
Hamartomy dutiny břišní a retroperitonea
– souhrnné sdělení doplněné kazuistikou
Hamartoma of the abdominal cavity and retroperitoneum
– a review and a case report
Methods:
We have reviewed recent as well as older literature with the intention of compiling a summary report on hamartomas of the abdominal cavity and retroperitoneum.
Introduction:
Hamartoma of the abdominal cavity and retroperitoneum is a rare condition which has received relatively little attention.
Results:
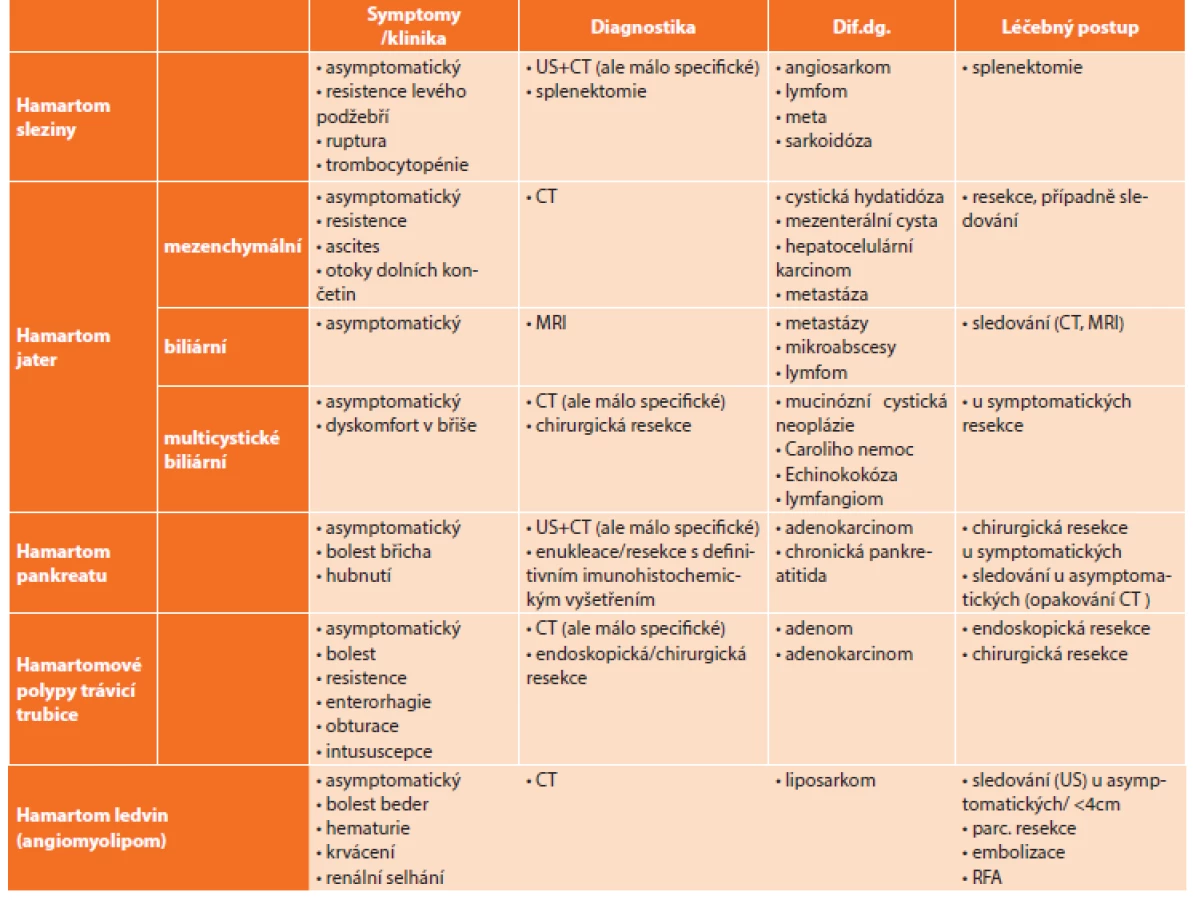
The most commonly affected organs are the liver, spleen, digestive tract, pancreas and kidneys. By its nature, hamartoma is a benign lesion with a good prognosis. However, neoplastic transformation may occur and hamartomas need to be regarded as potentially malignant.
Patients are very often asymptomatic. Signs and symptoms depend on the organ involved and are caused, to different degrees, by obstruction, surrounding tissue compression, rupture or tumor bleeding. In imaging diagnostics, it is usually not possible to safely distinguish between a hamartoma and a malignant lesion. Therefore, the patient is preferably indicated for surgery (or endoscopic removal) for the purpose of histological verification and definitive treatment.
Conclusion:
Hamartomas of the abdominal cavity and retroperitoneum are relatively rare, benign and often asymptomatic diseases which, however, may manifest as compression, obstruction, perforation, bleeding or even malignant transformation.
The recent case report of splenic hamartoma presented at the end of this review proves that surgical management is often the only rational option to obtain definitive confirmation of the diagnosis.
Key words:
splenic hamartoma − hamartoma of the liver − hamartoma of the pancreas − hamartomatous polyps of the colon − multiple hamartoma syndrome
:
A. Paclík; P. Dytrych
![]() ; D. Hoskovec; R. Jakša; Z. Krška
; D. Hoskovec; R. Jakša; Z. Krška
:
I. chirurgická klinika 1. LF Univerzity Karlovy a VFN v Praze
přednosta: prof. MUDr. Z. Krška, DrSc.
:
Rozhl. Chir., 2017, roč. 96, č. 9, s. 375-382.
:
Review
Metodika:
Provedli jsme revizi recentní i starší literatury s intencí sestavit souhrnné sdělení o hamartomech dutiny břišní a retroperitonea.
Úvod:
Hamartom v dutině břišní a retroperitoneu je vzácné onemocnění, jehož problematice je věnován relativně malý prostor.
Výsledky:
Nejčastěji postiženými orgány bývají játra, slezina, trávicí trubice, pankreas a ledviny. Svou biologickou povahou je hamartom benigní léze s dobrou prognózou. Může však dojít k neoplastické transformaci a je třeba jej pokládat za potenciálně maligní.
Pacienti bývají velmi často asymptomatičtí. Znaky a příznaky se odvíjejí od orgánové lokalizace a různou měrou se na nich podílejí obstrukce, útlak okolní tkáně nebo ruptura či krvácení z tumoru.
V diagnostice zobrazovacími metodami není ve většině případů možné bezpečně odlišit hamartom od maligní léze, proto bývá pacient raději indikován k chirurgické revizi (či endoskopickému odstranění) za účelem histologické verifikace a zároveň definitivní léčby.
Závěr:
Hamartomy dutiny břišní a retroperitonea jsou relativně vzácná, benigní a často asymptomatická onemocnění, která se ovšem mohou projevit útlakem, obstrukcí, perforací, krvácením a dokonce maligním zvratem. V závěru uvedená recentní kazuistika hamartomu sleziny dokazuje, že chirurgické řešení je často jedinou racionální možností definitivního potvrzení diagnózy.
Klíčová slova:
hamartom sleziny − hamartom jater − hamartom pankreatu − hamartogenní polypy střeva − syndrom mnohočetného hamartomu
Metodika
Informace byly získány formou retrospektivní analýzy článků v databázi PubMed. Přednost byla dána pracím vydaným v dané problematice recentně, nicméně vzhledem k absenci nových poznatků u některých hamartomů sahá naše analýza originálních prací v některých případech až do šedesátých let minulého století. Byla revidována souhrnná sdělení, klinické studie i kazuistiky. Jako hesla pro vyhledávání jsme použili hamartom sleziny, hamartom jater, hamartom pankreatu, hamartogenní polypy střeva, syndrom mnohočetného hamartomu a hamartom ledvin. Vycházeli jsme z informací článků publikovaných v českém a anglickém jazyce.
Úvod
Hamartom je benigní, tumoru podobná malformace. Je tvořen zralými buňkami normálně přítomnými v daném orgánu (tedy například hamartom sleziny se nachází pouze ve slezině a nikde jinde). Růst těchto buněk je však dezorganizovaný a nerespektuje fyziologickou stavbu orgánu. Nejedná se o neoplastický proces, hamartom nemetastazuje a neinfiltruje okolní struktury. Nejčastěji postiženými orgány jsou plíce, ledviny, játra a slezina [1]. Nejběžnější histologickou komponentou hamartomu je chrupavka, tuková, vaskulární, neuromezenchymální a myoepiteliální tkáň [2].
Vznik hamartomu je dáván do souvislosti s poruchou vývoje embryonální tkáně [3]. Etiologie však zůstává nejasná. Ačkoliv se hamartom nepovažuje za neoplastický proces, může během růstu dojít k neoplastické transformaci, a proto je třeba hamartom považovat za potenciálně maligní [4]. Hamartom je důležité odlišit od choristomu, což je benigní masa tvořená buňkami, které se za normálních okolností v dané tkáni nenacházejí (např. heterotopická tkáň pankreatu ve stěně duodena).
V diagnostice zobrazovacími metodami není ve většině případů možné bezpečně odlišit hamartom od maligní léze, a byť některé entity vykazují celkem sugestivní typické radiologické znaky, bývá pacient raději indikován k chirurgické revizi za účelem histologické verifikace a zároveň definitivní léčby. U polypů trávicí trubice je možno přistoupit k endoskopickému odstranění. V případě malého rizika záměny s malignitou jsou typicky některé hamartomy jater, ledvin a pankreatu často primárně indikovány k pečlivému sledování. Zejména u postiženého pankreatu hraje v rozhodování roli vyšší riziko pooperačních komplikací.
Chirurg se s hamartomem setkává buď v rámci elektivní operace, nebo v případě akutního výkonu. Plánovaně bývají operováni obvykle asymptomatičtí pacienti s nálezem ložiska nejisté biologické povahy. Další skupinou určenou k elektivnímu výkonu jsou pacienti s nespecifickými potížemi, jako jsou abdominální dyskomfort, nevolnost, pocit plnosti nebo chronické či recidivující krvácení do zažívacího traktu. Nejčastějším důvodem k akutní operaci bývá ruptura hamartomu spojená s výrazným krvácením nebo obstrukce zažívacího traktu.
Mezi nejčastěji postižené orgány patří játra a slezina. Prevalence hamartomových polypů zažívacího traktu je udávána asi 2 % v populaci [5].
Hamartomy se vyskytují buď sporadicky, nebo jsou součástí syndromů.
Nejčastější syndromy spojené s hamartomy
Cowdenův syndrom je autozomálně dominantně dědičná porucha způsobená mutací genu PTEN. Jeho incidence je zhruba 1 : 200 000 [6]. Projevuje se zvýšeným výskytem hamartomů zejména v kůži (90−100 %), štítné žláze (66 %), dále sliznici zažívacího traktu, kostní tkáni, CNS, očích a v urogenitálním traktu [7]. Cowdenův syndrom je součástí tzv. PTEN hamartoma tumor syndromu, kam se dále řadí Bannayan-Riley-Ruvalcaba syndrom, Proteus syndrom a Proteus-like syndrom. Jelikož v rámci tohoto syndromu je udáván větší výskyt karcinomu prsu u žen, karcinomu štítné žlázy a kolorektálního karcinomu u obou pohlaví [8], je důležitá včasná diagnóza tohoto onemocnění u lidí vykazujících multifokální postižení kůže.
Tuberózní skleróza je autozomálně dominantně dědičná porucha způsobená mutací genů TSC1 a TSC2. Je pro ni typická tvorba četných benigních ložisek v různých orgánech včetně mozku, kůže, srdce, ledvin, jater, sleziny a očí. V případě tuberózní sklerózy je vyšší riziko jejich malignizace. Nejčastějším prvním příznakem bývá kožní léze (hypopigmentované makuly, angiofibromy), která se opět objevuje téměř u všech pacientů. V retroperitoneu se tuberózní skleróza prezentuje tvorbou angiomyolipomů či benigních cyst [9].
Pro Peutz-Jeghersův syndrom jsou charakteristické hamartomatózní změny žlázového epitelu spolu s buňkami hladké svaloviny ve střevní sliznici. Dalším znakem jsou pigmentované mukokutánní papuly. Klinicky se syndrom začíná projevovat mezi 10. a 30. rokem života. Dominuje opakované krvácení s postupnou anemizací a dochází k intususcepcím, pro které jsou i opakovaně operováni. U rostoucích polypů může dojít po 30. roce věku k malignímu zvratu a pacienti jsou ohroženi vznikem karcinomu kolon a žaludku. Dále je prokázána asociace s karcinomem pankreatu, prsu a varlat. Incidence tohoto syndromu je udávána cca 1 : 8300 [10].
Juvenilní střevní polypóza je definována jako přítomnost 10 a více střevních polypů, což jsou hamartomy z lamina propria mucosae s přítomností dilatovaných cystických žlázek. Incidence je uváděna cca 1/100 000 obyvatel. V jedné třetině se objevuje onemocnění i u příbuzného 1. stupně, mluvíme pak o familiární juvenilní polypóze (FJP). Nejčastěji bývá postiženo tlusté střevo, méně pak duodenum a žaludek. FJP je spojena s vyšším rizikem vzniku kolorektálního karcinomu a karcinomu žaludku [11]. Polypy se objevují nejčastěji v první dekádě věku. Klinické příznaky, nejčastěji enterorhagie, meléna, prolabující rektální polyp nebo průjem, se projevují kolem 20. roku života. Solitární polypy jsou řešeny polypektomií, při difuzním postižení tračníku je vhodné zvážit preventivní proktokolektomii [12].
PŘEHLED HAMARTOMŮ V DUTINĚ BŘIŠNÍA RETROPERITONEU
Hamartom sleziny
Slezinné hamartomy jsou benigní vaskulární pseudotumory, poprvé popsány Rokitanským v roce 1861. Další používané názvy jsou splenom, splenoadenom a nodulární hyperplazie sleziny. Incidence splenomu je udávána asi 0,001 %. K roku 2014 bylo publikováno 190 případů [13].
Léze jsou velmi často asymptomatické a jejich diagnóza se většinou odvíjí od náhodného US nebo CT nálezu. Větší tumory se mohou projevit splenomegalií, bolestí v levém podžebří, rupturou sleziny, ještě méně často pak anémií nebo trombocytopenií. Hamartomy mohou být přítomny ve slezině v rámci tuberózní sklerózy nebo Wiskott-Aldrichova syndromu [14].
Splenický hamartom je ve většině případů vrozený, v etiopatogenezi se však uvažuje i o posttraumatickém mechanismu. Histologicky se jedná o anomální uspořádání normálních buněk červené pulpy. Obsahuje komplex dezorganizovaných cévních kanálků vystlaných endotelem a obklopených fibrotickými pruhy červené pulpy s nebo bez přítomnosti pulpy bílé. Je zde významná histologická podoba s hemangiomem, proto je k odlišení zapotřebí imunohistochemické dovyšetření. Imunofenotyp endoteliální výstelky u hamartomu je CD8 pozitivní [14]. Makroskopicky se jeví jako dobře ohraničená solidní léze červené barvy utlačující okolní parenchym [15].
Na ultrazvuku bývá patrný jako ohraničené homogenní lehce hyperechogenní ložisko se zvýšeným cévním průtokem při dopplerovském zobrazení. Někdy mohou být patrné heterogenity při přítomnosti krvácení nebo kalcifikace. Na CT se zobrazí jakožto izodenzní formace s heterogenní denzitou po aplikaci i.v. kontrastu [16]. Ačkoliv mohou být radiologické nálezy sugestivní, definitivní diagnózu vždy potvrzuje histologie. V diferenciální diagnostice je totiž kromě hemangiomu nutné odlišit tuto benigní lézi od angiosarkomu, lymfomu, metastázy, mykobakteriální infekce nebo sarkoidózy [17]. Aspirace tenkou jehlou není doporučována vzhledem k vysokému riziku krvácení a případné diseminace tumoru [18].
Diagnostickou a zároveň léčebnou metodou splenomu je tedy splenektomie.
Hamartom zažívacího traktu
Jedná se většinou o střevní polypy s převažující vaskulární a lymfatickou složkou v submukóze. V duodenu dominuje submukózní tuková metaplazie [19]. Ač nejsou považovány za neoplazii, může se jejich vývoj během růstu zvrhnout v neoplastický proces, a tedy střevní hamartom musí být považován za potenciálně maligní [20]. Prevalence hamartomových polypů se odhaduje na 2 % [21].
Projevuje-li se hamartom klinicky, bývá to nejčastěji abdominální bolest, hmatná rezistence, enterorhagie, střevní obstrukce nebo intususcepce [22].
Dle Sunovy studie [23] je incidence hamartomů tenkého střeva výrazně vyšší než tlustého (4 : 1). U hamartomů, nepřesahujících submukózu, je léčebnou metodou volby endoskopická submukózní excize. Ta je proveditelná i na tenkém střevu prostřednictvím enteroskopu [24]. K resekci přistupujeme i u větších hamartomů způsobujících intususcepci, střevní obstrukci nebo krvácení [25].
S hamartomatózními polypy se můžeme setkat v rámci chorob, jako jsou juvenilní střevní polypóza, Peutz-Jeghersův či Cowdenův syndrom (viz výše).
Hamartom Brunnerovy žlázy je proliferativní léze submukózní mucinózní žlázy duodena. Tato žláza produkuje zásaditý sekret a chrání tak stěnu dvanáctníku před kyselým sekretem žaludku. Doposud bylo popsáno asi 200 případů. Hamartom se většinou vyvine v polyp velikosti několika centimetrů a jen výjimečně dochází k malignímu zvratu [26]. Většina pacientů je asymptomatická a hamartom je náhodným nálezem [27]. 37 % ze symptomatických pacientů se projevuje krvácením. Většinou jde o chronické ztráty s postupnou anemizací. Dalším projevem je obstrukce. Byla zaznamenána i hemateméza s melénou při ulceraci hamartomu [28].
Diagnostika tohoto duodenálního hamartomu obvykle spadá až na terapeutickou endoskopickou polypektomii. Radiodiagnosticky (CT) lze popsat jen nespecifický útvar v lumen a případně vyloučit jeho extramurální propagaci [29]. Endoskopická biopsie klíšťkami je většinou nevýtěžná vzhledem k submukóznímu uložení útvaru [30].
Léčebnou metodou volby je tedy zmíněná endoskopická polypektomie prováděná zejména z důvodů prevence komplikací (krvácení, obstrukce, intususcepce, maligního zvratu).
Jaterní hamartom
V literatuře jsou popisovány jaterní hamartomy mezenchymální, biliární a od roku 2006 i multicystické biliární.
Mezenchymální hamartomy
S mezenchymálními hamartomy se nejčastěji setká pediatr nebo dětský chirurg. Tvoří 18−29 % benigní lézí jater a 95 % z nich je diagnostikováno do 5 let věku dítěte. V literatuře bylo popsáno asi 260 případů [31]. Jde o vrozenou abnormalitu vývoje základu žlučových cest lokalizovanou většinou v průběhu portální žíly. Histologicky patolog nalézá v tomto hamartomu nezralé mezenchymální buňky, ostrůvky hepatocytů, cévy a abnormální žlučové vývody v různém poměru. Progresivní zvětšování hamartomu je dáno akumulací tekutiny v cysticky změněné tkáni. Je udávána spojitost s polycystózou ledvin a kongenitální hepatální fibrózou. Klinicky se nemoc projevuje postupným narůstáním resistence v pravém podžebří spojeným s ascitem v případě útlaku portální žíly a s otokem dolních končetin v případě komprese dolní duté žíly [32].
Zobrazovací metody ukážou většinou cystickou formaci, méně často pak solidní, hypodenzní lézi. Solidní ložisko s menšími cystami uvnitř vytváří na CT obraz švýcarského sýru [33]. Diferenciálně diagnosticky je třeba uvažovat o echinokokóze, mezenteriální cystě a cystickém teratomu, v případě solidní léze pak o hepatocelulárním karcinomu či metastáze [34]. Vzhledem k popsané možné malignizaci někteří autoři doporučují exstirpaci [35], jiní dávají přednost konzervativnímu postupu a sledování vývoje [36].
Biliární hamartomy
Známé jsou též jako Von Meyenburgův komplex. Incidence je v literatuře udávána 0,69 %.
Mikroskopicky jsou ložiska tvořena deformovanými dilatovanými intrahepatálními žlučovody zavzatými do vazivového stromatu. Příčinou je pravděpodobně přerušení remodelace duktální ploténky za embryonálního vývoje [37]. Makroskopicky se biliární hamartom prezentuje většinou jako mnohočetné dobře ohraničené noduly v parenchymu velikosti od 1 mm do 1,5 cm, někdy však i jako větší solitární masa. Charakterem je ložisko benigní a asymptomatické, ale byl popsán i maligní zvrat v cholangiokarcinom [38]. Ultrazvuk zobrazí echo až hypoechogenní uzlíky rozeseté v parenchymu, CT prokáže hypodenzní drobná ložiska nesytící se i.v. kontrastem. Takovýto obraz však mohou poskytnout mnohočetné metastázy, mikroabscesy nebo lymfom [39]. Definitivní diagnózu ale stanoví magnetická rezonance [40]. V případě jakýchkoliv pochybností a zejména při současném záchytu extrahepatální malignity se doporučuje biopsie ložiska. Biliární hamartom léčbu nevyžaduje, ale doporučuje se sledování stavu lézí pomocí CT nebo magnetické rezonance [41].
Multicystické biliární hamartomy
Dle našeho zjištění bylo popsáno celkem pouze 10 biliárních multicystických lézí. Histologicky jde o cysticky dilatované žlučovody vystlané diferencovanými žlučovými epiteliálními buňkami, lemované pojivovou tkání místy s hepatocyty a cévami. Klinicky může být biliární hamartom němý nebo se projevuje nespecifickým abdominálním dyskomfortem. Radiologicky je prakticky nemožné odlišit tato ložiska od jiných cystických nálezů včetně těch maligních. Diferenciálně diagnosticky je nutné uvažovat o mucinózní cystické neoplazii, Caroliho nemoci, echinokokóze nebo jaterním lymfangiomu [42]. Všech 10 publikovaných pacientů podstoupilo resekci. Vzhledem k benigní povaze tumoru a absenci recidivy ve všech případech je zvažováno chirurgické řešení jen u symptomatických pacientů za předpokladu radiologického sledování [43].
Fokální nodulární hyperplazie
Fokální nodulární hyperplazie (FNH, hepatální hamartom, hamartomatózní cholangiohepatom) je nejčastější nevaskulární benigní tumorózní léze jater [44].
Častěji se objevuje u žen než u mužů (9 : 1), většinou mezi 20. a 50. rokem věku, prevalence se odhaduje na 0,9−3 % [45].
FNH byla dlouhou dobu považována patology za hamartom. V roce 1994 však Světový gastroenterologický kongres ustanovil patogenezi FNH jakožto hyperplastickou reparativní odpověď na hyperperfúzi danou anomální arterií nalezenou v centru nodulu [45]. Prokázaný vztah s hereditární teleangiektázií Rendu-Osler-Weber a jaterními hemangiomy nutí pomýšlet na FNH jako na kongenitální cévní anomálii [46,47].
Hamartom pankreatu
Případů tohoto vzácného onemocnění bylo dle našeho zjištění publikováno k lednu 2016 pouze 31. Necelá polovina pacientů neměla žádné subjektivní potíže. Ostatní pociťovali buď nespecifickou bolest v břiše, nebo ztráceli na váze [48].
Makroskopicky je možné tyto léze rozdělit na solidně-cystické nebo solidní. Typicky je složen z dobře diferencovaných, ale růstově dezorganizovaných buněk acinárních, duktálních i ostrůvkových. Imunohistochemicky vykazuje pozitivitu CD 117 a CD34 [49]. Sueyoshi a kolektiv [50] popisují případ transformace mnohočetných velkých cyst do mnohočetných menších cyst hamartomu se solidní komponentou. Tato časová morfologická změna by mohla být jedním ze znaků pankreatického hamartomu.
Na ultrazvuku je hamartom patrný jako hyperechogenní ložisko s/nebo bez přítomnosti cyst [51], CT zobrazuje dobře ohraničenou hypo nebo izodenzní lézi s heterogenním signálem při podání kontrastu. Žádná ze zobrazovacích metod nemůže jednoznačně odlišit benigní nález na slinivce od malignity [52].
Imitovat hamartom může i chronická pankreatitida s menším počtem acinárních buněk ve fibrotickém stromatu. K definitivnímu stanovení diagnózy je tedy nutný chirurgický výkon. Vzhledem k relativně častým komplikacím po resekci pankreatu se nyní chirurgické řešení doporučuje jen u symptomatických nemocných [53]. V případě dobře ohraničených lézí se přistupuje k enukleaci či resekci ložiska do zdravé tkáně. Na peroperační určení diagnózy vzhledem k potřebě imunohistochemické typizace se není možné spolehnout.
Renální hamartom
Renální hamartom je ložisko vzniklé proliferací epiteloidních buněk uložených kolem cévy. V posledních letech se na něj nahlíží jako na neoplazii z perivaskulárních epiteloidních buněk, tzv. angiomyolipom [54]. Vyskytuje se 4x častěji u žen, typicky mezi 20. a 40. rokem, prevalence je zhruba 0,44 % [55]. Ke klinickým projevům patří dyskomfort v jednom nebo obou bedrech, někdy i palpovatelná masa, a mikro nebo makroskopická hematurie [56]. Bylo popsáno i velmi vzácné spontánní krvácení do retroperitonea [57]. Okolo 20 % pacientů s renálním hamartomem mělo diagnostikováno tuberózní sklerózu (TS). U těchto pacientů častěji dochází k renálnímu selhání z důvodu komprese ledvinného parenchymu a vývoji renin-vázané hypertenze [58]. Histologicky rozlišujeme klasickou a epiteloidní variantu. V obou typech je přítomna tuková tkáň protkána cévní složkou, která se angiograficky dobře zobrazuje jako dilatované cévy procházející skrz tukovou masu, s četnými multisakulárními aneuryzmaty (připomínajícími hrozny) bez A-V zkratů. Ve venózní fázi dává obraz vrstev cibule [59]. Třetí histologickou komponentou bývají buňky hladké svaloviny. V epiteloidní formě jsou dále přítomny epiteloidní buňky a tato varianta má vyšší riziko maligního zvratu (větší v případě tuberózní sklerózy) než forma klasická [60]. CT vyšetření s i.v. kontrastem vzhledem k průkazu tukové masy lze pokládat za velmi spolehlivou diagnostickou metodu, i když odlišení od liposarkomu může být obtížné, pokud hamartom podlehl kalcifikaci nebo nekróze [61].
Pacienti s renálním hamartomem jsou nejčastěji pouze sledováni. Ložiska do 4 cm je vhodné sledovat na US v intervalu 6−12 měsíců. Pacienti symptomatičtí či s ložiskem větším než 4 cm nebo s aneuryzmaty většími než 5 mm jsou indikováni preventivně k intervenci. Snahou je vždy zachovat ledvinu, a to i u multifokálních lézí. Metodou volby je tedy parciální resekce. Selektivní arteriální embolizace je využívána v případě nepříznivého uložení hamartomu znemožňujícího parciální resekci nebo v případě akutního krvácení. Radiofrekvenční ablaci lze využít k ložiskům velikosti pod 3 cm [62].
Výčet uvedených hamartomů je uveden v přehledné tabulce společně s porovnáním klinických projevů, diagnostikou, diferenciálně diagnostickou rozvahou a možnostmi léčby (Tab. 1).
Kazuistika Hamartom sleziny
34letý muž byl v roce 2009 vyřazen z programu dárcovství krve pro záchyt trombocytopenie. Naměřená hodnota trombocytů činila 115x109/l, příčina trombocytopenie však nebyla dále vyšetřována. Muž kromě dlouhodobě průjmovité stolice neudával žádné subjektivní obtíže.
V osobní anamnéze neměl žádná další sledovaná onemocnění, z operací podstoupil apendektomii v dětském věku. Závažnější úraz v anamnéze pacient neutrpěl až do roku 2010, kdy prý přeletěl přes řídítka při pádu z kola. Incident se však stal až po zjištěné trombocytopenii. Jeho bratr měl diagnostikovánu Crohnovu chorobou, jinak byla rodinná anamnéza bez pozoruhodností. Pacient byl nekuřák, profesí údržbář a průvodce v jeskyních.
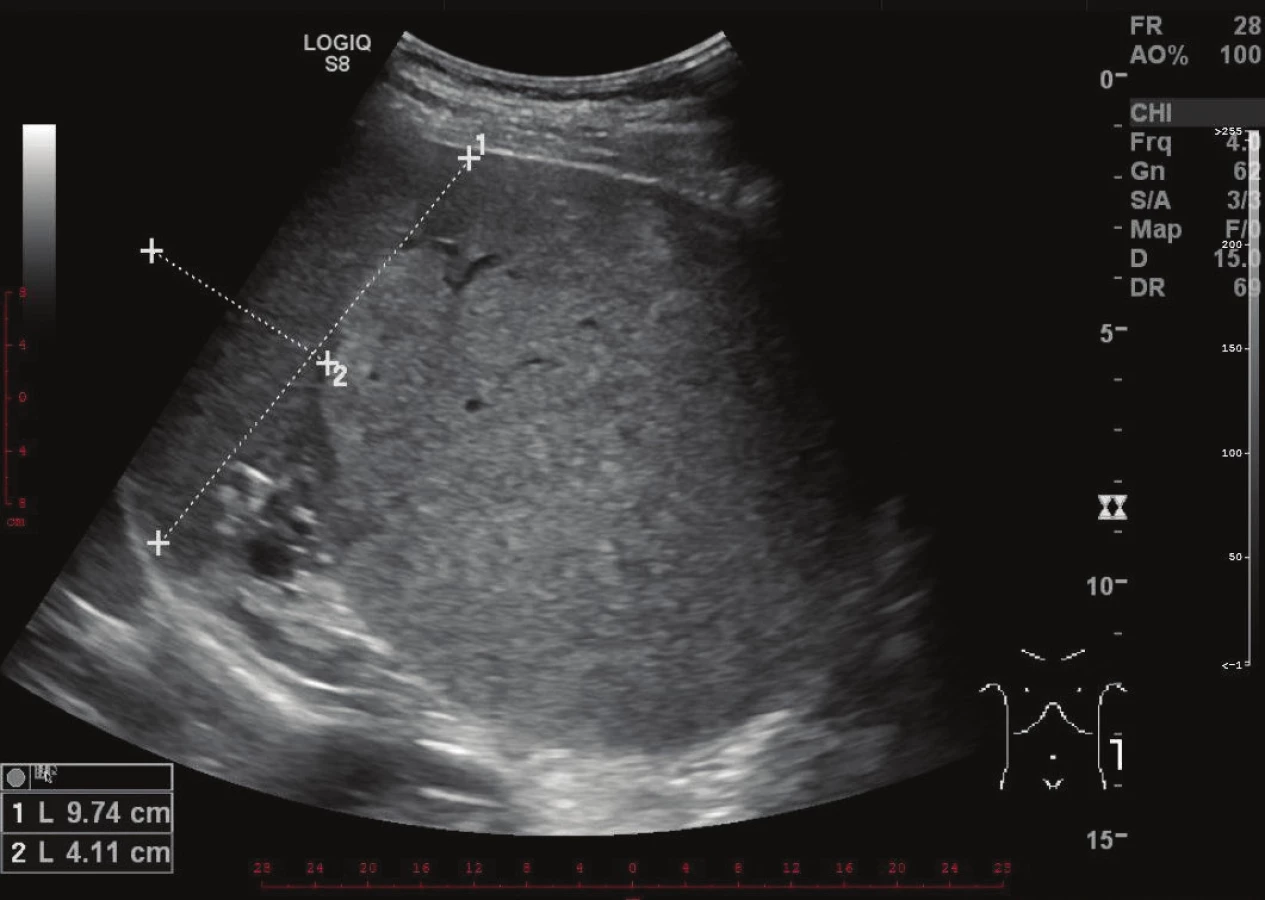
Při kontrolním vyšetření krevního obrazu v létě roku 2016 byla zjištěna prakticky stacionární hodnota trombocytopenie a v září téhož roku podstoupil vyšetření na hematologii. Lékař vyhmatal nebolestivou rezistenci pod levým obloukem žeberním v poloze na pravém boku. Uzliny klinicky zvětšeny nebyly. Hematolog indikoval ultrasonografii břicha a nálezem byla kulovitá expanze v levém hypochondriu velikosti cca 12x12 cm. Sonografista popsal, že nález téměř jistě nesouvisí s levou ledvinou a že vztah k dalším orgánům není jasný. Slezina se zdála být útvarem odtlačena, ale bylo možné, že z ní útvar vycházel. Ložisko mělo vyšší echogenitu než slezina a vykazovalo vyšší perfúzi. Nadledvina na ultrazvuku nebyla vůbec patrná (Obr. 1).
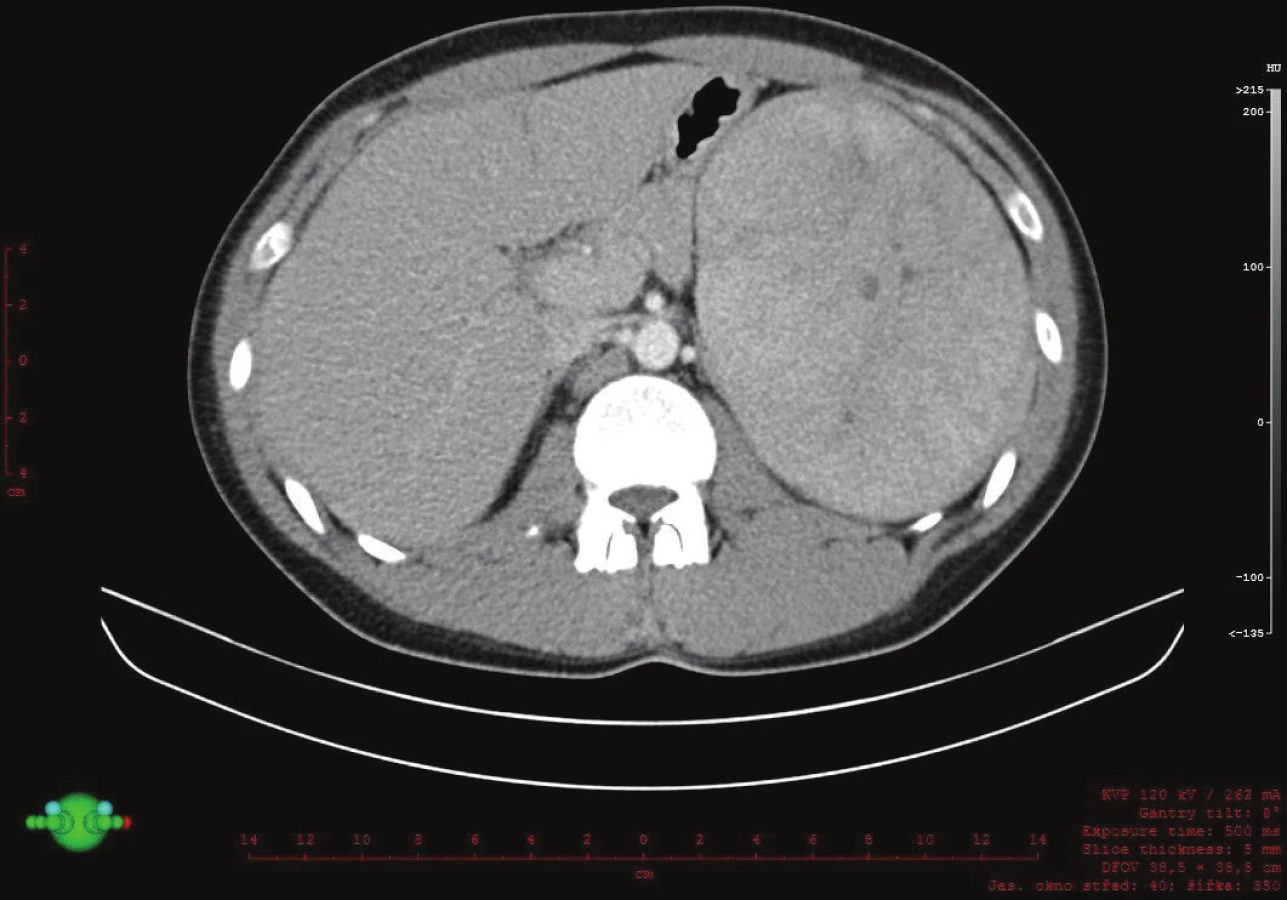
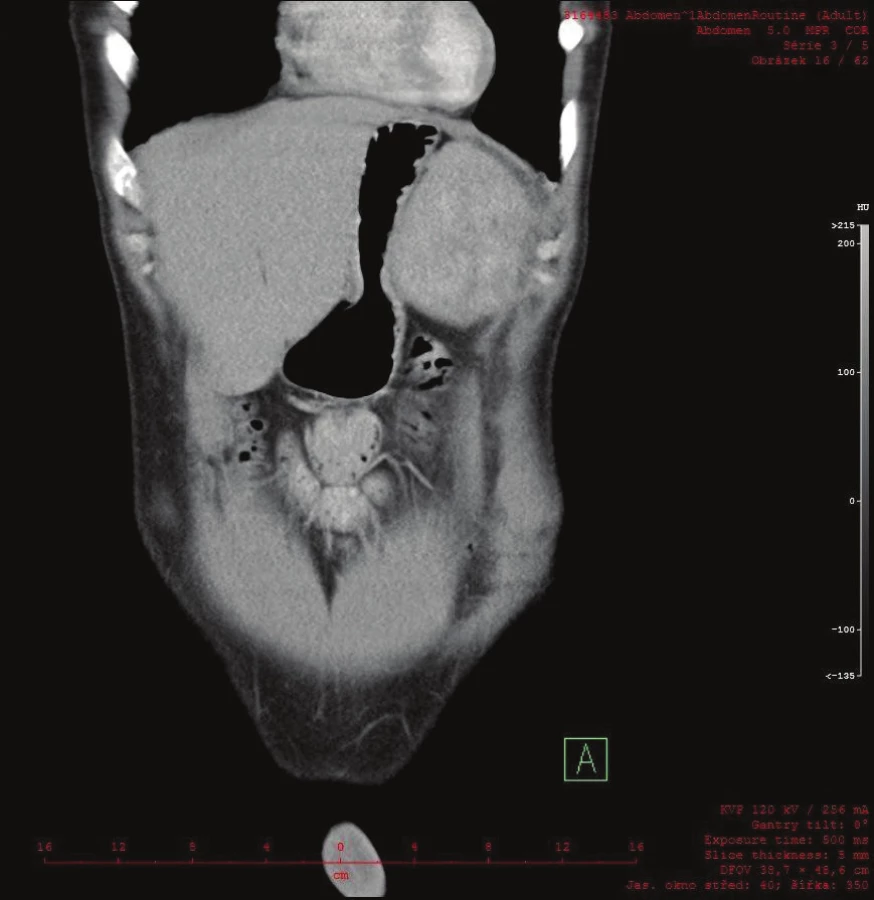
Vyšetřování pokračovalo doplněním CT břicha s kontrastem, které zobrazilo hladce ohraničenou nehomogenní expanzi velikosti 16x12x12 cm. Struktura byla na postkontrastních snímcích lehce hyperdenzní s několika hypodenzními okrsky odpovídajícími denzitou tuku. Ložisko naléhalo na nadledvinu, odtlačovalo levou ledvinu směrem kaudálním a budilo dojem, že se vtlačuje do sleziny. Hranice se slezinou však nebyla patrná. Radiolog uzavřel nález jako suspektní myelolipom levé nadledviny (Obr. 2, 3, 4).
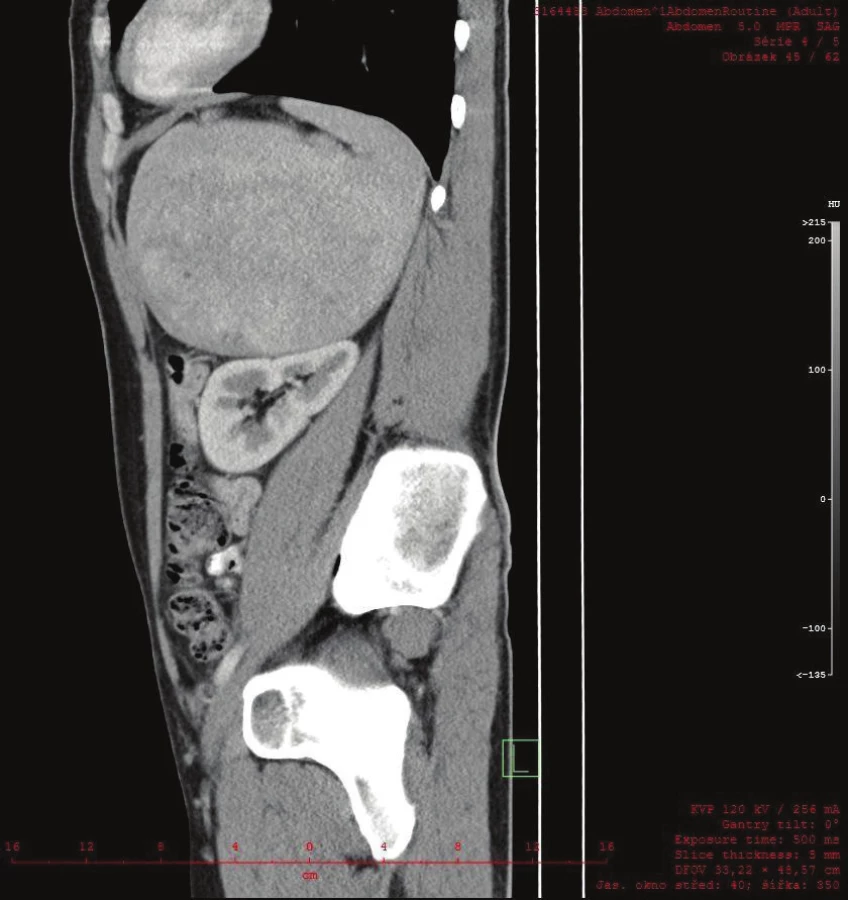
Vzhledem k diagnostickým rozpakům byl pacient odeslán na naši kliniku a indikován chirurgem k operační revizi. Předoperační příprava proběhla bez nutnosti substituce trombocytů.
Kvůli výše uvedenému (velikost ložiska, nejasné origo tumoru a vztah k okolním orgánům, neznámá biologická povaha) bylo přistoupeno primárně k otevřené operaci. Do dutiny břišní bylo proniknuto z levého subkostálního řezu. Levému hypochondriu dominovala zvětšená slezina, naopak levá ledvina i nadledvina nevykazovaly žádnou makroskopickou patologii. Bylo přistoupeno ke splenektomii (Obr. 5). Krevní ztráta byla nevýznamná a výkon proběhl bez komplikací.
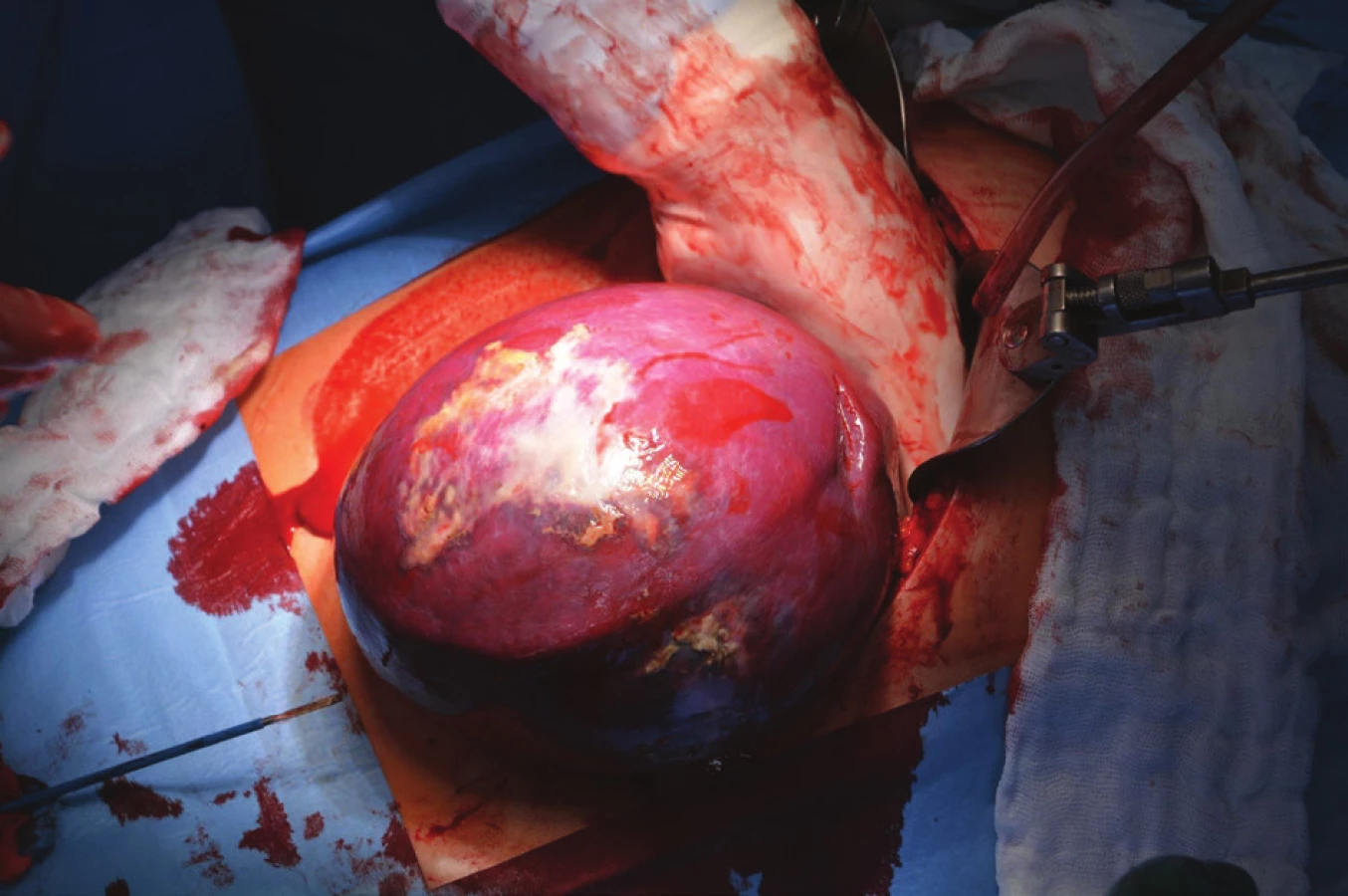
Slezina vážila 1500 gramů a byla odeslána k histologickému vyšetření nativně.
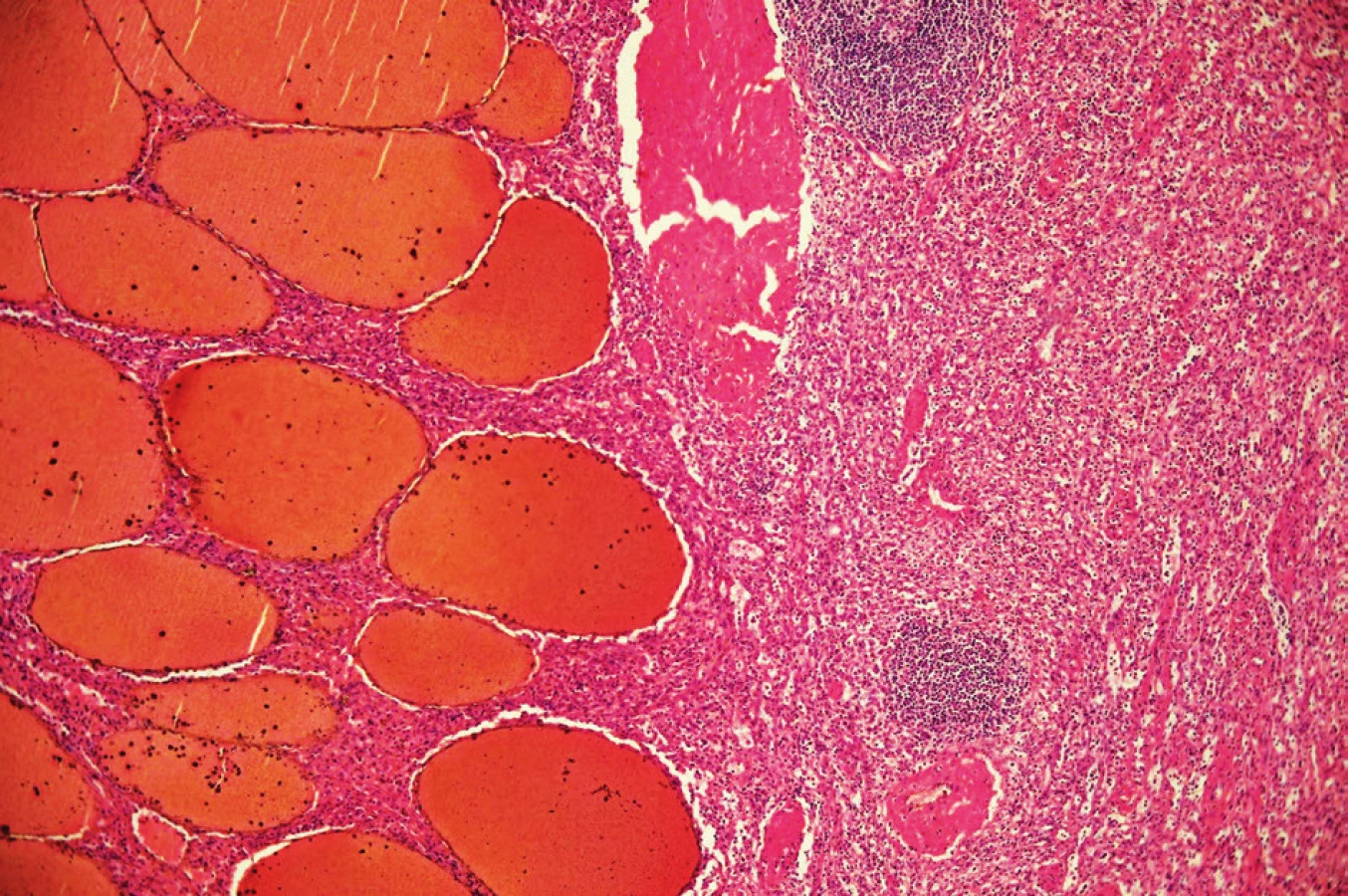
Patolog na řezu slezinou popsal dobře ohraničený solidní prokrvácený uzel s rozměry 130x110x95 mm, místy voštinovité struktury. Pouzdro bylo ložiskovitě ztluštělé. Zbytek sleziny měl parenchym přiměřený. Histologicky byl nález hodnocen jako benigní léze charakteru splenického hamartomu (Obr. 6, 7).

Laparotomie se po výkonu zahojila per primam a operant byl bez teplot. Opakovaná ultrasonografie z důvodu frenikového příznaku sice ozřejmila zmenšující se hematom v lůžku, ale pacientovy obtíže spontánně ustoupily. Již 6. pooperační den se vyvinula postsplenektomická trombocytóza s hodnotami až 825x109/l. Do dlouhodobé medikace byl tedy nasazen anopyrin v dávce 100 mg denně.
Jelikož pacient nebyl před splenektomií očkován, infektolog dodatečně provedl vakcinaci proti meningokoku, pneumokoku a haemophilu influenzae.
K březnu 2017 byl muž zcela bez obtíží, hladina trombocytů byla 511x109/l a chronické průjmy po operaci ustaly. Po tříměsíční pracovní neschopnosti je muž plně začleněn do pracovního procesu. Etiologie chronických průjmů (typických např. pro neuroendokrinní tumory) zůstává v případu hamartomu sleziny nezodpovězena. Dle dostupných poznatků nebyla nikdy prokázána sekrece endokrinních působků těmito nádory. Výskyt průjmů a jeho možná spojitost s přirozeným obsahem serotoninu a dalších biogenních aminů v trombocytech nebyla v literatuře popsána ani v případech jiných onemocnění spojených se zvýšenou degradací trombocytů (hypersplenismus a podobně). V případě našeho pacienta nebylo imunohistochemické vyšetřování nádoru provedeno. Při pátrání po etiologii průjmů by v úvahu mohl připadat efekt mechanického tlaku zvětšené sleziny, čímž by docházelo k urychlení pasáže tračníkem.
Závěr
Hamartomy orgánů dutiny břišní a retroperitonea jsou až na výjimky (biliární hamartomy) dosti vzácně se vyskytující tumory benigního charakteru, a tedy relativně na okraji chirurgického zájmu. Jejich histologická klasifikace se neustále vyvíjí a u některých lézí, které byly dříve řazeny mezi hamartomy, byl zjištěn odlišný původ. Ačkoliv je většina hamartomů asymptomatických, je nutno zdůraznit, že jako jakýkoliv jiný rostoucí nádor se mohou i tyto tumory projevit útlakem, obstrukcí, perforací, krvácením a dokonce maligním zvratem. Symptomatologie je pak dána velikostí tumoru, rychlostí růstu a zejména jeho lokalizací. To dokazuje i kazuistika z našeho pracoviště, kdy hamartom sleziny byl zjištěn až při vyšetřování důvodu trombocytopenie. Tento případ také demonstruje, že chirurgické řešení je často jedinou racionální možností definitivního potvrzení diagnózy. Navzdory nízké incidenci jsou hamartomy nedílnou součástí diferenciálně diagnostické rozvahy.
Seznam zkratek:
PTEN − phosphatase and tensin homolog
CNS − centrální nervový systém
TSC gen 1,2 − Tuberous sclerosis gen 1, 2
FJP − familiární juvenilní polypóza
CT − computed tomography – počítačová tomografie
CD − cluster of differentiation
FNH − fokální nodulární hyperplazie
TS − tuberózní skleróza
A-V zkraty − arteriovenózní zkraty
US − ultrazvuk
Dif. dg. − diferenciálně diagnostické
Konflikt zájmů
Autoři článku prohlašují, že nejsou v souvislosti se vznikem tohoto článku ve střetu zájmů a že tento článek nebyl publikován v žádném jiném časopise.
MUDr. Aleš Paclík
Hrubínova 640/22
143 00 Praha 4
e-mail: ales.paclik@vfn.cz
Sources
1. Manxhuka-Kerliu S, Sahatciu-Meka V, Kerliu I, et al. Small intestinal gastrointestinal stromal tumor in a young adult woman: a case report and review of the literature. J Med Case Rep 2014;8 : 321.
2. Jelsig AM, Qvist N, Brusgaard K, et al. Hamartomatous polyposis syndromes: a review. Orphanet J Rare Dis 214;9 : 101.
3. Tanaka N, Seya T, Onda M, et al. Myoepithelial hamartoma of the small bowel: report of a case. Surg Today 1996;26 : 1010−13.
4. Theodosiou E, Voulalas G, Salveridis N, et al. Neuromesenchymal hamartoma of small bowel-an extremely rare entity: a case report. World J Surg Oncol 2009;7 : 92.
5. Brosens LA, Langeveld D, van Hattem WA, et al. Juvenile polyposis syndrome. World J Gastroenterol 2011;17 : 4839–44.
6. Uppal S, Mistry D, Coatesworth AP. Cowden disease: a review. Int J Clin Pract 2007;61 : 645–52.
7. Lloyd KM, Dennis M. Cowden disease. A possible new symptom complex with multiple system involvement. Ann Intern Med 1963;58 : 136.
8. Kato M, Mizuki A, Hayashi T, et al. Cowden’s disease diagnosed through mucocutaneous lesions and gastrointestinal polyposis with recurrent hematochezia, unrevealed by initial diagnosis. Intern Med 2000;39 : 559.
9. Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet 2008;372 : 657.
10. Van Lier MG, Wagner A, Mathus-Vliegen EM, et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 2010;105 : 1258.
11. Veale AM, McColl I, Busey HJ. Juvenile polyposis coli. J Med Genet 1966;3 : 5–16.
12. Grotsky HW, Rickert RR, Smith WD, et al. Familial juvenile polyposis coli. A clinical and pathologic study of a large kindred. Gastroenterology 1982;82 : 494.
13. Soto-Medina CA, Mier-Escurra EA, Treviño-Garza F, et al. Splenic hamartoma. Case report. Cir Cir 2014;82 : 328−31.
14. Lee H, Maeda K. Hamartoma of the spleen. Arch Pathol Lab Med. 2009;133 : 147–51.
15. Sankar S, Thanka J, Jagdishchandrabose S, et al. Splenic hamartoma: A rare vascular space occupying lesion of the Spleen. Indian J Pathol Microbiol 2011;54 : 223−5.
16. Zissin R, Lishner M, Rathaus V. Case report: Unusual presentation of splenic hamartoma; computed tomography and ultrasonic findings. Clin Radiol 1992;45 : 410−1.
17. Wang JH, Ma XL, Ren FY, et al. Multi-modality imaging findings of splenic hamartoma: a report of nine cases and review of the literature. Abdom Imaging 2013;38 : 154–62.
18. Namikawa T, Kitagawa H, Iwabu J, et al. Laparoscopic splenectomy for splenic hamartoma: case management and clinical consequences. World J Gastrointest Surg 2010;2 : 147–52.
19. Tan CL, Tan SH, So JB, et al. Muco-submucosal elongated polyps of the gastrointestinal tract: a case series and a review of the literature. World J Gastroenterol 2013;19 : 1845−9.
20. Manxhuka-Kerliu S, Sahatciu-Meka V, Kerliu I, et al. Small intestinal gastrointestinal stromal tumor in a young adult woman: a case report and review of the literature. J Med Case Rep 2014;8 : 321.
21. Brosens LA, Langeveld D, van Hattem WA, et al. Juvenile polyposis syndrome. World J Gastroenterol 2011;17 : 4839–44.
22. Jiangang Sun, Yongshun Gao, Bo Yang, et al. Intestinal obstruction caused by giant ileal hamartoma: a case report. Ann Transl Med 2016;4 : 138.
23. Sun JG, Qi J, Yang B, et al. The clinical characteristics and treatment of intestinal hamartomas. Lasers Med Sci 2016;31 : 1761.
24. Marques M, Antunes J, Coelho R, et al. Single-balloon enteroscopy efficacy and degree of concordance with noninvasive evaluation of small bowel. Endosc Int Open. 2017;5: E96–E102.
25. Akahoshi K, Akahane H, Motomura Y, et al. A new approach: endoscopic submucosal dissection using the clutch cutter (R) for early stage digestive tract tumors. Digestion 2012;85 : 80–4.
26. Brookes MJ, Manjunatha S, Allen CA, et al. Malignant potential in a Brunner’s gland hamartoma. Postgrad Med J 2003;79 : 416–7.
27. Botsford TW, Crowe P, Croker DW. Tumors of the small intestine. A review of experience with 115 cases including a report of a rare case of malignant hemangio-endothelioma. Am J Surg 1962;103 : 358–65.
28. Levine JA, Burgart LJ, Batts KP, et al. Brunner’s gland hamartomas: clinical presentation and pathological features of 27 cases. Am J Gastroenterol 1995;90 : 290–4.
29. Merine D, Jones B, Ghahremani GG, et al. Hyperplasia of Brunner glands: the spectrum of its radiographic manifestations. Gastrointest Radiol 1991;16 : 104–8.
30. Perez A, Saltzman JR, Carr-Locke DL, et al. Benign nonampullary duodenal neoplasms. J Gastrointest Surg 2003;7 : 536–41.
31. Debraj Sen, Y.S. Gulati, Anusree Majumder. Hepatic cystic mesenchymal hamartoma. Med J Armed Forces India 2015; 71(Suppl 2): S574–7.
32. Chung EM, Cube R, Lewis RB, et al. Pediatric liver masses: radiologic-pathologic correlation part 1. Benign tumors. Radiographics 2010;30 : 801–26.
33. Ros P, Goodman Z, Ishak KG, et al. Mesenchymal hamartoma of the liver: radiologic-pathologic correlation. Radiology 1986;158 : 619–24.
34. Chung EM, Lattin GE, Jr, Cube R, et al. From the archives of the AFIP: Pediatric liver masses: radiologic-pathologic correlation. Part 2. Malignant tumors. Radiographics 2011;31 : 483–507.
35. Ramanujam TM, Ramesh JC, Goh DW, et al. Malignant transformation of mesenchymal hamartoma of the liver: case report and review of the literature. J Pediatr Surg 1999;34 : 1684–6.
36. Barnhart DC, Hirschl RB, Garver KA, et al. Conservative management of mesenchymal hamartoma of the liver. J Pediatr Surg 1997;32 : 1495–8.
37. Desmet VJ. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol 2004;19(Suppl 7):S356–60.
38. Kin HK, Jin SY. Cholangiocarcinoma arising in von Meyenburg complexes. Korean J Hepatol. 2011;17 : 161–4.
39. Zheng RQ, Zhang B, Kudo M, et al. Imaging findings of biliary hamartomas. Wolrd J Gastroenterol 2005;11 : 6354–9.
40. Semelka RC, Hussain SM, Marcos HB, et al. Biliary hamartomas: solitary and multiple lesions shown on current MR techniques including gadolinium enhancement. J Magn Reson Imaging. 1999;10 : 196–201.
41. Sinakos E, Papalavrentios L, Chourmouzi D et al. The clinical presentation of Von Meyenburg complexes. Hippokratia 2011;15 : 170–3.
42. Zen Y, Terahata S, Miyayama S, et al. Multicystic biliary hamartoma: a hitherto undescribed lesion. Hum Pathol 2006;37 : 339–44.
43. Beard R, Yee E, Mortele K, et al. Multicystic biliary hamartoma: A report of a rare entity and a review of the literature. Int J Surg Case Rep. 2014;5 : 919–23.
44. Craig J, Peters R, Edmundson H. Tumors of the liver and intrahepatic bile ducts, Fasc 26, 2nd ed, DC Armed Forces Institute of Pathology, Washington DC 1989.
45. Wanless IR, Mawdsley C, Adams R. On the pathogenesis of focal nodular hyperplasia of the liver. Hepatology 1985;5 : 1194.
46. Haber M, Reuben A, Burrell M, et al. Multiple focal nodular hyperplasia of the liver associated with hemihypertrophy and vascular malformations. Gastroenterology 1995;108 : 1256.
47. Wanless IR, Gryfe A. Nodular transformation of the liver in hereditary hemorrhagic telangiectasia. Arch Pathol Lab Med 1986;110 : 331.
48. Matsushita D, Kurahara H, Mataki Y, et al. Pancreatic hamartoma: a case report and literature review. BMC Gastroenterol 2016;16 : 3.
49. Nagata S, Yamaguchi K, Inoue T, et al. Solid pancreatic hamartoma. Pathol Int 2007;57 : 276–80.
50. Sueyoshi R, Okazaki T, Lane GJ, et al. Multicystic adenomatoid pancreatic hamartoma in a child: Case report and literature review. Int J Surg Case Rep 2013;4 : 98–100.
51. Wu SS, Vargas HI, French SW. Pancreatic hamartoma with Langerhans cell histiocytosis in a draining lymph node. Histopathology 1998;33 : 485–7.
52. Pauser U, Kosmahl M, Kruslin B, et al. Pancreatic solid and cystic hamartoma in adults: characterization of a new tumorous lesion. Am J Surg Pathol 2005;29 : 797–800.
53. Beger HG, Siech M, Poch B, et al. Limited surgery for benign tumours of the pancreas: A systematic review. World J Surg 2015;39 : 1557–66.
54. Jinzaki M, Silverman SG, Akita H. Renal angiomyolipoma: a radiological classification and update on recent developments in diagnosis and management. Abdom Imaging 2014;39 : 588–604.
55. Fittschen A, Wendlik I, Oeztuerk S, et al. Prevalence of sporadic renal angiomyolipoma: a retrospective analysis of 61,389 in - and out-patients. 1. Abdom Imaging 2014;39 : 1009–13.
56. Becker JA, Kinkhabwala M, Pollack H, et al. Angiomyolipoma (Hamartoma) - angiographic review. Acta Radiol 1973;14 : 561−8.
57. Allen TD, Risk W. Renal angiomyolipoma. J Urol 1965;94 : 203−7.
58. Clark ER, Palubinskas AJ. The angiographic spectrum of renal hamartoma. Am J Roentgenol Radium The Nucl Med 1972;114 : 715−21.
59. Khrlnam M, Wolf BS. Hamartoma of the kidney, clinical and roentgenographic manifestation. Am Roentgenol Radium Ther Nucl Med 1961;86 : 830−41.
60. Aydin H, Magi-Galluzzi C, Lane BR. Renal angiomyolipoma: clinicopathologic study of 194 cases with emphasis on the epithelioid histology and tuberous sclerosis association. Am J Surg Pathol 2009;33 : 289-97.
61. Vít V, Pacík D, Čermák A, et al. Je CT vyšetření dostatečně spolehlivé při hodnocení neoplasmatických procesů ledvin? Čes urol 2004;3 : 17–20.
62. Sooriakumaran P, Gibbs P, Coughlin G, et al. Angiomyolipomata: challenges, solutions, and future prospects based on over 100 cases treated. BJU Int 2010;105 : 101.
Labels
Surgery Orthopaedics Trauma surgeryArticle was published in
Perspectives in Surgery

2017 Issue 9
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
-
Hamartoma of the abdominal cavity and retroperitoneum
– a review and a case report - Pneumoperitoneum after colonoscopy – “to cut or not to cut”
- Current application and future perspectives of capsule colonoscopy
- Diagnosis of early pancreatic cancer and precursor lesions