
X-vázaná myotubulární myopatie u dvou bratrů v důsledku nové mutace v MTM1 genu – kazuistiky
X-linked Myotubular Myopathy: a Novel Mutation in the MTM-1 Gene – Case Reports
Mutations in the MTM1 gene cause X-linked myotubular myopathy (XLMTM). This is a serious, often lethal, condition. We report a family with two affected children – dizygotic twins. Boys were born in the 28th gestational week. Severe hypotonia and muscle weakness were present at birth and necessitated ventilatory support. Intensive care was terminated 26 and 35 days after birth, respectively, due to cardiac and renal failure. Hypotrophic muscle fibers, fetal myotubes and centrally located nuclei were present in muscle sections. All the available information led us to the diagnosis of XLMTM. The MTM1 gene was sequenced and a novel mutation c.82delA in exon 3 was identified. The mother of the patients is a heterozygous carrier of the mutation. This might well be the first report of genetically confirmed XLMTM in the Czech Republic. DNA test is now available and prenatal or preimplantation diagnosis might be offered to the family.
Key words:
MTM1 gene – myotubular myopathy – frameshift mutation
Autoři:
P. Laššuthová 1; V. Sebroň 2; J. Zámečník 3; J. Haberlová 1; A. Baxová 4; P. Seeman 1
Působiště autorů:
DNA laboratoř Kliniky dětské neurologie 2. LF UK a FN v Motole, Praha
1; Novorozenecké oddělení Gynekologicko-porodnické kliniky 1. LF UK a VFN v Praze
2; Ústav patologie a molekulární medicíny 2. LF UK a FN v Motole, Praha
3; Ústav biologie a lékařské genetiky 1. LF UK a VFN v Praze
4
Vyšlo v časopise:
Cesk Slov Neurol N 2013; 76/109(2): 241-245
Kategorie:
Kazuistika
Souhrn
Mutace v MTM1 genu způsobují X-vázanou myotubulární myopatii (XLMTM). Jedná se o závažné, většinou časně letální onemocnění. Popisujeme dva chlapce, dvojčata s molekulárně geneticky prokázanou XLMTM. Chlapci se narodili ve 28. týdnu gravidity, nápadná byla těžká hypotonie a slabost s areflexií, od narození byla nutná intenzivní péče včetně umělé plicní ventilace. Progredující oběhová a renální nedostatečnost byla u obou důvodem ukončení péče po 26, resp. 35 dnech přežívání. Ve svalové excizi byla nalezena hypotrofická svalová vlákna a fetální myotuby spolu s četnými centrálně lokalizovanými jádry. To vše vedlo k podezření na XLMTM, které jsme následně potvrdili molekulárně geneticky nálezem mutace c.82delA ve třetím exonu MTM1 genu. Stejná mutace, v heterozygotním stavu, byla následně prokázána i u matky pacientů. Pravděpodobně se jedná o první případ tohoto onemocnění molekulárně geneticky potvrzeného v ČR. Rodině můžeme nabídnout cílenou genetickou prevenci v podobě prenatální nebo preimplantační diagnostiky. Zavedení vyšetření MTM1 genu může být využito u dalších rodin s buď již dříve klinicky stanovenou dg. XLMTM, nebo pro nově diagnostikované rodiny.
Klíčová slova:
MTM1 gen – myotubulární myopatie – mutace vedoucí k posunu čtecího rámce
Úvod
Centronukleární myopatie (CNM) je skupina kongenitálních strukturálních myopatií, pro které jsou typickým nálezem četná, centrálně ve svalovém vláknu uložená jádra ve svalové biopsii.
Rozlišujeme několik forem CNM s různým typem dědičnosti, různou závažností a odpovědným genem [1,2].
Autozomálně dominantně dědičná, klasická forma centronukleární myopatie je způsobena mutacemi v genu pro dynamin 2 (DNM2) [2,3]. Pacienti s mutacemi v DNM2 genu mají mírnější postižení, nemoc má i pozdější začátek a riziko opakování nemoci u potomstva je 50 %.
Autozomálně recesivní forma je způsobena mutacemi v genu pro amphiphysin 2 (BIN2) [1]. Riziko opakování nemoci u potomstva postiženého je jen velmi nízké.
X-vázaně recesivní forma (XLMTM) je zapříčiněna mutacemi v genu pro myotubularin 1 (MTM1). Postižení jsou potomci mužského pohlaví. Ženy jsou přenašečky a nemají klinické obtíže. Většina dětí umírá v perinatálním období, i když jsou publikovány práce, které popisují pacienty přežívající do období dětství [4]. Velký počet pacientů má i dechovou slabost vedoucí k dechové nedostatečnosti a potřebuje ventilační podporu [5]. Typický je i kryptorchizmus [6]. Mentální vývoj je u přežívajících pacientů normální [4].
Protože část pacientů s klinickým i bioptickým obrazem centronukleární myopatie nemá mutaci ani v jednom z těchto genů, lze předpokládat, že i jiné lokusy jsou vázány s tímto onemocněním, nebyly však dosud popsány [2].
Gen MTM1 – jehož mutace jsou příčinou XLMTM – je lokalizován na dlouhém raménku chromozomu X, má 15 exonů, z toho 14 kódujících. Dlouhý transkript je exprimován povšechně, ve svalu je však specificky přítomen kratší transkript [4]. Ten si zachovává stejný čtecí rámec, liší se na 3` konci, kde je kratší. Specializovanou funkcí tohoto kratšího transkriptu je role v maturaci svalových buněk. Mutace v genu MTM1, které vedou ke ztrátě funkce proteinu, pak způsobují poruchu svalového vyzrávání, svalová vlákna tak zůstanou ve svém vývoji zastavena ve stadiu fetálním [7]. Tomu pak odpovídá i vzhled fetálních myotub ve svalové biopsii. Odsud je odvozen i název onemocnění – myotubulární myopatie.
Soubor a metodika
Pacienti
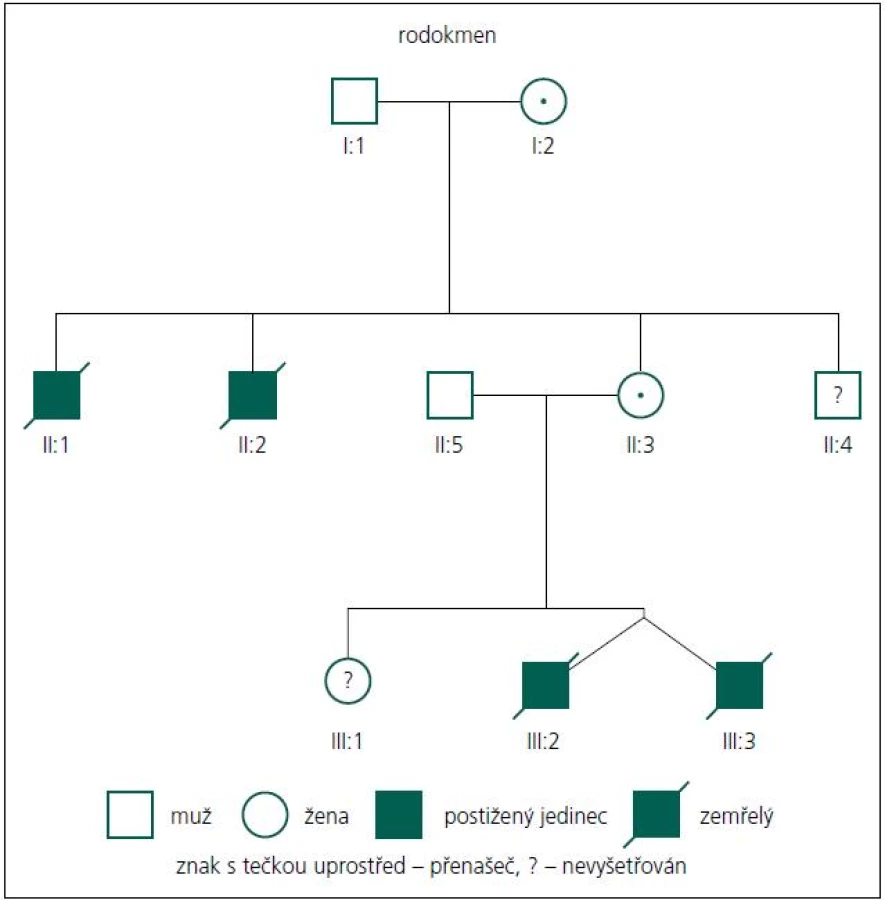
Rodiče pacientů podepsali informovaný souhlas s DNA vyšetřením. Popisujeme dva chlapce, dvojvaječná dvojčata. Chlapci se narodili akutním císařským řezem ve 28. týdnu těhotenství pro tíseň plodu. Rodokmen je uveden na obr. 1.
Dvojče A
Klinický nález
Chlapec po porodu s výrazným periferně hypotonickým syndromem, spontánně nedýchal, byl areaktivní k zevním podnětům. Apgarové skóre 2-4-6. Po porodu byl podporován distenční terapií CPAP maskou s prodechy. Pro přetrvávající nulovou dechovou aktivitu byl ve 20. minutě života intubován a byl aplikován intratracheálně surfaktant a následně byl chlapec převezen na novorozeneckou jednotku intenzivní péče, kde byl trvale závislý na umělé plicní ventilaci.
Zde byla zahájena parenterální výživa, později byly podávány i stimulační dávky mateřského mléka. Docházelo k výrazně negativní vodní bilanci. Byla progrese bilirubinemie a ikteru. V prvním týdnu došlo i k mozkovému intraventrikulárnímu krvácení II.–III. stupně, oboustranně, s následnou progresí do parenchymu a rozvojem posthemoragického hydrocefalu. Ve druhém týdnu života se objevila i volná tekutina v pleurální dutině oboustranně – nejspíše chylothorax. Následně se rozvinulo renální selhání. I přes restriktivní vodní bilanci došlo k rozvoji hypostatických a hypalbuminických edémů v podkoží. Hybné projevy byly stále výrazně omezeny na občasný pohyb víček, očních bulbů nebo rtů. Během 5.–6. týdne progredovalo renální selhání, chlapec byl oligourický až anurický, progredovaly výrazné edémy. Vzhledem ke klinickému stavu – nulová dechová aktivita, závislost na přístrojích, nemožnost krmení a selhávání všech vitálních funkcí – byla po dohodě s rodiči ukončena intenzivní péče 35. postnatální den.
U obou pacientů byl od narození hemodynamicky významný, na farmakologickou léčbu nereagující, perzistující ductus arteriosus – PDA. Oba pacienti měli kryptorchizmus, který je typický pro XLMTM.
Pomocná vyšetření
Základní metabolické, biochemické a hematologické nálezy nevysvětlily původ těžké hypotonie a byly přiměřené těžké nezralosti. Novorozenecký skríning neprokázal žádnou odchylku od normy. Skríning poruch metabolizmu – provedený z krve, moči a mozkomíšního moku – 7. den života neprokázal poruchu metabolizmu aminokyselin, organických kyselin, beta oxidace mastných kyselin a karnitinů. U pacienta nebyla prokázána Pompeho choroba (GSD II).
Cytogenetické vyšetření prokázalo normální mužský karyotyp 46, XY. Bylo provedeno molekulárně genetické vyšetření na syndrom Prader-Willi, kongenitální myotonickou dystrofii (DM1), myotonickou dystrofii typ 2 (DM2) a spinální svalovou atrofii typ I. (SMA 1) (m. Werdnig-Hoffmann) – vše bez průkazu kauzální mutace.
EMG vyšetření 5. postnatální den neprokázalo známky periferní neuropatie, neurogenní léze, ani poruchu nervosvalového přenosu, ani jednoznačné známky myogenní léze. Hodnoty CK v séru byly normální.
Dvojče B
Průběh onemocnění byl u druhého dvojčete obdobný. Apgarové skóre bylo 5-6-7 bodů. Chlapec byl intubován v 10. minutě života, po intratracheální aplikaci surfaktantu byl extubován s nutností distenční léčby CPAP a prodýchávání pomocí masky. Pro nedostatečnou dechovou aktivitu následovala reintubace ve 20. minutě života s následnou dlouhodobou umělou plicní ventilací. Rozvoj pleurálních výpotků byl výraznější než u bratra. Chylothorax byl prokázán po zavedení pleurální drenáže, cytologickým a biochemickým vyšetřením. Další průběh onemocnění i výsledky pomocných vyšetření byly podobné jako u bratra. Intenzivní péče byla – vzhledem ke klinickému stavu – po dohodě s rodiči ukončena 26. postnatální den.
Vyšetření svalové excize
Vzhledem k maximální snaze určit správnou diagnózu byly při úmrtí dvojčete A provedeny odběry svalu, kůže, srdce a jater. Kosterní sval byl u dvojčete A odebrán z m. deltoideus a m. triceps surae. Nálezy z histologického vyšetření svalu jsou na obr. 2. Kosterní sval z m. deltoideus a m. triceps surae byl odebrán i při úmrtí dvojčete B. Nález byl prakticky shodný jako u bratra.
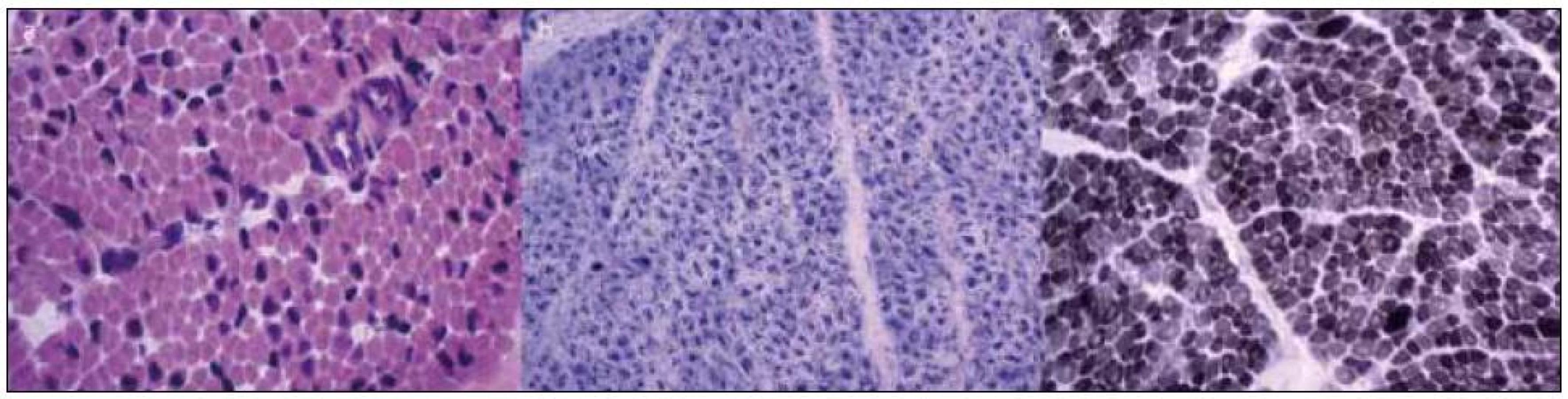
V diferenciálně diagnostické úvaze byl celkový stav a obraz u chlapců uzavírán jako nejspíše X-vázaná myotubulární myopatie, a to na základě výsledků histologického nálezu ve svalu, klinického stavu – výrazně hypotonický syndrom u plodu mužského pohlaví, nutnosti dechové podpory, kryptorchizmu, normálních hladin CK, dle výsledků EMG vyšetření a rodinné anamnézy – časné úmrtí dvou bratrů matky.
Matka pacientů
Anamnesticky neudává žádná neuromuskulární onemocnění. Průběh těhotenství byl normální – biochemický skríning v těhotenství byl v normě, ultrazvukové vyšetření v těhotenství neprokázalo vrozené vady ani jiné odchylky od normálního stavu.
U matky bylo také provedeno elektrofyziologické vyšetření, které prokázalo normální rychlosti vedení vzruchu periferním nervem. Nález v jehlové EMG u matky pacientů byl nejednoznačný, se známkami lehké smíšené léze: ve svalech m. TBA a svalech extenzorové skupiny předloktí byl identický nález, byly zachyceny četné komplexní repetitivní výboje nejasného významu, dále ojedinělé motorické jednotky o vyšší amplitudě a delší době trvání (amplituda MUP 1990 uV a doba trvání okolo 16 ms), byly zachyceny i jednotky o kratší době trvání (5–6 ms), průměrné hodnoty MUP však byly v mezích normy. Příčina četných komplexních repetitivních výbojů je nejasná, možná nespecifická, ale vzhledem k zjištěnému přenašečství vlohy pro X-MTM se dá spekulovat, že by se mohlo jednat o změnu excitability sarkolemy, může jít ovšem i o zcela jinou příčinu, jednoznačné potvrzení dle EMG není možné. Matka má zcela normální neurologický topický nález, je bez známek jakékoli svalové slabosti.
Molekulárně genetické analýzy
DNA od obou pacientů a jejich zdravých rodičů a maternální babičky byla izolována z periferní krve běžným postupem.
Ze vzorků DNA od postižených pacientů bylo pomocí polymerázové řetězové reakce (PCR) amplifikováno všech 14 kódujících exonů a přilehlých intronových oblastí MTM1 genu. Sekvence použitých PCR primerů jsou k dispozici u korespondujícího autora na vyžádání. Primery pro vyšetřování byly navrženy programem ExonPrimer [8].
PCR produkty pro jednotlivé exony byly sekvenovány s použitím sekvenačního kitu BDTv3.1 a analyzovány na automatickém genetickém analyzátoru ABI 3130 (vše Applera – Applied Biosystems, Foster City, CA, USA). Získané sekvence byly porovnány s referenční sekvencí NM_000252.2 [9]. DNA od rodičů a maternální babičky byla vyšetřena pouze na přítomnost nalezené mutace c.82delA, a to amplifikací a sekvenováním pouze druhého kódujícího exonu (exonu 3) MTM1 genu.
Výsledky
U dvou českých pacientů, dvojčat, jsme na základě klinického obrazu, histologického nálezu ve svalu a EMG vyšetření klinicky diagnostikovali X-vázanou myotubulární myopatii. Proto jsme se pokusili o molekulárně genetické potvrzení klinické diagnózy a objasnění příčiny vyšetřením MTM1 genu.
Sekvenováním všech 14 kódujících exonů genu MTM1 u dvou pacientů s těžkou formou kongenitální myopatie jsme prokázali novou, dosud nepopsanou mutaci c.82delA (p.N28I fs X 43) v druhém kódujícím exonu (exonu 3) MTM1 genu (obr. 3). Tato delece 1 báze (A), vede k posunu čtecího rámce a zařazení předčasného stop kodonu, a tím k předčasné terminaci proteinu, jde proto nesporně o mutaci velmi závažnou a tedy kauzální.
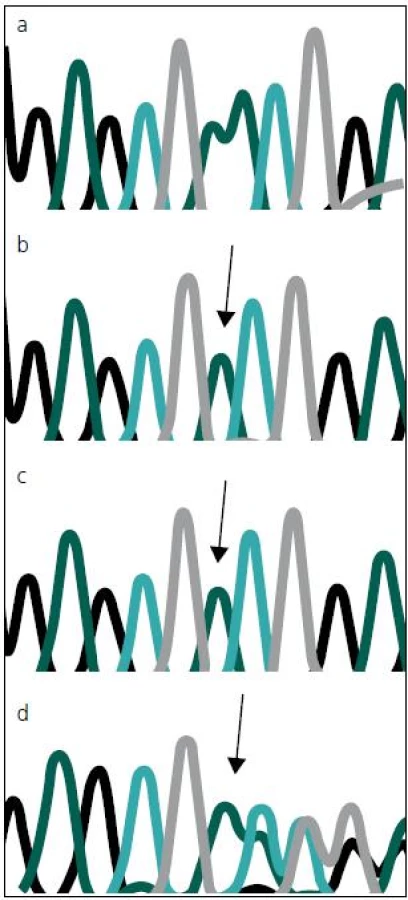
Stejná mutace, v heterozygotním stavu byla následně prokázána i u matky a maternální babičky obou zemřelých chlapců. Obě tyto ženy jsou tudíž zdravými přenašečkami uvedené mutace. To odpovídá X-vázanému typu onemocnění. Obě ženy mají, resp. měly 50% pravděpodobnost, že tuto mutaci předají svým potomkům. Mutace, podle očekávání, nebyla nalezena u zdravého otce pacientů.
Diskuze
Popisujeme nález nové mutace v MTM1 genu u dvou českých pacientů – dvojčat – a klinický i histologický obraz myotubulární myopatie u obou bratrů. Nalezená mutace c.82delA sice dosud nebyla popsána, ale vzhledem k charakteru mutace – delece 1 báze, vedoucí k posunu čtecího rámce a předčasnému stop kodonu – není pochyb o jejím patogenním a kauzálním charakteru pro XLMTM. Dalším důležitým faktem je, že tato mutace v rodině segreguje s onemocněním. Mutace, kterou popisujeme, se nachází ve druhém kódujícím exonu (exonu 3) genu MTM1 a predikuje posun čtecího rámce. Studie, jež zkoumaly vztah mutace a závažnosti postižení u pacientů s XLMTM, ukázaly, že mutace s posunem čtecího rámce mohou způsobovat závažnější fenotyp než missense mutace [6]. To je v souladu s pozorovaným průběhem nemoci u našich pacientů.
Průkaz mutace v genu MTM1 u této rodiny je důležitý také proto, že rodině nyní může být nabídnuta cílená genetická prevence ve formě prenatální nebo nověji i preimplantační diagnostiky, a to buď pro matku zemřelých chlapců, nebo v budoucnu možná i sestru pacientů. Domníváme se také, že nález mutace c.82delA v genu MTM1 u probandů, ale také u jejich matky a maternální babičky, které jsou přenašečky, vysvětluje úmrtí dvou bratrů matky pacientů (II:1, II:2, obr. 1). Jeden zemřel intrauterinně cca v 6.–7. měsíci těhotenství, druhý z bratrů zemřel časně postnatálně. Dosud se obě úmrtí vysvětlovala jako pupečníková komplikace blíže neurčená. Dnes je zřejmé, že se v jejich případě velmi pravděpodobně jednalo také o X-vázanou myotubulární myopatii. Matka má ještě jednoho mladšího zdravého bratra (II:4), ten tedy s vysokou pravděpodobností není nosičem mutace c.82delA, nebyl ale k dispozici k vyšetření na DNA úrovni. Sestra obou pacientů (III:1) je taktéž zdráva a zatím (vzhledem k věku) nebyla vyšetřována na přítomnost uvedené mutace. Toto vyšetření však určitě doporučujeme v období před plánováním potomstva pro vysoké riziko 12,5 % pro postižení svých synů také XLMTM. V případě, že by se prokázalo, že je také přenašečkou uvedené mutace (pravděpodobnost 50 %), může jí být rovněž nabídnuta cílená genetická prevence.
Závěr
Dle našich vědomostí se jedná o první případy XLMTM, které byly molekulárně geneticky prokázány v ČR. Toto vyšetření MTM1 genu, které dosud nebylo v ČR dostupné, lze nyní provést v naší DNA laboratoři i pro další rodiny. Doporučujeme XLMTM zahrnout do diferenciální diagnostiky, když se jedná o závažné onemocnění kosterních svalů u chlapců v novorozeneckém věku, kteří mají generalizovanou svalovou slabost a hypotonii vyžadují ventilační podporu a mají normální hodnoty CK v séru. Důležitý je také nález ve svalové biopsii s nálezem centrálně uložených jader.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 25. 6. 2012
Přijato do tisku: 25. 10. 2012
MUDr. Petra Laššuthová, Ph.D.
DNA laboratoř
Klinika dětské neurologie
2. LF UK a FN v Motole
V Úvalu 84
150 06 Praha 5
e-mail: petra.lassuthova@gmail.com
Práce byla podpořena grantem IGA MZ NS 11521-4 a projektem koncepčního rozvoje výzkumné organizace 00064203.
Zdroje
1. Romero NB, Bitoun M. Centronuclear myopathies. Semin Pediatr Neurol 2011; 18(4): 250–256.
2. Romero NB. Centronuclear myopathies: a widening concept. Neuromuscul Disord 2010; 20(4): 223–228.
3. Bijarnia S, Puri RD, Jain M, Kler N, Roy S, Urtizberea JA et al. Mutation studies in X-linked myotubular myopathy in three Indian families. Indian J Pediatr 2010; 77(4): 431–433.
4. Tosch V, Vasli N, Kretz C, Nicot AS, Gasnier C, Dondaine N et al. Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul Disord 2010; 20(6): 375–381.
5. Tachi N, Kozuka N, Chiba S, Miyaji M, Watanabe I. A double mutation in a patient with X-linked myotubular myopathy. Pediatr Neurol 2001; 24(4): 297–299.
6. McEntagart M, Parsons G, Buj-Bello A, Biancalana V, Fenton I, Little M et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord 2002; 12(10): 939–946.
7. Tanner SM, Schneider V, Thomas NS, Clarke A, Lazarou L, Liechti-Gallati S. Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients. Neuromuscul Disord 1999; 9(1): 41–49.
8. ExonPrimer [online]. Cited 2010-09-10. Available from: http://ihg.gsf.de/ihg/ExonPrimer.html.
9. PubMed [online]. Cited 2011-09-10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/.
Štítky
Detská neurológia Neurochirurgia NeurológiaČlánok vyšiel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 2
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Tramadol a paracetamol v tlumení poextrakční bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
Najčítanejšie v tomto čísle
- Creutzfeldtova-Jakobova choroba
- Spinocerebelární ataxie typ 7 (SCA7) – kazuistika
- Lymeská borelióza jako příčina bilaterální neuroretinitidy s výraznou jednostrannou hvězdicovitou makulopatií u osmileté dívky
- Elektrofyziologické vyšetření pánevního dna