
Idiopatická plicní fibróza: Nastal čas opět změnit doporučený postup diagnostiky a léčby?
Idiopathic pulmonary fibrosis: Does the time for change of diagnostic and therapeutic recommendations come?
Idiopathic pulmonary fibrosis (IPF) belongs to the most severe and difficult-to-treat lung diseases, with median survival 2–3 years if not treated. The disease arises in genetically disposed individuals of middle and older age based on repeated alveolar injuries. Fibroproliferative healing of these lesions form the pathogenetic background of the disease, leading to the end-stage pulmonary fibrosis.
Our knowledge of pathogenesis, diagnosis and treatment of IPF increases dynamically since the beginning of new millennium. Progressive breathlessness, crepitus and clubbed fingers are typical for diagnosis of IPF. Radiologic picture of IPF in high resolution CT imaging is characterized by usual interstitial pneumonia, however nearly half of the patients do not have this typical pattern. The careful evaluation of clinical picture, radiologic findings and additional investigations as bronchoalveolar lavage differential cell count and immunologic results, by multidisciplinary team is essential in these cases. In minority of the patients lung biopsy, either surgical or cryobiopsy, is recommended.
Only last 7 years causal treatment of IPF with antifibrotic drugs is available. Nevertheless, antifibrotic treatment only slows down the disease progression. Minor part of the patients with end-stage-diseased is referred for lung transplant. Palliative and symptomatic care is an integral part of IPF treatment.
Key words:
idiopathic pulmonary fibrosis, typical and atypical radiologic findings, cryobiopsy, antifibrotic treatment, lung transplantation, palliative care
Autori:
Martina Vašáková
Pôsobisko autorov:
Pneumologická klinika 1. LF UK a Thomayerovy nemocnice, Praha
Vyšlo v časopise:
Čas. Lék. čes. 2018; 157: 237-243
Kategória:
Přehledový článek
Súhrn
Idiopatická plicní fibróza (IPF) patří mezi nejobtížněji léčitelné a zároveň prognosticky nejzávažnější plicní choroby, se středním přežitím 2–3 roky, pokud není léčena. Onemocnění vzniká u geneticky disponovaných jedinců středního a staršího věku na podkladě opakovaných poranění plicních sklípků. Patogenetickým podkladem nemoci je fibroproliferativní hojení těchto alveolárních lézí, jež ústí v ireverzibilní plicní fibrózu.
Od počátku nového tisíciletí se poměrně dynamicky vyvíjí poznání IPF ruku v ruce s rozvojem nových diagnostických a léčebných možností. Základem pro diagnostiku je typický klinický obraz progredující dušnosti, paličkovitých prstů a krepitu. K podpoření definitivní diagnózy napomůže CT hrudníku s vysokou rozlišovací schopností s nálezem obvyklé intersticiální pneumonie. Nicméně téměř polovina pacientů nemá typický radiologický nález. Zde je pak důležité posouzení klinického obrazu a dalších nálezů, jako je rozpočet buněk v bronchoalveolární laváži a imunologické vyšetření, v rámci multidisciplinárního týmu, což případně vede k indikaci plicní chirurgické biopsie nebo nově kryobiopsie.
Až posledních 7 let máme k dispozici kauzální léčbu IPF antifibrotickými léky. Ta však nevede k vyléčení, jedná se pouze o zpomalení progrese nemoci. Jen pro část nemocných je potom vhodná transplantace plic. Nedílnou součástí péče o nemocné s IPF je paliativní péče a symptomatická léčba.
Klíčová slova:
idiopatická plicní fibróza, typický a netypický radiologický nález, kryobiopsie, antifibrotická léčba, transplantace plic, paliativní péče
ÚVOD
Idiopatická plicní fibróza (IPF) patří mezi nejobtížněji léčitelné a zároveň prognosticky nejzávažnější plicní nemoci, se středním přežitím bez léčby 2–3 roky. Od počátku nového tisíciletí se poměrně dynamicky vyvíjí poznání patogeneze a klinicko-patologicko-radiologických typů IPF ruku v ruce s rozvojem léčebných možností této dříve neléčitelné nemoci. Nicméně dosavadní možnosti léčby dokáží pouze zpomalit průběh onemocnění a pokles plicních funkcí, a tak IPF i nadále vyžaduje další zkoumání a nacházení nových terapeutických možností.
Zatím poslední mezinárodní prohlášení o diagnostice a léčbě této nemoci je z roku 2011, s následným prohlášením k léčbě antifibrotiky v roce 2015 (1, 2). Nicméně tato doporučení neodrážejí nové poznatky o IPF, jako je kupříkladu nový názor na radiologickou diagnostiku a méně invazivní typy plicní biopsie, a proto je již očekáván nový konsenzus, který by tyto novinky zařadil do diagnostického a léčebného standardu.
DEFINICE
Dle definice z roku 2015 je IPF specifickou formou chronické progredující fibrotizující intersticiální pneumonie nejasné etiologie, objevující se primárně u dospělých jedinců, postihující pouze plíce a spojené s histopatologickým a/nebo radiologickým obrazem obvyklé intersticiální pneumonie (UIP) (2).
EPIDEMIOLOGIE
Prevalence IPF je podle výsledků epidemiologických studií celosvětově odhadována na 13–20/100 000 a incidence na 6,8–16,3/100 000 obyvatel; IPF je tedy postiženo přibližně 5 000 000 lidí na celém světě.
Americké a evropské údaje se značně liší. V USA je dle klinických studií prevalence udávána 14–27,9 případů/100 000 obyvatel při užití volnějších kritérií; při užití přísnějších kritérií pak 42,7–63/100 000. Obdobně je incidence v USA udávána 16,3–17,4/100 000 při aplikaci volnějších kritérií a 6,8–8,8/100 000 při užití přísnějších kritérií. V Evropě je prevalence udávána 1,25–25,6/100 000 a incidence 0,22–7,56/100 000 obyvatel. IPF se vyskytuje s o něco větší četností u mužů než u žen. Incidence onemocnění stoupá s věkem. Pacienti jsou nejčastěji ve středním věku, v rozmezí od 45 do 70 let. Přibližně dvě třetiny jich jsou starší 60 let.
IPF se obvykle vyskytuje sporadicky, familiární případy jsou vzácné. Z toho důvodu je i relativně málo prací zkoumajících familiární plicní fibrózu. Navíc v jedné rodině často najdeme několik subtypů idiopatických intersticiálních pneumonií, což by podporovalo domněnku o vlivu prostředí na fenotyp.
ETIOPATOGENEZE
Patogenetickým podkladem IPF je uniformní patologická odpověď plicní tkáně na různá infekční i neinfekční agens inzulty u geneticky disponovaného jedince středního a staršího věku s genetickou dispozicí k fibroproliferativnímu hojení plicní tkáně (7). Z vyvolávajících faktorů, jež způsobují léze alveolární výstelky, je důležité zmínit hlavně virové infekce, kouření, expozici prachům a také pravděpodobně extraezofageální reflux, který je v subklinické podobě přítomen až u 90 % pacientů s IPF (8).
Na genetické dispozici k IPF se zřejmě podílí řada genů. Jsou to jednak geny, jejichž mutace způsobují familiární intersticiální pneumonie/fibrózy, které se mohou projevit již v dětství, a pak geny, které jsou vázány převážně se sporadickou formou dospělých. Z genů podílejících se jak na dětském typu plicní fibrózy, tak na sporadické IPF je nejčastěji mutován gen pro surfaktantový protein C (SP-C) – 5 z 20 pacientů s familiární plicní fibrózou (FPF) jsou nositelé mutovaného genu. Mutace surfaktantových genů vede k produkci vadného surfaktantového proteinu, což způsobuje dysfunkci alveolárních epiteliálních buněk II. typu. Z dalších genů, jejichž mutace s podílí na poškození epitelových buněk, a to mechanismem jejich předčasného stárnutí, jsou geny pro telomerázy (hTERT, hTER). Mutace genů pro telomerázy se uplatňují hlavně u FPF. Mezi geny, jejichž polymorfismy se uplatňují hlavně u sporadické IPF, je v poslední době nejdiskutovanějším gen pro mucin, a to jmenovitě jeho variace v promotorové oblasti MUC5B. V souvislosti s tím byla prokázána i vyšší exprese MUC5B v bioptických materiálech plic u pacientů s IPF. Genetické variace genu pro TOLLIP, které ovlivňují mechanismy vrozené imunity, také zřejmě zvyšují pravděpodobnost vzniku IPF (9).
Na začátku patogenetického procesu IPF dochází k mnohočetnému poškození alveolárního epitelu, kdy je obnažena bazální membrána (BM) a spuštěna kaskáda produkce cytokinů, chemokinů a enzymů, jež se pak podílejí na dalším rozvoji a udržování procesu fibroprodukce a jizvení plicní tkáně. Samotná BM je procesem poškozena také, a tak dochází k proliferaci pneumocytů II. typu a endotelových buněk v terénu „nedokonalé“ extracelulární matrix (ECM), a nemůže proto dojít k restituci původní architektoniky alveolu. Dalším krokem je pak nábor (recruitment) fibroblastů a myofibroblastů s další depozicí ECM a progrese k nevratné fibróze alveolu. Vyvíjejí se tzv. fibroblastické fokusy, jež jsou mezi sebou navzájem spojeny pojivovou tkání a vytvářejí jakousi trojrozměrnou síťovitou strukturu, která vytlačuje a nahrazuje spojitou architektoniku respiračních lobulů v plicích. Množství fibroblastických fokusů v plicích pravděpodobně ovlivňuje prognózu pacientů s IPF.
Hlavními cytokiny, které určují vzorec patologického hojení plicní tkáně směrem k fibróze, jsou interleukiny IL-4 a IL-13. Oba tyto cytokiny podporují růst fibroblastů a produkci kolagenu a zvyšují produkci transformujícího růstového faktoru beta (TGF-β2) lidskými bronchiálními epiteliemi. TGF-β2 indukuje transkripci kolagenu typu I a fibronektinu ve fibroblastech. V endotelu navíc reguluje produkci a aktivitu růstového faktoru fibroblastů (FGF-2), který má výrazný mitogenní vliv na pneumocyty II. typu. To má za následek poruchu fyziologické reepitalizace drobných alveolárních lézí s následným vznikem prefibrotických okrsků.
Existuje několik teorií o původu fibroblastů a myofibroblastů v plicní tkáni u pacientů s IPF. Jedna z nich předpokládá, že jde o rezidentní kmenové buňky, které se aktivují a proliferují v odpověď na poranění. Je možný i jejich původ z buněk alveolárního epitelu, které projdou takzvanou epitelově-mezenchymovou tranzicí (EMT). Dalším možným zdrojem myofibroblastů jsou mezotelové buňky, které mohou vcestovávat do plíce a vykazovat rysy myofibroblastické diferenciace. Svým způsobem by to vysvětlovalo i subpleurální predominanci změn u IPF. Některé teorie a studie však podporují spíše hypotézu o mimoplicním původu fibroblastů, jež jsou původem z kostní dřeně, chovají se jako mezenchymové kmenové buňky a jsou chemotakticky atrahovány do míst tkáňového poranění, kde přispívají k fibrotizaci plicní tkáně. Je pravděpodobné, že fibroblasty u jedinců s IPF mají i sníženou schopnost apoptózy a jsou proti apoptóze mediované receptory Fas chráněny antiapoptotickými faktory ze skupiny inhibitorů apoptózy (IAP).
V patogenezi sporadické IPF nepochybně hraje roli rovněž stárnutí, neboť IPF se prakticky nevyskytuje u jedinců do 45 let věku. Vliv senescence lze vysvětlit ztrátou integrity epitelu na podkladě opakovaných poranění epitelových buněk, sníženou schopností normální regenerace alveolárního epitelu, kumulací oxidativního stresu a sklonem k fibroproliferativnímu hojení ve stáří. Ztráta schopnosti regenerace epitelu je pravděpodobně způsobena poruchou autofagie, která je důležitá pro normální obrat subcelulárních organel a proteinů, a dysfunkcí mitochondrií, jež mohou mediovat apoptózu alveolárních epitelových buněk u IPF. Význam má také imunosenescence, kdy s věkem klesá imunokompetence jedince, objevuje se zvýšené zastoupení CD4+ CD8+ T lymfocytů a zvyšuje se tvorba autoprotilátek, jež se pravděpodobně mohou také účastnit na patogenezi plicní fibrózy. Svou úlohu má jistě i dlouhotrvající mechanické namáhání plicní tkáně (komprese, relaxace, střižné síly) při dechových exkurzích, kdy nejvíce namáhané části plíce – tedy partie v zadních kostofrenických úhlech – jsou fibrózou postiženy nejvíce (7).
DIAGNOSTIKA
Stanovení diagnózy IPF je postaveno na klinických symptomech, fyzikálním vyšetření a typickém radiologickém nálezu na CT hrudníku s vysokou rozlišovací schopností (HRCT). Bronchoalveolární laváž (BAL) je pomocným vyšetřením, které především umožňuje odlišit intersticiální plicní procesy (IPP) exogenního původu, hlavně chronickou exogenní alergickou alveolitidu či azbestózu. Klinickým a laboratorním (autoprotilátky) revmatologickým screeningem je třeba vyloučit systémové nemoci pojiva.
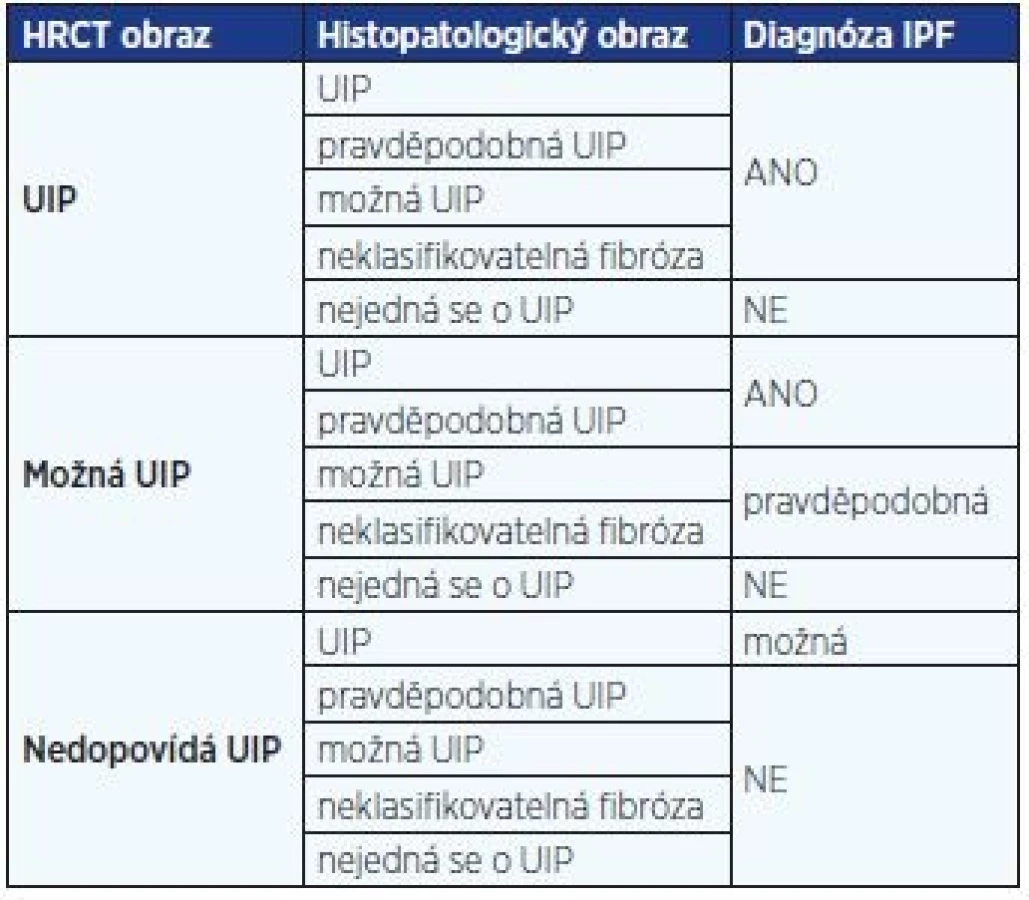
Diagnóza IPF by měla být stanovena multidisciplinárním týmem sestávajícím z pneumologa – odborníka na IPP, radiologa zběhlého v diagnostice IPP a případně patologa. Plicní biopsii potřebujeme pouze v případech, kdy klinicko-radiologický obraz nemoci není typický. Diagnóza IPF je pak stanovena na základě kombinace radiologických a histopatologických změn v kontextu klinického obrazu (tab. 1) (1, 10, 11).
Klinický obraz
Idiopatická plicní fibróza by měla být zvažována jako možná příčina v diferenciální diagnostice dušnosti u jedinců středního a staršího věku. Dušnost obvykle pozvolna progreduje, pokud se však objeví akutní exacerbace, pak je změna dušnosti výrazná. Dalšími symptomy jsou kašel a snadná unavitelnost, v pozdních stádiích i cyanóza. U 75 % pacientů se vyskytují typické fenotypové projevy nemoci, a to paličkovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomén krepitu slyšitelný nad plicními bazemi (2).
Radiologický nález
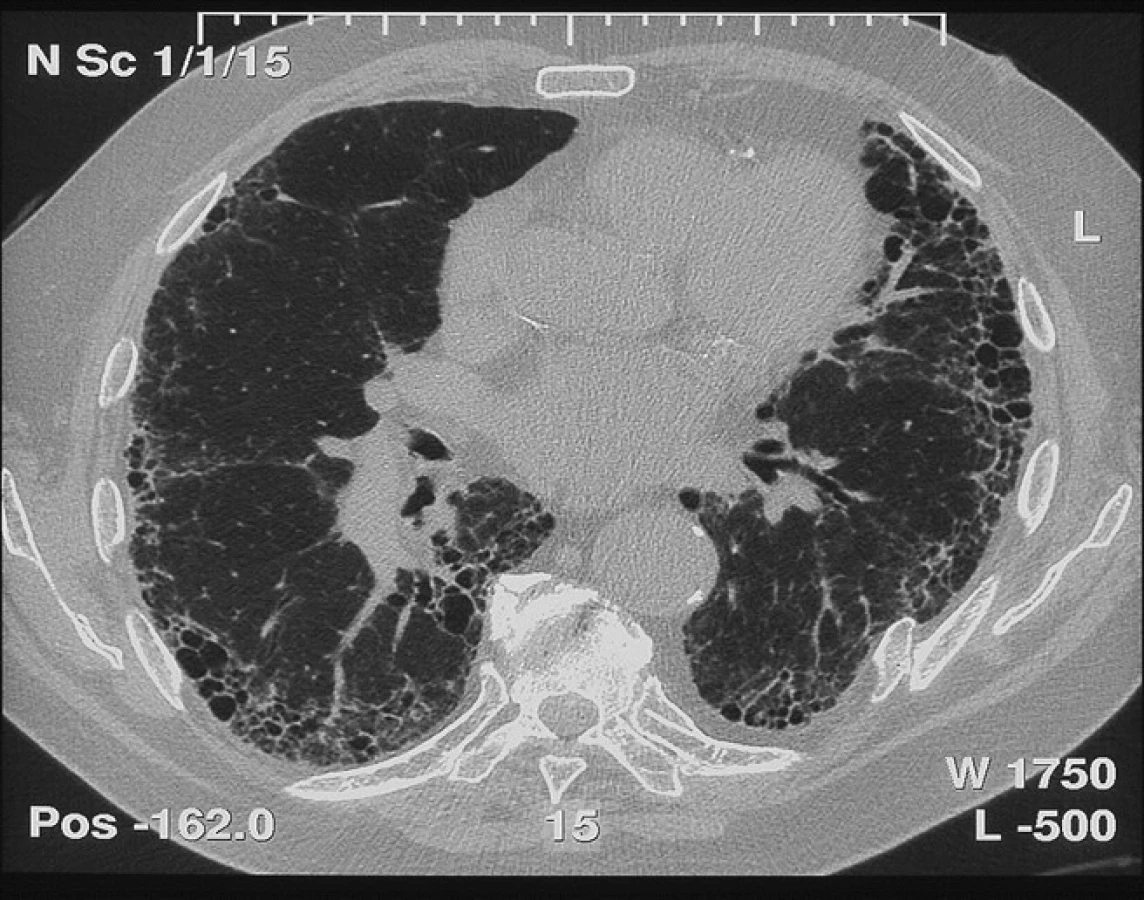
Typickým radiologickým nálezem IPF na HRCT hrudníku jsou intersticiální změny charakteru retikulací, cystických lézí (voštinovitá plíce) a trakčních bronchiektázií s časovou heterogenitou a subpleurální dominancí, obvykle s maximem v pravém dolním laloku dorzobazálně (obr. 1). Toto je typický radiologický obraz IPF. Nicméně tento obraz tzv. definitivní UIP v radiologickém obraze chybí dle některých studií téměř u poloviny jedinců s IPF. Navíc řada pacientů s netypickým radiologickým nálezem není schopna plicní biopsie, jak to vyžaduje doporučený postup z roku 2011 (obr. 2) (1).
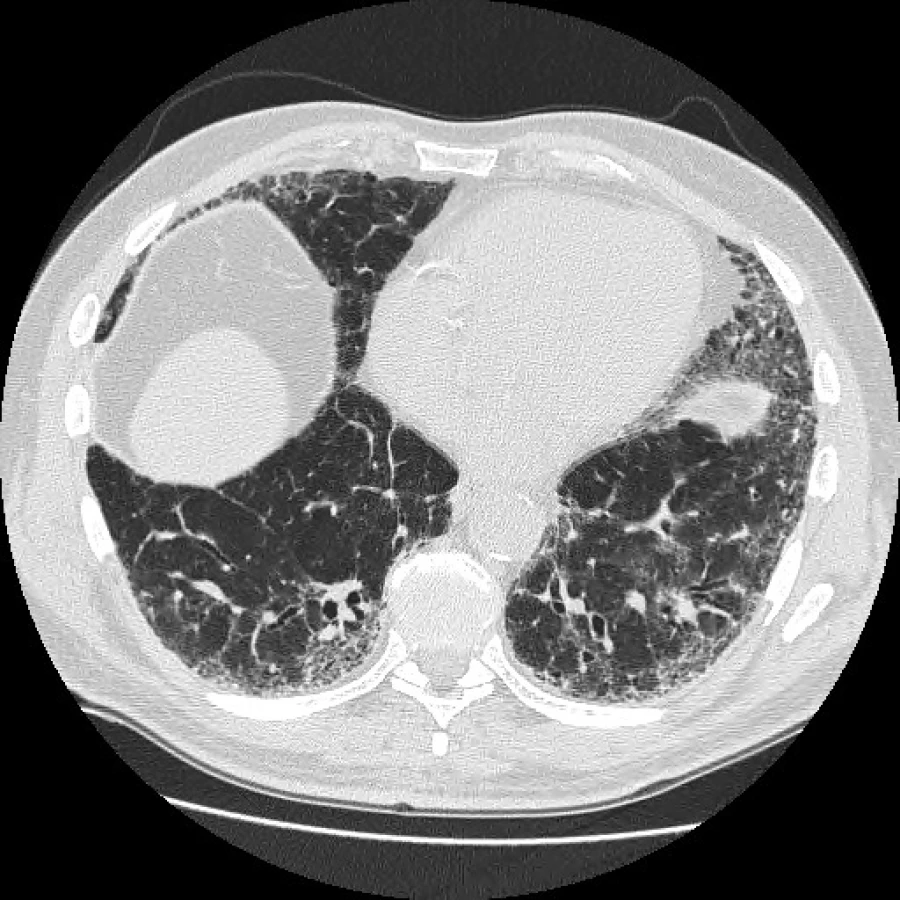
Fleischnerova společnost proto v minulém roce navrhla změnu v radiologické diagnostice IPF, kdy za definitivní diagnózu IPF může být považován nejen případ s klinickým obrazem odpovídajícím IPF a s radiologickým obrazem definitivní UIP, ale navíc nově i s obrazem pravděpodobné UIP, který je novým podtypem možné UIP (fibróza bez voštinovité plíce) s obrazem trakčních bronchiektasií, a to bez nutností plicní biopsie. Hrubá heterogenní fibróza (pravděpodobná UIP) tedy také odpovídá IPF/UIP, i když zde není voštinovitá plíce (obr. 1, 2) (12).
Funkční vyšetření plic
Funkční vyšetření plic samo o sobě nemá přínos z hlediska diagnózy, ale je důležitým nástrojem pro určení závažnosti nemoci a sledování v čase včetně stanovení efektu léčby. Typickou funkční poruchou je u pacientů s IPF restrikční ventilační porucha (RVP) s poruchou difuzní kapacity – transferfaktoru (TLCO) – a snížením plicní poddajnosti. V počínajících fázích nemoci může být ovšem vitální kapacita (VC) zcela v normě a může být jen mírně snížená difuzní kapacita.
Nedílnou součástí je vyšetření krevních plynů, kdy je u pacientů s IPF v úvodu onemocnění patrný pokles saturace krve kyslíkem (SaO2) a parciálního tlaku kyslíku (paO2) v tepenné krvi pouze při zátěži a teprve s progresí onemocnění dochází i ke klidové hypoxemii. Na základě hodnot krevních plynů je pak u pacientů s IPF zvažováno přidělení suplementární léčby kyslíkem, a to buď cestou koncentrátoru kyslíku, nebo kapalného kyslíku. Dobrou informaci o funkčním stavu pacienta nám dá 6minutový test chůzí (6MWT), který je také nedílnou součástí vyšetření pro případnou indikaci přidělení kapalného kyslíku (10).
Bronchoalveolární laváž
Navzdory tomu, že doporučený postup ATS/ERS/JRS/ALAT z roku 2011 je proti rutinně prováděné BAL, sekce IPP ČPFS se nadále domnívá (v souladu s řadou především evropských pracovišť), že BAL je cenným diferenciálně diagnostickým nástrojem umožňujícím zejména odlišení exogenní alergické alveolitidy (EAA), případně plicního postižení při systémových nemocech pojiva a polékovém poškození plic (14, 15). Pro IPF je typické zmnožení granulocytů obvykle s malou příměsí eozinofilů v tekutině získané pomocí BAL (BALTe), lymfocyty bývají zvýšené minimálně. Pokud jsou lymfocyty v BALTe zvýšeny nad 20 %, významně stoupá pravděpodobnost, že se jedná o EAA.
Plicní biopsie
Plicní biopsii by měli podstoupit pouze pacienti s možnou UIP s radiologicky neurčeným podtypem možné UIP a případně nálezem nekonzistentním s UIP. Pokud pacienti nejsou schopni biopsii podstoupit, lze v rámci multidisciplinární diskuse rozhodnout o definitivní diagnóze dle klinického chování nemoci (16–18).
V zásadě pokud je klinický obraz kompatibilní s IPF, HRCT nález odpovídá UIP a vyloučili jsme exogenní příčiny IPP a systémové nemoci pojiva, není plicní biopsie indikována. Je třeba si navíc uvědomit, že chirurgická plicní biopsie u pacientů s IPF starších 65 let a s TLCO < 35 % je zatížena signifikantní mortalitou, a měli bychom se jí tedy pokud možno vyhnout. Nově přináší bezpečnější alternativu získání vzorku plicní tkáně pro diagnostiku IPF transbronchiání kryobiopsie, která má slušnou výtěžnost a pravděpodobně menší riziko akutní exacerbace spojené s výkonem (19) (obr. 3).
Histopatologický obraz
Základním histomorfologickým rysem při malém zvětšení je ložiskové postižení plicního parenchymu. Oblasti postižené chronickým intersticiálním zánětlivým procesem a fibrózou, v níž jsou přítomná ložiska fibroblastů, se střídají s částmi relativně normálního nepostiženého plicního parenchymu. Fibroticky změněná plicní tkáň je prostoupena poměrně dobře formovanou fibrózou, jež je tvořena jen velmi málo buněčným, hyalinně transformovaným hustým kolagenním pojivem. Bezprostředně na tuto starou fibrotizaci však navazují části granulační tkáně s nakupením fibroblastů s myxoidním stromatem. Právě výrazná proměnlivost stáří fibrotických změn, popisovaná jako „časová heterogenita“, je typická pro tento typ postižení (10).
LÉČBA
Léčba IPF by měla být časná a komplexní, zahrnující kromě kauzální farmakoterapie i kyslíkovou léčbu, rehabilitaci a paliativní a symptomatickou léčbu.
Farmakoterapie
Základem farmakoterapie je od roku 2011 kauzální antifibrotická léčba (14). Efekt v léčbě IPF prokázaly a pro léčbu jsou schváleny dva antifibroticky působící léky, pirfenidon a nintedanib. Stran volby antifibrotika není žádná preference jednoho či druhého pro 1. či 2. linii léčby. Obvykle probereme s pacientem nežádoucí účinky jednotlivých preparátů a jejich dávkování a navrhneme lék i na základě pacientových preferencí. Switch z jednoho léku na druhý je při intoleranci možný, při dysefektu jednoho z léků však není dostatek důkazů, že druhý bude efektivní, a plátci péče se proto tyto switche zdráhají hradit.
Pirfenidon snižuje proliferaci fibroblastů a produkci s fibrózou asociovaných proteinů a cytokinů pravděpodobně inhibicí TGF-β a destičkového růstového faktoru (PDGF – platelet-derived growth factor), a to cestou blokády nukleární translokace proteinu Smad.
Klinická účinnost léčby pirfenidonem již byla prokázána v řadě studií, zejména ve společné analýze multinárodních studií CAPACITY 004 a 006, v japonské studii Shionogi (SP3) a následně ve studii ASCEND (20–23). Pirfenidon ve studii ASCEND prokazatelně zpomaloval progresi onemocnění, a to zpomalením poklesu plicních funkcí. Ve skupině pacientů léčených pirfenidonem bylo méně těch, u kterých došlo k poklesu VC o ≥ 10 % a více těch, kteří neměli žádný pokles VC, pirfenidon navíc statisticky významně snižoval i pokles 6MWD a prodlužoval přežití bez progrese onemocnění. Společnou analýzou studií CAPACITY 004 a 006 a ASCEND byla navíc prokázána redukce mortality ze všech příčin i na IPF ve skupině léčené pirfenidonem oproti placebu (20).
V ČR byl pirfenidon dostupný pro pacienty s IPF od roku 2011 nejprve v pacientském programu, od roku 2012 pak na individuální úhradu mimořádně dováženého léku a od července 2014 byl lék hrazen z prostředků zdravotního pojištění jako vysoce inovativní léčebný prostředek (VILP). Od 1. 12. 2017 má stanovenu řádnou úhradu.
Je hrazen z prostředků zdravotního pojištění pro nekouřící pacienty s IPF s VC mezi 50 a 90 % náležitých hodnot (NH) a TLCO > 35 % NH, a to v centrech pro diagnostiku a léčbu intersticiálních plicních procesů garantovaných ČPFS. Bohužel trvá omezení úhrady týkající se pacientů s poklesem plicních funkcí v 6měsíčních intervalech mezi jednotlivými měřením, kdy z úhrady vypadávají jedinci s poklesem VC > 10 % a TLCO > 15 % oproti předchozím hodnotám. Toto úhradové omezení odporuje medicíně založené na důkazech, neboť nemocní, jejichž plicní funkce klesají rychleji, se mohou po vysazení antifibrotik ještě výrazněji zhoršit.
Pacienti indikovaní k léčbě pirfenidonem zahajují 1.–7. den 3× 1 kapslí po 267 mg, 8.–15. den pak zvyšují dávku na 3× 2 kapsle a od 16. dne užívají 3× 3 kapsle (2403 mg pirfenidonu/den). Nemocní, kteří mají výrazné nežádoucí gastrointestinální účinky (obvykle nauzeu a nechutenství), mohou mít redukovanou dávku pirfenidonu na nejvyšší tolerovanou (kupříkladu 3× 2 nebo pouze 3× 1 kapsle po 267 mg/den). Také v případě fotosenzitivní reakce nebo významné elevace transamináz je zpravidla nutná přechodná redukce dávky léku, eventuálně jeho úplné vysazení na přechodnou dobu.
Pacient musí být při zahájení léčby seznámen s fototoxicitou léku a s nutností soustavné ochrany proti slunečnímu záření (celoročně krémy s vysokým ochranným faktorem, tzv. sun-blockery, na nechráněná místa kůže, pokrývka hlavy a ochrana těla oděvem při riziku oslunění). Také musí vědět o gastrointestinálních nežádoucích účincích, hlavně o nevolnosti a nechuti k jídlu a o možnosti snížit dávku léku při jejich výskytu. Musí být poučen i o možnosti poruchy funkce jater při léčbě pirfenidonem a nutnosti opakovaných odběrů krve v průběhu léčby, s kontrolou jaterních testů. Vzhledem k tomu, že tabákový kouř je induktorem CYP1A2, čímž významně ovlivňuje farmakokinetiku a tím biologickou dostupnost (expozici) pirfenidonu, by pacienti užívající tento lék neměli kouřit. Navíc nekouření je samo o sobě jednou z podmínek úhrady pirfenidonu v ČR. Zvýšené opatrnosti je třeba při současné léčbě léky snižujícími aktivitu CYP1A2, kdy může dojít až k významné toxicitě pirfenidonu (např. fluvoxamin, který je při léčbě pirfenidonem kontraindikovaný, chinolony). Naopak léky zvyšující aktivitu CYP1A2 mohou snižovat účinnost pirfenidonu (rifampicin, omeprazol).
Nintedanib je dalším lékem, který je dostupný pro léčbu IPF v ČR, a to doposud v rámci VILP. Jedná se o trikinázový inhibitor (inhibice receptoru pro růstový faktor fibroblastů /FGF – fibroblast growth factor/, vaskulární endotelový růstový faktor /VEGF – vascular endothelial growth factor/ a PDGF – platelet derived growth factor), který v dávce 150 mg 2 × denně perorálně prokazatelně snižuje pokles plicních funkcí u pacientů s IPF.
Efektivita a bezpečnost nintedanibu v léčbě IPF byly prokázány ve dvou velkých studiích fáze III INPULSIS-1 a INPULSIS-2, do nichž bylo zařazeno 1066 pacientů s IPF (24, 25). Nintedanib prokázal v obou těchto studiích efektivitu vyjádřenou významnou redukcí poklesu VC a navíc v INPULSIS-2 i benefit ve smyslu redukce počtu a doby do výskytu akutních exacerbací IPF (26).
Nintedanib je v ČR hrazen u pacientů s VC 50–90 % NH a TLCO > 30 % NH. I pro nintedanib platí ukončení úhrady léku při poklesu plicních funkcí, tak jako pro pirfenidon. Lék je pro perorální užití, ve formě kapslí, užívá se 2 × 1 kapsle po 150 mg denně (přibližně v intervalu 12 hodin). Při intoleranci je možné dávku snížit na 2× 100 mg. Nejčastějšími nežádoucími účinky léčby jsou průjmy a elevace jaterních enzymů. Frekvenci stolic lze snížit podáním loperamidu. Při elevaci transamináz lze při významném vzestupu přechodně přerušit léčbu, častěji však postačí většinou přechodné snížení dávky na 2× 100 mg denně.
Úloha N-acetylcysteinu (NAC) v léčbě IPF doposud není vyjasněna, je však pravděpodobné, že přinejmenším u části nemocných s genovými polymorfismy TOLLIP může být přínosná. Nicméně plošné testování pacientů s IPF na genové polymorfismy TOLLIP není nyní dostupné ,tudíž nelze léčbu NAC pro pacienty s IPF plošně doporučit. Pokud bychom však znali TOLLIP status, pak genotyp TT na pozici rs3750920 disponuje ke zlepšení přežití u pacientů s IPF a bylo by možné lék k terapii přidat (9).
Podávání inhibitorů protonové pumpy (PPI) jako léčba asymptomatického gastroezofageálního a extraezofageálního refluxu není plošně doporučeno. PPI nemají vliv n snížení poklesu plicních funkcí, navíc mohou změnou mikrobiomu spíše nepříznivě ovlivnit průběh nemoci. Také interferují s metabolismem pirfenidonu. Lze je doporučit pouze při symptomatickém refluxu, a to raději esomeprazol, kde je interference relativně nízká (8, 25).
Dlouhodobá domácí oxygenoterapie
Je doporučena pro všechny pacienty s IPF s klidovou či zátěží indukovanou hypoxemií, kteří splní kritéria ČPFS.
Transplantace plic
Je doporučena pro vhodně vybrané pacienty s IPF. Pětileté přežití po transplantaci plic u pacientů s IPF je odhadováno na 50–56 %. Transplantace plic u adekvátně indikovaných pacientů s IPF významně snižuje riziko úmrtí v pátém roce po transplantaci. Navíc pacienti s IPF mají lepší dlouhodobé přežití po transplantaci plic než ti transplantovaní pro jinou diagnózu (28). Pacienti s IPF by při respiračním selhání způsobeném IPF neměli být paušálně invazivně uměle ventilováni, pokud nejsou zařazeni na čekací listinu transplantace plic a není rozhodnuto o bridgingu mimotělním oběhem. Mortalita při umělé plicní ventilaci u pacientů s IPF dosahuje 96 % (29).
Paliativní péče a symptomatická léčba
Měla by být součásti péče o pacienty s IPF prakticky od stanovení diagnózy. Spočívá v tlumení obtěžujících symptomů. Pro tlumení kašle lze použít systémové kortikosteroidy, a to obvykle v minimální dávce (obvykle do 20 mg prednisonu denně a dávku postupně snižovat), která dosáhne zmírnění úporného kašle. Opioidy indikujeme u těžké dušnosti s kašlem či bez něj.
Plicní rehabilitace
Všichni pacienti s IPF by měli být motivováni k plicní rehabilitaci. Je to jedna z možností jak zlepšit kvalitu života pacienta, funkční výkonnost a zmírnit dušnost. Rehabilitace musí být v případě IPF komplexní, zahrnující učení, poradu a behaviorální techniky ke zlepšení sebeobsluhy, redukci symptomů a optimalizaci funkční kapacity (30).
DISPENZARIZACE
Interval sledování je doporučen 3–6měsíční. Kontroly spočívají ve vyšetření pacienta – funkčním (spirometrie, transferfaktor, krevní plyny) a klinickém. HRCT hrudníku není třeba zhotovovat při každé z těchto kontrol, pokud nedojde k významné změně klinického obrazu a funkčních parametrů. Pokud však máme podezření na tzv. akutní exacerbaci IPF, je provedení HRCT hrudníku na prvním místě. V případě podezření na infekci či nádor v terénu IPF jsou další vyšetřovací postupy včetně radiologického vyšetření samozřejmostí.
PROGNÓZA
Prognóza pacientů s nově stanovenou diagnózou IPF je do značné míry závislá na pokročilosti a rozsahu fibrózních změn, rychlosti progrese a přítomnosti akutní exacerbace (31). Také charakter HRCT změn ovlivňuje prognózu. Pacienti, kteří mají na HRCT změny zcela typické pro IPF, mají nejhorší prognózu. Ti, kteří mají obraz HRCT spíše podobný nespecifické intersticiální pneumonii (NSIP) nebo necharakteristický, mají prognózu lepší.
Z funkčních parametrů mají prognostický význam změny VC a TLCO v čase. Pokles VC o více než 10 % a TLCO o více než 15 % za 6 měsíců svědčí pro časnou mortalitu pacienta. Také vzdálenost ujitá při 6minutovém testu chůzí (6MWD) < 250 m a snížení 6MWD za 24 týdnů o > 50 m je spojeno s 3× vyšší mortalitou. Pro určení prognózy je možné použít rovněž složené indexy, kupříkladu GAP (Gender-Age-Physiology), který je také dobrým prediktorem mortality pacientů s IPF (32).
U pacientů s IPF jsou ve zvýšené míře pozorovány komorbidity jako bronchogenní karcinom, ischemická choroba srdeční a žilní trombóza. Antikoagulační léčba z jakýchkoliv důvodů významně zhoršuje prognózu pacientů s IPF, a to pravděpodobně nezávisle na diagnóze, pro kterou byla zavedena. Nicméně mechanismus tohoto dysefektu není přesně znám (33).
ZÁVĚR
Idiopatická plicní fibróza je závažné onemocnění plic s prognózou srovnatelnou s rakovinou plic. Poznání patogenetického podkladu této nemoci nám umožňuje hledat nové léčebné metody, jež mohou v budoucnu vést nejen ke zpomalení progrese nemoci, ale i k jejímu zastavení nebo regresi změn.
Pro zvyšování povědomí o IPF za účelem zvýšení počtu časných diagnóz IPF a pro klinický výzkum IPF je velmi důležitá organizace specializované péče o pacienty s IPF a registry IPF. V České republice je etablována síť Center pro diagnostiku a léčbu IPP, kde jsou pacienti odborně vyšetřeni, sledováni a léčeni.
Registr EMPIRE (European MultiPartner IPF Registry), který vznikl v ČR, je navíc světově největším registrem IPF. Poskytuje řadu informací o této vážné nemoci, a to jak pro výzkum či nábor pacientů do nových klinických studií, tak také v neposlední řadě pro jednání o úhradách nových léků (www.pneumologie.cz, záložka Sekce IPP; http://empire.registry.cz/index-en.php) (34).
Čestné prohlášení
Autorka práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Seznam zkratek
6MWD vzdálenost ujitá při šestiminutovém testu chůzí
6MWT šestiminutový test chůzí
BAL bronchoalveolární laváž
BALTe tekutina získaná pomocí BAL
BM bazální membrána
EAA exogenní alergická alveolitida
ECM extracelulární matrix
EMT epitelově-mezenchymová tranzice
FGF růstový faktor fibroblastů
FPF familiární plicní fibróza
HRCT CT hrudníku s vysokou rozlišovací schopností
IAP antiapoptotické faktory ze skupiny inhibitorů apoptózy
IL interleukin
IPF idiopatická plicní fibróza
IPP intersticiální plicní procesy
NAC N-acetylcystein
NH náležitá hodnota
NSIP nespecifická intersticiální pneumonie
paO2 parciální tlak kyslíku
PDGF růstový faktor z destiček
PPI inhibitory protonové pumpy
RVP restrikční ventilační porucha
SaO2 saturace krve kyslíkem
TGF-b transformující růstový faktor beta
TLCO transferfaktor
UIP obvyklá intersticiální pneumonie
VC vitální kapacita
VEGF vaskulární endotelový růstový faktor
VILP vysoce inovativní léčebný prostředek
Adresa pro korespondenci:
prof. MUDr. Martina Vašáková, Ph.D.
Pneumologická klinika 1. LF UK
Thomayerova nemocnice
Vídeňská 800, 140 59 Praha 4
Tel.: 261 082 372
e-mail: martina.vasakova@ftn.cz
Zdroje
- Raghu G, Collard HR, Egan JJ et al., on behalf of ATS/ERS/JRS/ALAT; Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6): 788–824.
- Raghu G, Rochwerg B, Zhang Y et al., on behalf of ATS, ERS, JRS and ALAT. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis: executive summary. Am J Respir Crit Care Med 2015; 192(2): e3–e19.
- Fernández Pérez ER, Daniels CE, Schroeder DR et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest 2010; 137(1): 129–137.
- Hopkins RB, Burke N, Fell C et al. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J 2016; 48(1): 187–195.
- Olson AL, Swigris JJ. Idiopathic pulmonary fibrosis: diagnosis and epidemiology. Clin Chest Med 2012; 33(1): 41–50.
- Agabiti N, Porretta MA, Bauleo L et al. Idiopathic pulmonary fibrosis (IPF) incidence and prevalence in Italy. Sarcoidosis Vasc Diffuse Lung Dis 2014; 31(3): 191–197.
- Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am J Respir Crit Care Med 2014; 189: 1161–1172.
- Savarino E, Carbone R, Marabotto E et al. Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients. Eur Respir J 2013; 42(5): 1322–1331.
- Oldham JM, Ma SF, Martinez FJ et al; IPFnet Investigators. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015; 192(12): 1475–1482.
- Vašáková M. Idiopatická plicní fibróza. In Vašáková M., Polák J., Matěj R. Intersticiální plicní procesy (2., rozšířené vyd.). Maxdorf, Praha, 2016.
- Travis WD, Costabel U, Hansell DM et al.; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188(6): 733–748.
- Lynch DA, Sverzellati N, Travis WD et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med 2018; 6(2): 138–153.
- Lynch DA, Godwin JD, Safrin S et al.; Idiopathic Pulmonary Fibrosis Study Group. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med 2005; 172(4): 488–493.
- Vašáková M., Šterclová M, Sekce IPP ČPFS. Kapitola 6.1. Idiopatická plicní fibróza (Doporučený postup pro diagnózu léčbu a sledování). In: Kolek V a kol. Doporučené postupy v pneumologii. Maxdorf, Praha, 2017.
- Ohshimo S, Bonella F, Cui A et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11): 1043–1047.
- Chung JH, Lynch DA. The value of a multidisciplinary approach to the diagnosis of usual interstitial pneumonitis and idiopathic pulmonary fibrosis: radiology, pathology, and clinical correlation. AJR Am J Roentgenol 2016; 206(3): 463–471.
- Wells AU. Managing diagnostic procedures in idiopathic pulmonary fibrosis. Eur Respir Rev 2013; 22(128): 158–162.
- Xaubet A, Behr J, Bendstrup E et al. Review of IPF diagnosis and management recommendations in Europe. Sarcoidosis Vasc Diffuse Lung Dis 2013; 30(4): 249–261.
- Casoni GL, Tomassetti S, Cavazza A et al. Transbronchial lung cryobiopsy in the diagnosis of fibrotic interstitial lung diseases. PLoS One 2014; 9(2): e86716.
- Noble PW, Albera C, Bradford WZ et al.; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779): 1760–1769.
- King TE Jr, Bradford WZ, Castro-Bernardini S et al.; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 22: 2083–2092.
- Nathan SD, Albera C, Bradford WZ et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1): 33–41.
- Hilberg O, Simonsen U, du Bois R et al. Pirfenidone: significant treatment effects in idiopathic pulmonary fibrosis. Clin Respir J 2012; 6(3): 131–143.
- Richeldi L, du Bois RM, Raghu G et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl. J Med 2014; 370: 2071–2082.
- Richeldi L, du Bois RM, Raghu G et al.; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2071–2082.
- Suissa S, Ernst P. The INPULSIS enigma: exacerbations in idiopathic pulmonary fibrosis. Thorax 2015; 70(5): 508–510.
- Kreuter M, Wuyts W, Renzoni E et al. Antacid therapy and disease outcomes in idiopathic pulmonary fibrosis: a pooled analysis. Lancet Respir Med 2016; 4(5): 381–389.
- Thabut G, Christie JD, Ravaud P et al. Survival after bilateral versus single-lung transplantation for idiopathic pulmonary fibrosis. Ann Intern Med 2009; 151(11): 767–774.
- Mooney JJ, Raimundo K, Chang E et al. Mechanical ventilation in idiopathic pulmonary fibrosis: a nationwide analysis of ventilator use, outcomes, and resource burden. BMC Pulm Med 2017; 17: 84.
- Nishiyama O, Kondoh Y, Kimura T et al. Effects of pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Respirology 2008; 13(3): 394–399.
- Maher TM, Whyte MK, Hoyles RK et al. Development of a consensus statement for the definition, diagnosis, and treatment of acute exacerbations of idiopathic pulmonary fibrosis using the Delphi technique. Adv Ther 2015; 32(10): 929–943.
- Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4): 431–440.
- Kreuter M, Wijsenbeek MS, Vašáková M et al. Unfavourable effects of medically indicated oral anticoagulants on survival in idiopathic pulmonary fibrosis. Eur Respir J 2016; 47(6): 1776–1784.
- Doubková M, Švancara J, Svoboda M et al. EMPIRE Registry, Czech Part: Impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J 2018; 12(4): 1526–1535.
Štítky
Adiktológia Alergológia a imunológia Angiológia Audiológia a foniatria Biochémia Dermatológia Detská gastroenterológia Detská chirurgia Detská kardiológia Detská neurológia Detská otorinolaryngológia Detská psychiatria Detská reumatológia Diabetológia Farmácia Chirurgia cievna Algeziológia Dentální hygienistkaČlánok vyšiel v časopise
Časopis lékařů českých

- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Antidepresivní efekt kombinovaného analgetika tramadolu s paracetamolem
- Kombinace metamizol/paracetamol v léčbě pooperační bolesti u zákroků v rámci jednodenní chirurgie
- 100 let s metamizolem: jaké je jeho současné postavení v léčbě bolesti
Najčítanejšie v tomto čísle
- Karcinom plic
- Vznik a vývoj tukové kapénky a její role ve zdraví a nemoci
- Idiopatická plicní fibróza: Nastal čas opět změnit doporučený postup diagnostiky a léčby?
- Alkohol z pohledu veřejného zdraví v ČR: fakta a souvislosti