1016/j.jcmg.2017.02.012.
- Ferreira VM, Piechnik SK, Robson MD, et al. Myocardial tissue characterization by magnetic resonance imaging: novel applications of T1 and T2mapping. J Thorac Imaging. 2014;29 : 147–154. https://doi.org/10.1097/RTI.0000000000000077.
- Kotecha T, Martinez-Naharro A, Treibel TA, et al. Myocardial Edema and Prognosis in Amyloidosis. J Am Coll Cardiol 2018;71 : 2919–2931. https://doi.org/10.1016/j.jacc.2018.03.536
- Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46 : 1076–1084. https://doi.org/10.1016/j.jacc.2005.05.073.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133 : 2404–2412. https://doi.org/10.1161/CIRCULATIONAHA.116.021612.
- Hutt DF, Quigley AM, Page J, et al. Utility and limitations of 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in systemic amyloidosis. Eur Heart J Cardiovasc Imaging. 2014;15 : 1289–1298. https://doi.org/10.1093/ehjci/jeu107.
- Hutt DF, Fontana M, Burniston M, et al. Prognostic utility of the Perugini grading of 99mTc-DPD scintigraphy in transthyretin (ATTR) amyloidosis and its relationship with skeletal muscle and soft tissue amyloid. Eur Heart J Cardiovasc Imaging. 2017;18 : 1344–1350. https://doi.org/10.1093/ehjci/jew325.
- Cappelli F, Gallini C, Costanzo EN, et al. Lung uptake during 99mTc-hydroxymethylene diphosphonate scintigraphy in patient with TTR cardiac amyloidosis: An underestimated phenomenon. Int J Cardiol. 2018;254 : 346–350. https://doi.org/10.1016/j.ijcard.2017.10.027.
- Gertz MA, Brown ML, Hauser MF, et al. Utility of technetium Tc 99m pyrophosphate bone scanning in cardiac amyloidosis. Arch Intern Med 1987;147 : 1039–1044.
- Bokhari S, Castano A, Pozniakoff Tet al. (99 m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013;6 : 195–201.
- Rapezzi C, Quarta CC, Guidalotti PL, et al. Role of (99 m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc imaging 2011;4 : 659–470.
- Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99 mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005;46 : 1076–1084.
- Dorbala S, Vangala D, Semer J, et al. Imaging cardiac amyloidosis: a pilot study using 18Fflorbetapir positron emission tomography. Eur J Nucl Med Mol Imaging 2014;41 : 1652–1662. https://doi.org/10.1007/s00259-014-2787-6.
- Antoni G, Lubberink M, Estrada S, et al. In vivo visualization of amyloid deposits in the heart with 11C-PIB and PET. J Nucl Med 2013;54 : 213–220. https://doi.org/10.2967/jnumed.111.102053.
- Lee SP, Lee ES, Choi H, et al. 11CPittsburgh B PET imaging in cardiac amyloidosis. JACC Cardiovasc Imaging 2015;8 : 50–59.
- Galat A, Rosso J, Guellich A, et al. Usefulness of (99m)Tc-HMDP scintigraphy for the etiologic diagnosis and prognosis of cardiac amyloidosis. Amyloid 2015;22 : 210–220.
- Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018;39 : 2799–2806. https://doi.org/10.1093/eurheartj/ehx589.
- Grogan M, Scott CG, Kyle RA, et al. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J Am Coll Cardiol 2016;68 : 1014–1020. https://doi.org/10.1016/j.jacc.2016.06.033.
- Knight DS, Zumbo G, Barcella W, et al. Cardiac Structural and Functional Consequences of Amyloid Deposition by Cardiac Magnetic Resonance and Echocardiography and Their Prognostic Roles. JACC Cardiovasc Imaging 2019 : 12 : 823–833. https://doi.org/10.1016/j.jcmg.2018.02.016.
- Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2018. Blood Cancer J 2018;8 : 44. https://doi.org/10.1038/s41408-018-0080-9.
- Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers 2018;4 : 1–19. https://doi.org/10.1038/s41572-018-0034-3.
- Ruberg FL. Cardiac Amyloidosis: A Zebra Hiding in Plain Sight? Circ Cardiovasc Imaging. 2017;10:e006186. https://doi.org/10.1161/CIRCIMAGING.117.006186.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379 : 22–31. https://doi.org/10.1056/NEJMoa1716793.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018;379 : 1007–1016. https://doi.org/10.1056/NEJMoa1805689.
- Adams D, Gonzalez-Duarte A, O‘RiordanWD, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379 : 11–21. https://doi.org/10.1056/NEJMoa1716153.
- Lang O. Možnosti scintigrafických metod u pacientů se srdečním selháním a zachovanou ejekční frakcí. NuklMed 2020;9 : 49-58
Labels
Nuclear medicine Radiodiagnostics RadiotherapyArticle was published in
Nuclear Medicine

2020 Issue 4
Most read in this issue
- Srdeční amyloidóza v zobrazovacích metodách – pohled kardiologa
- 99Mo99mTc generátor: výroba a využití v nukleární medicíně 1. část
- Náhodný záchyt nemoci COVID-19 při FDG-PET/CT
-
Studentský den nukleární medicíny -
sborník abstraktů