
Vrozené vývojové vady vnitřního ucha
Congenital malformations of the inner ear
Congenital malformations of the inner ear consist of many different anomalies of the labyrinth. They often cause hearing loss, mostly of the sensorineural type. Eighty percent of hearing loss is caused by an anomaly of the membranous labyrinth, and 20% by an anomaly of the bone labyrinth. The role in pathogenesis is played by hereditary factors and influence of the environment. The treatment depends on the severity of the hearing loss, abnormalities of the external and middle ear, associated defects, and presence and function of the auditory nerve. We have modern hearing aids or implantable systems. Another options include a sign language and mouth-reading. The article includes a retrospective analysis of patients with congenital inner ear malformations at our tertiary center in 2010–2020. In conclusion, our patients clearly prove that even children with profound hearing loss are successfully implanted and restored hearing can be achieved in most of them.
Keywords:
congenital ear malformation – inner ear – hearing loss – anomalies of bony labyrinth – anomalies of membranous labyrinth – rehabilitation
Authors:
S. Šikolová 1
![]() ; Milan Urík 1
; Milan Urík 1
![]() ; J. Jančíková 1
; J. Jančíková 1
![]() ; D. Hošnová 1
; D. Hošnová 1
![]() ; R. Katra 2
; R. Katra 2
![]()
Authors‘ workplace:
Klinika dětské otorinolaryngologie LF MU a FN Brno
1; Klinika ušní, nosní a krční 2. LF UK a FN v Motole, Praha
2
Published in:
Otorinolaryngol Foniatr, 70, 2021, No. 3, pp. 167-173.
Category:
Review Article
doi:
https://doi.org/10.48095/ccorl2021167
Overview
Vrozené vývojové vady vnitřního ucha zahrnují celou řadu rozmanitých postižení labyrintu. Jsou jedním z důvodů těžkého poškození sluchu, převážně senzorineurálního typu. Přibližně 80 % nedoslýchavosti je následkem poškození membranózního labyrintu, zbývajících 20 % připadá na anomálie kostěného labyrintu. Projevit se mohou dědičné faktory a vliv prostředí. Vyskytují se samostatně, nebo jsou součástí syndromů. Léčba závisí na závažnosti poruchy sluchu, na případných anomáliích zevního a středního ucha, na přidružených vadách a na přítomnosti sluchového nervu. Podle situace volíme z výběru několika naslouchacích přístrojů a implantabilních systémů. Pokud je u dítěte z jakéhokoliv důvodu implantace kontraindikována nebo ji rodiče odmítají, lze dítě naučit znakové řeči a odezírání z úst. Součástí článku je přehled pacientů s vrozenými vadami vnitřního ucha na našem pracovišti v letech 2010–2020. Hodnotili jsme četnost výskytu pacientů s příslušnou diagnózou, četnost výskytu vad v závislosti na věku a pohlaví a následnou rehabilitaci. Naše výsledky dokazují, že i děti s těžkými sluchovými vadami podstupují na naší klinice úspěšnou kochleární implantaci a většině z nich lze sluch navrátit.
Klíčová slova:
vrozené vývojové vady – vnitřní ucho – nedoslýchavost – anomálie kostěného labyrintu – anomálie membranózního labyrintu – rehabilitace
Úvod
Vrozené vady vnitřního ucha dělíme na anomálie kostěného labyrintu a membranózního labyrintu. Kostěné anomálie diagnostikujeme pomocí počítačové tomografie (CT), membranózní lze potvrdit pouze histologicky. Pokud se tedy při senzorineurální poruše sluchu vyloučí kostěná anomálie podle CT, může se jednat o anomálii membranózní [1–3].
Embryologie vnitřního ucha
Vývoj vnitřního ucha začíná ve 3.–4. týdnu vývoje plodu ze sluchové plakody, která vzniká ztluštěním ektodermu na každé straně rombencefala. Vchlípení plakody dává základ sluchovému váčku (otocysta), který je rozdělen na ventrální (sacculus a ductus cochlearis) a dorzální část (utriculus, ductus semicirculares a ductus endolymphaticus), jak ilustruje obr. 1. Společně vytváří tyto struktury blanitý labyrint, kolem něhož se diferencuje labyrint kostěný. Základem je spirálovité prorůstání ductus cochlearis okolním mezenchymem, který se kolem duktu diferencuje v chrupavku, v jejímž pouzdře se zformují dva perilymfatické prostory – scala vestibuli a scala tympani. Epitelové buňky v duktu vytvoří vnitřní (limbus spiralis) a vnější val (organum Corti). Osifikace kochley je ukončena v 25. týdnu a vzruchy z Cortiho orgánu jsou přenášeny do ganglion cochleare a dále vlákny nervus cochlearis do jader rombencefala. Sluchové podněty začíná plod vnímat v 5. měsíci těhotenství [1, 4, 5].
a, b) dorzální část, c–e) ventrální část.
Fig. 1. Development of the otic vesicle [1].
a, b) dorsal part, c–e) ventral part.
![Vývoj sluchového váčku [1].<br>
a, b) dorzální část, c–e) ventrální část.<br>
Fig. 1. Development of the otic vesicle [1].<br>
a, b) dorsal part, c–e) ventral part.](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/ba311e721775bac10ab18edf2e3ecfd6.png)
Anomálie kostěného labyrintu
Kostěné malformace jsou vzácné a tvoří asi 20 % vývojových anomálií vnitřního ucha. Vývojové abnormality kochley můžeme rozdělit na čtyři skupiny podle Jacklera et al. [6]:
1. úplná aplazie,
2. společná dutina,
3. kochleární hypoplazie,
4. neúplné vytvoření závitů.
Na základě možné implantace i u hypoplastické kochley vznikla nová klasifikace kochleovestibulárních malformací podle Sennaroglu a Saatci [7]:
1. kochleární malformace: Michelova deformita, kochleární aplazie a hypoplazie, neúplné vytvoření kochley typ I a typ II, společná dutina,
2. vestibulární malformace: chybějící nebo hypoplastické a dilatované vestibulum,
3. malformace semicirkulárních kanálků: chybějící, hypoplastické nebo dilatované kanálky,
4. abnormality aqueductus cochlearis a vestibularis: dilatované akvadukty,
5. malformace vnitřního zvukovodu: chybějící, zúžený nebo rozšířený zvukovod.
Kochleární malformace
Michelova deformita
Tato anomálie patří mezi nejzávažnější z kochleárních malformací. Jedná se o úplnou aplazii labyrintu podmíněnou hypoplazií pyramidy spánkové kosti a chyběním kochleovestibulárních struktur (obr. 2). Z hlediska embryonálního vývoje se tak musí stát před 3. týdnem vývoje, tedy ještě před založením sluchové plakody. Může být asociována se srdečními vadami, se zúžením vnitřního zvukovodu a chyběním n. VIII. Zevní a střední ucho mohou být normálně vyvinuté [1].
Fig. 2. Agenesis of the inner ear on the left (marked by an
arrow), MRI, transverse projection.
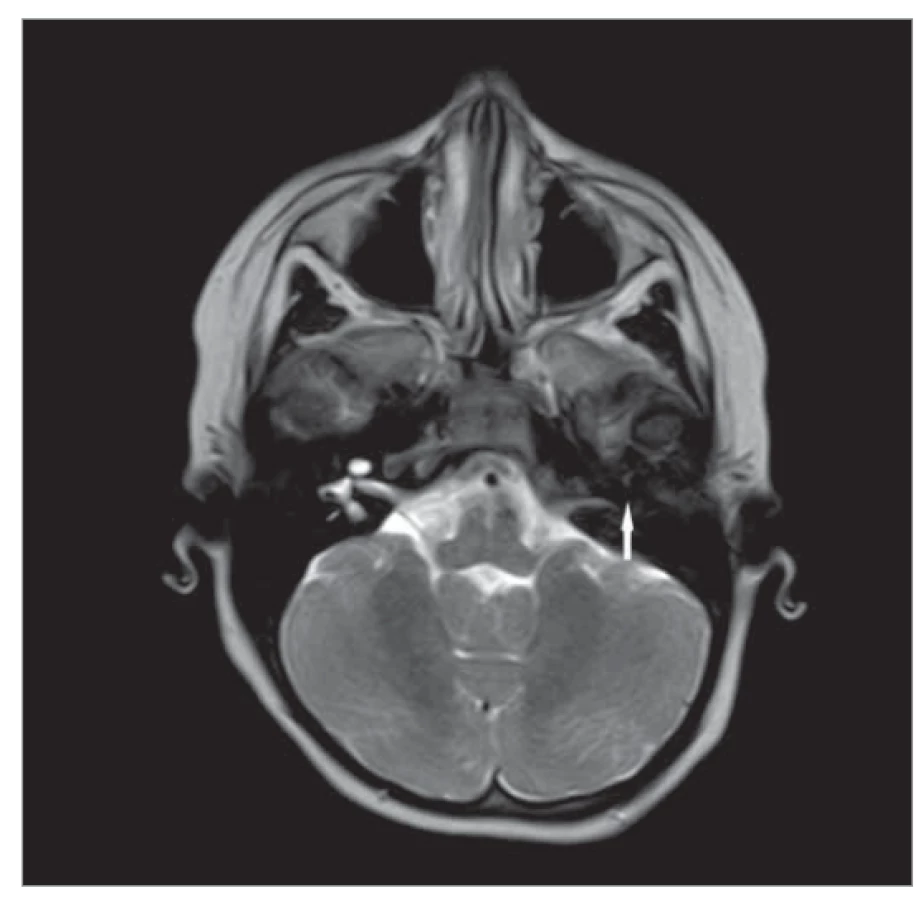
Při aplazii sluchového nervu je přítomna anakuze, jež není řešitelná kochleární implantací. V takovém případě je možnost implantace kmenové neuroprotézy, která přináší novou éru v rehabilitaci sluchu u pacientů, kde je kochleární implantace kontraindikována nebo není úspěšná, zejména z hlediska nedostatečného rozvoje řeči [8].
Kochleární aplazie a hypoplazie
V průběhu 6.–8. týdne embryogeneze dochází ke zvětšování a prodlužování ductus cochlearis, který je později rozdělen do jednotlivých závitů. Stupeň kochleární deformity závisí na tom, kdy došlo ve vývoji kochley k zabrzdění. Ve většině případů k tomu dochází v 6. týdnu, kdy je kochlea přítomna ve formě malého zárodku dotýkajícího se vestibula. Pokud se vývoj zabrzdí později kolem 8. týdne, projeví se kochleární hypoplazie mírněji jako neúplné vytvoření závitů.
Neúplné vytvoření kochley typ I
Postižení představuje 20 % kochleárních malformací charakterizované cystickou kochleou, která zcela postrádá modiolus a dilatovaným cystickým vestibulem.
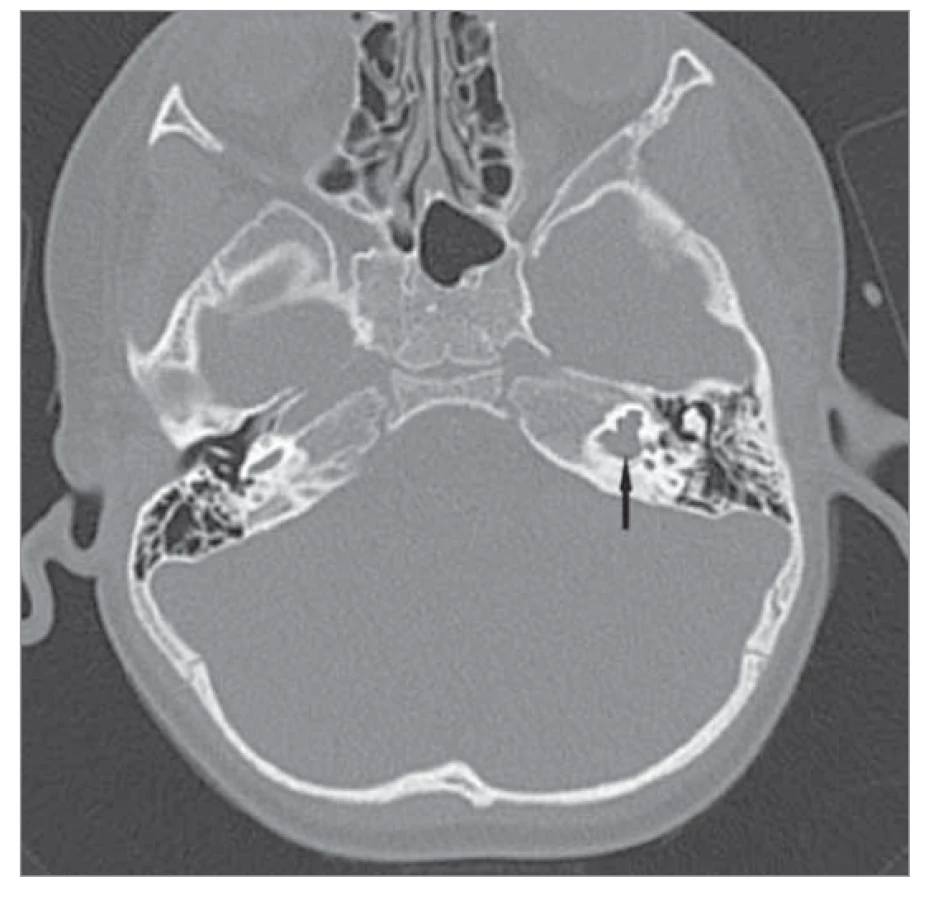
Neúplné vytvoření kochley typ II
Nejčastější vnitřní malformací a zároveň druhou nejčastější příčinou vrozené hluchoty u dětí je tzv. Mondiniho malformace, kdy je kochlea rozdělena na méně než dva a půl závitu. Představuje přibližně 30 % kochleárních malformací. Nejčastějším obrazem na CT (obr. 3) je normální bazální závit kochley s cystickým apexem nahrazujícím střední a apikální závity, minimální dilatace vestibula a prodloužený vestibulární akvadukt. Vývoj Cortiho orgánu je přitom variabilní, z čehož vyplývá i různý stupeň poškození sluchu. Vada je velmi úspěšně korigována kochleární implantací. Podle anatomického utváření kochley volíme z několika různých délek elektrod [1, 9, 10].
Fig. 3. Mondini malformation on the left (marked by an
arrow), CT image, axial projection.
Společná dutina (common cavity)
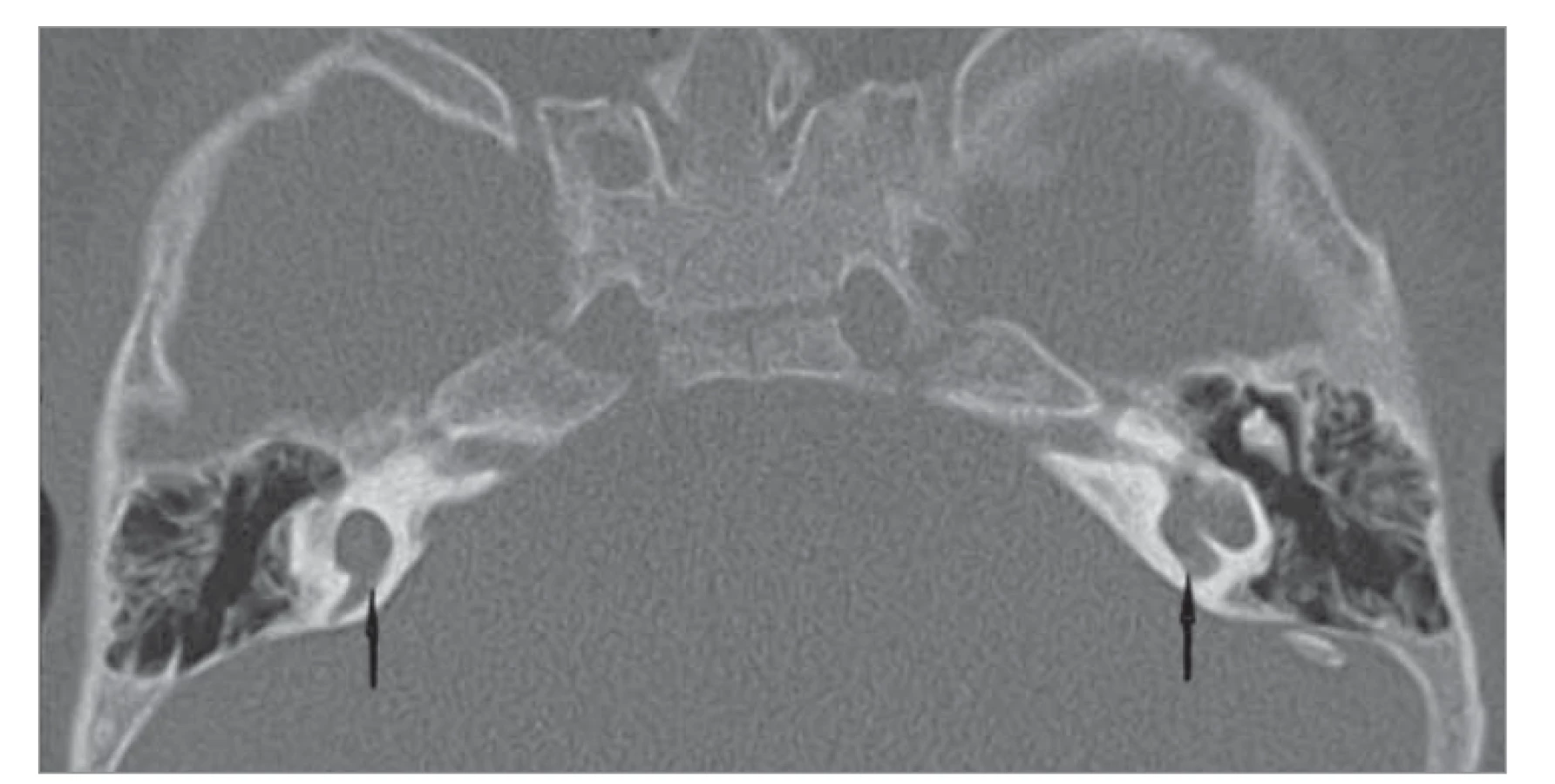
Tato anomálie vzniká při zastavení embryonálního vývoje ve 4. týdnu, kdy se ze sluchové plakody vyvine otocysta a vnitřní ucho zůstane tvořené jedinou dutinou (obr. 4). Cystická dutina tedy prezentuje kochleu i vestibulum bez jejich diferenciace [6]. Při této anomálii je přítomna těžká percepční nedoslýchavost. Ještě donedávna se vedly spory o významu kochleární implantace, ale některé práce a kazuistiky jasně dokazují její úspěšnost [11, 12]. Na naší klinice byla pacientovi s oboustrannou společnou dutinou úspěšně provedena implantace, nejdříve v necelých 2 letech života na pravou stranu a o rok později na stranu levou [13].
Fig. 4. Common cavity on both sides (marked with an arrow), CT image, axial
projection.
Malformace semicirkulárních kanálků
Aplazie a hypoplazie semicirkulárních kanálků jsou nejčastější anomálií vestibulárního aparátu. Mohou se vyskytovat samostatně, častěji je ale nacházíme společně s anomálií kochley nebo souvisí s CHARGE syndromem, kde je přítomna kombinace několika vážných poruch orgánů, zejména porucha sluchu a zraku, srdeční vady a psychomotorická retardace. Nejčastěji bývá postižen kanálek laterální, který se vyvíjí jako poslední. Porucha sluchu může být převodní, percepční nebo smíšená.
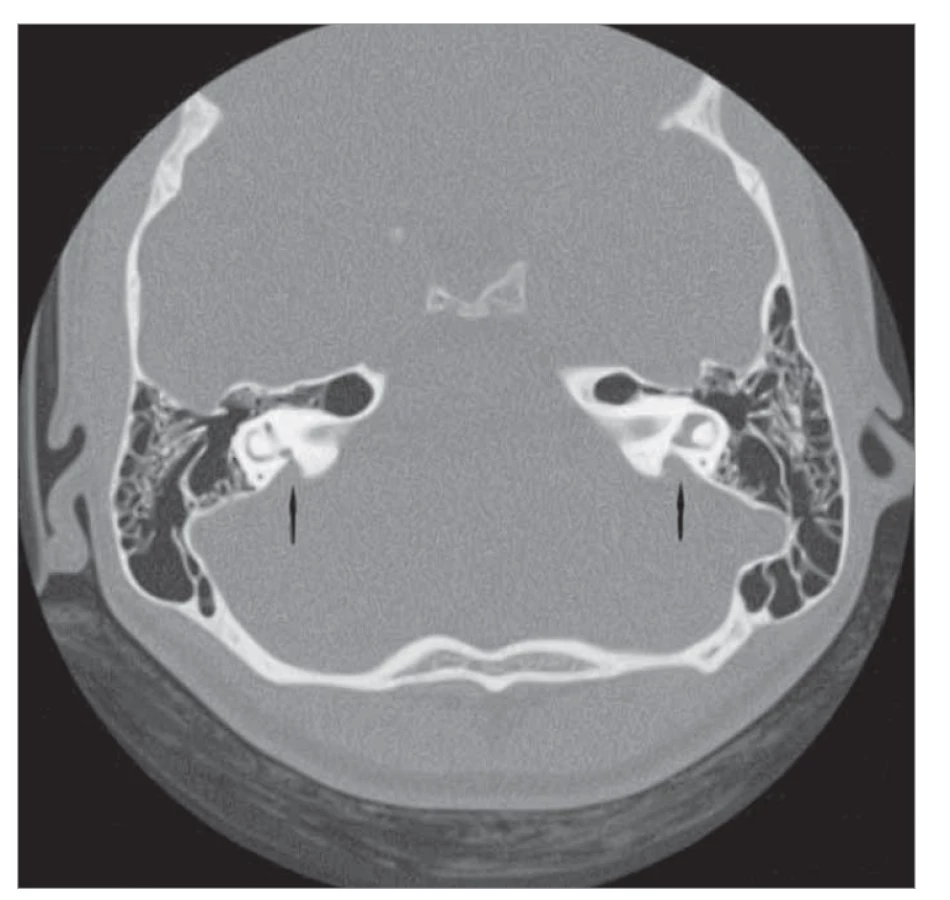
Dilatace vestibulárního duktu (EVA – enlarged vestibular aqueductus)
Jedná se o patologii, při které je průměr ductus vestibularis větší než 1,5 mm a současně s ním může být rozšířen i saccus endolymphaticus. Dilataci duktu prokážeme pomocí CT vyšetření (obr. 5), dilataci saku pouze pomocí MR. Tato malformace se může vyskytovat samostatně, nebo současně s anomálií kochley, často bývá také asociována s dalšími syndromy. Porucha sluchu bývá těžká až u 40 % pacientů a může být percepčního nebo smíšeného typu. Někdy ale není nedoslýchavost odhalena ihned po narození, dítě může normálně slyšet a dochází k postupné progresi či fluktuaci onemocnění. U pacientů s EVA syndromem a percepční poruchou sluchu je implantace běžně úspěšná. Během operace je často přítomen profuzní výtok nitroušních tekutin, tzv. gusher [1, 14, 15].
Fig. 5. EVA on both sides (marked with an arrow), CT image, axial projection.
Anomálie vnitřního zvukovodu
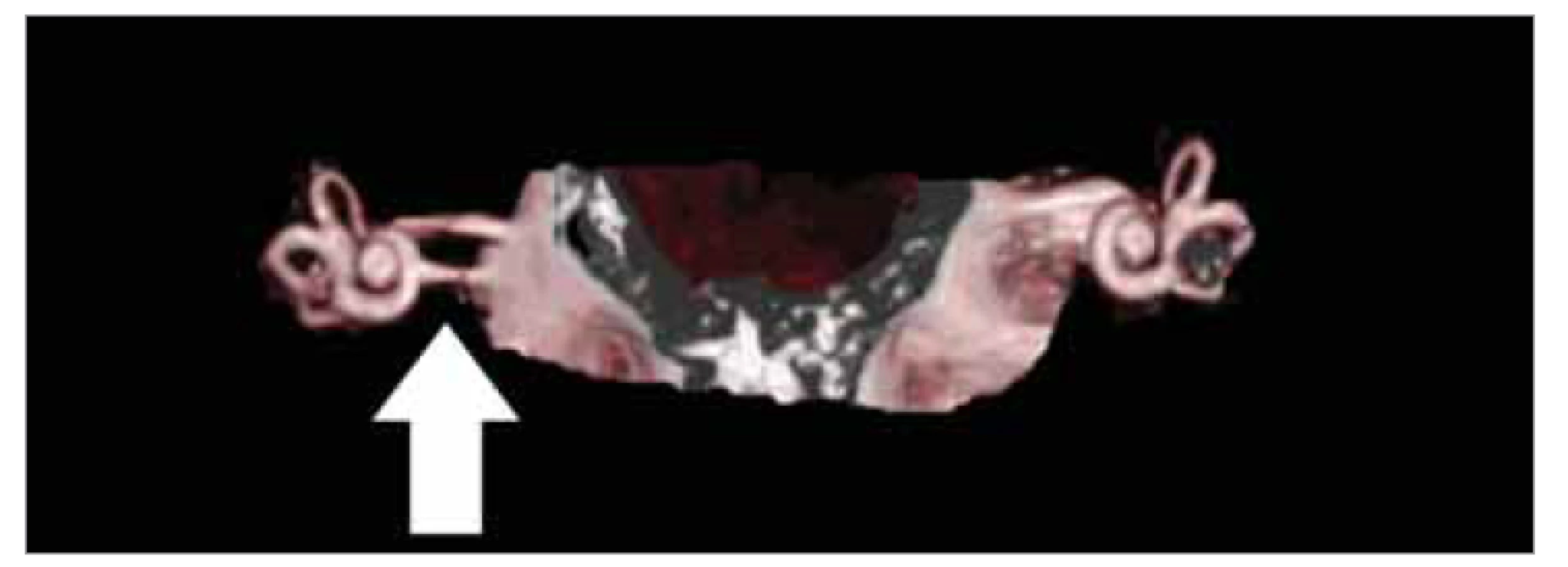
Průměr vnitřního zvukovodu je přibližně 4 mm a může být patologicky rozšířen nebo zúžen. Je-li průměr menší než 2 mm, označujeme stav jako stenózu, prokazatelnou na CT snímku. Při tomto stavu může být přítomna aplazie či hypoplazie n. VIII (vzácněji i n. VII), prokazatelná pouze pomocí MR. Anomálie se projeví vrozenou hluchotou na postižené straně, jež může být řešitelná kochleární implantací. Podle některých autorů je ovšem implantace při oboustranné stenóze vnitřního zvukovodu kontraindikována, jinými autory je hodnocena úspěšně, pokud se elektroda dotýká neurosenzitivní tkáně a jsou přítomny elektrické potenciály z mozkového kmene (EABR – evoked auditory brainstem response) alespoň na jedné straně [1, 16, 17]. Vzácnou anomálii prezentuje rozštěp pravého vnitřního zvukovodu (obr. 6).
Fig. 6. Cleft of the internal auditory canal on the right (marked by an arrow), MRI
reconstruction.
Anomálie membranózního labyrintu
Membranózní malformace jsou daleko častější než kostěné, představují zhruba 80 % vývojových vad vnitřního ucha. Jsou závislé na genech, jedná se tedy o patologii na buněčné úrovni.
Bingova-Siebenmannova malformace (cochleosaccular dysplasia)
Sem řadíme aplazie nebo hypoplazie membranózního labyrintu ve vyvinutém kostěném labyrintu. Často se vyskytují u Jervellova-Langeova-Nielsenova syndromu a Usherova syndromu. Je přítomna těžká percepční vada sluchu.
Scheibeho malformace
Jedná se o nejčastější malformaci membranózního labyrintu, kdy dochází k defektu vývoje kochley a sakulu. Cortiho orgán je chabě diferencován, je přítomna malformace tektoriální membrány a kolaps Reissnerovy membrány. Podobně jako u předchozí malformace bývá asociována s jinými syndromy a je přítomna těžká percepční nedoslýchavost.
Alexandrova malformace
Při této deformitě je dysplastický membranózní bazální závit hlemýždě a rozšířený vestibulární aqadukt. Jedná se o nejlehčí malformaci labyrintu, při které je přítomna percepční nedoslýchavost ve vysokých frekvencích, někdy i závratě a poruchy rovnováhy. Často se vyskytuje při Pendredově syndromu [1, 15, 18].
Nejčastější syndromy asociované s anomálií vnitřního ucha
Pendredův syndrom
Tento syndrom patří mezi autozomálně recesivní onemocnění způsobené mutací v genu SLC26A4, který kóduje aniontový transportér pendrin. Jedná se o protein zodpovědný za metabolizmus jodu a při jeho deficitu je postižena především štítná žláza a vnitřní ucho. Postižení štítnice se manifestuje nejčastěji ve druhé dekádě života jako eutyroidní či hypotyroidní struma, vzácněji dyshormonogenezí již po narození, která je diagnostikována novorozeneckým screeningem pro kongenitální hypotyreózu. Ušní malformace se nejčastěji projeví jako rozšířený vestibulární akvadukt a/nebo Mondiniho či Alexandrova malformace. Je přítomna závažná progredující senzorineurální porucha sluchu, jež vyžaduje užití sluchových pomůcek či možnost kochleární implantace, která přináší dobré výsledky i přes možné anomálie vnitřního ucha.
Pendredův syndrom je nejčastější syndromovou hluchotou a gen SLC26A4 je zřejmě druhým nejčastějším genem zodpovědným za autozomálně recesivní (AR) nesyndromovou ztrátu sluchu, hned po genu GJB2 pro Connexin 26, který je zodpovědný až za 50 % AR nesyndromové hluchoty [1, 19].
CHARGE syndrom
Tato anomálie se vyskytuje sporadicky nebo vykazuje autozomálně dominantní dědičnost. Je charakterizována několika diagnostickými kritérii (coloboma iris, heart defect, atresia choanae, retardation growth and mental development, genitourinary, ear abnormality). Mezi ušní anomálie patří nejčastěji atrezie zevního zvukovodu, různé anomálie sluchových kůstek a semicirkulárních kanálků, popsána je i Mondiniho dysplazie. Podle postižení je porucha sluchu převodního, percepčního nebo smíšeného typu. Časté jsou i abnormality hlavových nervů. Z ORL příznaků jsou závažné především atrezie jícnu, tracheo-ezofageální píštěl a rozštěp hrtanu, dále různé anomálie tváře, dysfagie a anosmie [1, 20].
Usherův syndrom
Jedná se o autozomálně recesivní syndrom charakterizovaný percepční poruchou sluchu a progresivní ztrátou zraku z retinitis pigmentosa. Nejčastěji bývá přítomna Scheibeho malformace či Bingova-Siebenmannova malformace a podle stupně poškození sluchu rozlišujeme 4 typy. Typ I – kongenitální poškození sluchu s absencí vestibulárních funkcí, ztráta zraku se projeví prepubertálně. Typ II – kongenitální poškození sluchu ve vysokých frekvencích s normální vestibulární funkcí, retinitida začíná v pubertě. Typ III – progresivní porucha sluchu, vestibulární funkce mohou být poškozeny nebo zachovány, počátek ztráty zraku je variabilní. Typ IV – klinicky podobný typu II, ale s X-vázanou dědičností. Zatímco ztrátu sluchu lze korigovat sluchovými pomůckami či kochleární implantací, na postupnou ztrátu zraku zatím neexistuje účinná terapie. Ve výzkumu hraje roli genová terapie [1, 15, 21].
Treacherův-Collinsův syndrom
Autozomálně dominantní postižení, které je u dítěte patrné ihned po narození typickou malformací obličeje. Dominuje zúžená tvář, kolobom dolních víček a antimongoloidní postavení očí. Pro hypoplazii mandibuly a rozštěp patra je syndrom častou příčinou dýchacích potíží u novorozence. Ušní anomálie charakterizuje kombinace poškození zevního, středního a vnitřního ucha. Přítomna bývá mikrotie boltce s atrezií zevního zvukovodu, ušní přívěsky a fistuly, anomálie středoušních kůstek a dysplastické semicirkulární kanálky s normální funkcí kochley. Porucha sluchu je kombinací převodní a senzorineurální nedoslýchavosti [15, 18].
Waardenburgův syndrom
Tento autozomálně dominantní syndrom se vyskytuje ve 4 formách, společným znakem je percepční nedoslýchavost a pigmentové anomálie. Postižení membranózního labyrintu se nejčastěji projeví Scheibeho malformací. Typy I a III mají navíc lehké kraniofaciální anomálie, typ II anomálie končetin a typ IV je asociován s Hirschprungovou chorobou. U 20–30 % postižených je přítomen charakteristický bílý pramen vlasů nad čelem. Variabilně shledáváme hypoplazii nosních křídel, spojené obočí, bílé řasy, heterochromia iridis, předčasné šedivění vlasů a hypopigmentaci kůže ve formě bílých skvrn.
Jervellův-Langeův-Nielsenův syndrom
Syndrom s autozomálně recesivní dědičností podmíněný mutací draslíkového kanálu, jež je zodpovědný za správnou funkci srdečních buněk a kochley. Charakteristický je prodloužený QT interval na EKG a těžká percepční nedoslýchavost, jež je nejčastěji podmíněna Scheibeho malformací nebo Bingovou-Siebenmannovou malformací. Děti s tímto syndromem jsou ohroženy fibrilací komor až náhlou smrtí [1, 6, 15].
Přehled pacientů s vrozenými vadami vnitřního ucha na našem pracovišti v letech 2010–2020
V tomto 10letém období činil celkový počet dětí s vrozenou vadou vnitřního ucha 24. Věkový průměr pacientů v době diagnostiky byl 6,7 roku a převažovalo ženské pohlaví nad mužským v poměru 14 : 10. Jedenáct pacientů bylo úspěšně rehabilitováno, a to 5 sluchadlem, 5 kochleární implantací a 1 pacient obdržel sluchadlo a následně mu byla provedena implantace. Zbývajících 13 pacientů zůstalo bez rehabilitace, nejčastěji pro jednostranné postižení sluchu. Přesto náš soubor jasně dokazuje, že i děti s těžkými sluchovými vadami podstupují na naší klinice úspěšnou kochleární implantaci s dobrým sluchovým ziskem.
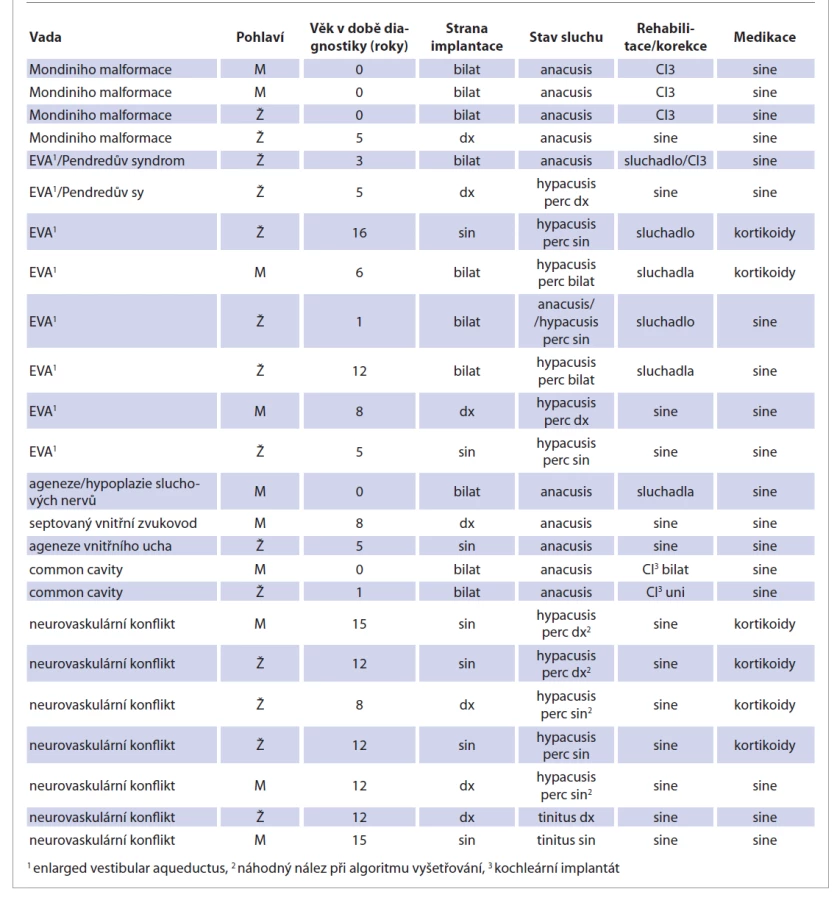
Přehled pacientů s jednotlivými diagnózami, anamnestickými údaji a následnou rehabilitací shrnuje tab. 1.
Tab. 1. An overview of patients with congenital inner ear defects at our workplace in 2010–2020.
Závěr
Vrozené vývojové vady ucha se vyskytují v rozmezí 1/10 000 až 1/15 000 narozených dětí. Na každé tisící dítě připadá různý stupeň percepční nedoslýchavosti, jejíž příčinou jsou přibližně ve 20 % anomálie vnitřního ucha. Poruchu sluchu lze diagnostikovat již v porodnici měřením OAE a dále pomocí dalších objektivních audiologických metod. U každé vrozené poruchy sluchu je indikováno vyšetření zobrazovací metodou a genetické vyšetření. Léčba závisí na závažnosti poruchy sluchu a dalších anatomických a klinických okolnostech. V současné době máme k dispozici sofistikované metody rehabilitace sluchu, od indikace naslouchacích přístrojů přes kochleární implantaci až po kmenovou neuroprotézu. Alternativou implantabilního systému je nauka znakové komunikace a vizuální percepce řeči.
Grantová podpora
Tato práce byla podpořena grantem: Ztráta sluchu v dětském věku – příčiny, diagnostika, možnosti rehabilitace IV (MUNI/A/0847/2019).
Prohlášení
Autorka práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i spoluautorů.
ORCID autorů
S. Šikolová ORCID 0000-0001-6687-6839,
M. Urík ORCID 0000-0002-2872-185X,
J. Jančíková ORCID 0000-0003-2590-3567,
D. Hošnová ORCID 0000-0001-9692-1178,
R. Katra ORCID 0000-0001-8503-1348.
Přijato k recenzi: 30. 6. 2020
Přijato k tisku: 14. 10. 2020
MUDr. Soňa Šikolová
Klinika dětské otorinolaryngologie
LF MU a FN Brno
Černopolní 9
613 00 Brno
Sources
1. Jakubíková J. Vrozené anomálie hlavy a krku. Praha: Grada; 2012.
2. Rodriguez K, Shah RK, Kenna M. Anomalies of the middle and inner ear. Otolaryngol Clin North Am 2007; 40 (1): 81–96, vi. Doi: 10.1016/j.otc.2006.10.006.
3. Yiin RS, Tang PH, Tan TY. Review of congenital inner ear abnormalities on CT temporal bone. Br J Radiol 2011; 84 (1005): 859–863. Doi: 10.1259/bjr/18998800.
4. Anson BJ, Davies J. Embryology of the ear: developmental anatomy of the ear. In: Otolaryngology. 2nd ed. Edited by Paparella MM, Shumrick DA. Philadelphia: WB Saunders 1980 : 3–25.
5. Peck JE. Development of hearing. Part II. Embryology. J Am Acad Audiol 1994; 5 (6): 359–365.
6. Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope 1987; 97 (3 Pt 2 Suppl 40): 2–14. Doi: 10.1002/lary.5540971301.
7. Sennaroglu L, Saatci I. A new classification for cochleovestibular malformations. Laryngoscope 2002; 112 (12): 2230–2241. Doi: 10.1097/00005537-200212000-00019.
8. Sennaroglu L, Sennaroglu G, Atay G. Auditory brainstem implantation in children. Curr Otorhinolaryngol Rep 2013; 1 (2): 80–91. Doi: 10.1007/s40136-013-0016-7.
9. Arellano B, Ramirez-Camacho R, Trinidad A et al. Inner ear malformations: Mondini‘s dysplasia. ORL J Otorhinolaryngol Relat Spec 1999; 61 (6): 360–363. Doi: 10.1159/000027700.
10. Ramírez LV, Cano HD, Lubinus FG. Congenital malformations of the inner ear. Rev Colomb Radiol 2018; 29 (3): 4481–4485.
11. McElveen JT, Jr., Carrasco VN, Miyamoto RT et al. Cochlear implantation in common cavity malformations using a transmastoid labyrinthotomy approach. Laryngoscope 1997; 107 (8): 1032–1036. Doi: 10.1097/00005537-199708000 - 00005.
12. Zhang L, Qiu J, Qin F et al. Cochlear implantation outcomes in children with common cavity deformity; a retrospective study. J Otol 2017; 12 (3): 138–142. Doi: 10.1016/j.joto.2017.03. 004.
13. Urík M, Šlapák I, Hošnová D et al. Bilateral cochlear implantation in children with common cavity. Acta Oto-Laryngologica Case Reports 2020; 5 (1): 38–41. Doi: 10.1080/23772484. 2020.1756820.
14. Pyle GM. Embryological development and large vestibular aqueduct syndrome. Laryngoscope 2000; 110 (11): 1837–1842. Doi: 10.1097/00005537-200011000-00014.
15. Wetmore RF, Muntz HR, McGill TJ. Pediatric Otolaryngology: Principles and Practise Pathways. Stuttgart: Thieme 2000.
16. Yan F, Li J, Xian J et al. The cochlear nerve canal and internal auditory canal in children with normal cochlea but cochlear nerve deficiency. Acta Radiol 2013; 54 (3): 292–298. Doi: 10.1258/ar.2012.110596.
17. Kumakawa K, Takahashi M, Takeda H. Is a narrow internal auditory meatus a contraindication to cochlear implants? Cochlear Implants Int 2004; 5 (Suppl 1): 80–82. Doi: 10.1179/cim. 2004.5.Supplement-1.80.
18. Šlapák I, Urík M. Dětská otorinolaryngologie. Praha: Mladá Fronta 2019.
19. Katra R, Pourová R, Dytrych P. Pendredův syndrm v České republice. Otorinolaryngol Foniatr 2011; 60 (2): 103–111.
20. Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis 2006; 1 : 34. Doi: 10.1186/ 1750-1172-1-34.
21. Moller CG, Kimberling WJ, Davenport SL et al. Usher syndrome: an otoneurologic study. Laryngoscope 1989; 99 (1): 73–79. Doi: 10.1288/ 00005537-198901000-00014.
Labels
Audiology Paediatric ENT ENT (Otorhinolaryngology)Article was published in
Otorhinolaryngology and Phoniatrics

2021 Issue 3
Most read in this issue
- Congenital malformations of the inner ear
- Acute vertigo in the ENT emergency
- Ultrasound-guided percutaneous ethanol injection therapy of neck cysts as an alternative to surgery
- Evaluation of speech in patients after cochlear implantation – Motol Speech Scale