
Strategie diagnostiky, terapie a dispenzarizace pacientů s hemangioblastomy v CNS z pohledu neurochirurga
A Strategy for Diagnosis, Therapy and Follow-up of Patiens with CNS Haemangioblastoma from the Perspective of a Neurosurgeon
The aim of our study
was to establish a strategy of diagnosis, treatment and follow-up of patients after surgery for CNS haemangioblastoma (HB) and to evaluate its use in our group of patients. Primarily, it was necessary to distinguish between patients with sporadic HBs and those with von Hippel-Lindau disease (mVHL). Originally, the diagnosis of this genetically transmitted disease was made based on clinical findings only, including a combination ofHB presence and mVHL-associated tumours and both familiar and multilocal or recurrent HBs. This diagnostic approach resulted in many false positive as well as false negative conclusions. The introduction of VHL gene DNA analysis into clinical practice facilitated unequivocal identification of patients with mVHL. We then focused on patient screening. If mVHL was not proved and post-surgical MRI was negative, no further follow-up was indicated. In the presence of positive germinal VHL gene mutation and absence of other CNS lesions, MRI was repeated every 12 months. In case of newly identified CNS HBs not indicated for surgery, MRI is performed and surgical treatment reconsidered every six months. It depends on the patient’s clinical condition, cyst and solid tumour volume and, in particular, on tumour location. Clinical signs are also influenced by neoplasm growth rate and character. This strategy has been extrapolated to mVHL-associated extraneuronal tumours. CNS HB was the only manifestation in a substantial number of patients with confirmed VHL gene mutation. Surgical approach tumour location and characteristics are of major importance. A DNA analysis-based follow-up strategy was designed and applied to all patients with histologically verified HB. The goal is to detect other signs of mVHL before they are manifested clinically.
Key words:
haemangioblastoma – von Hippel‑ Lindau disease – genetic testing
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autori:
O. Kalita 1; M. Vaverka 1; L. Hrabálek 1; M. Zlevorová 2; A. Šantavá 3; A. Křepelová 4; J. Ehrmann Jr 5; E. Čecháková 6
Pôsobisko autorov:
Neurochirurgická klinika LF UP a FN Olomouc
1; Onkologická klinika LF UP a FN Olomouc
2; Oddělení klinické genetiky LF UP a FN Olomouc
3; Ústav biologie a lékařské genetiky 2. LF UK a FN v Motole, Praha
4; Laboratoř molekulární patologie, Oddělení patologie LF UP a FN Olomouc, Ústav molekulární a translační medicíny (BIOMEDREG)
5; Radiologická klinika LF UP a FN Olomouc
6
Vyšlo v časopise:
Cesk Slov Neurol N 2013; 76/109(5): 623-629
Kategória:
Krátké sdělení
Súhrn
Cílem práce bylo stanovit strategii diagnostiky, léčby a dispenzarizace pacientů po operaci v CNS lokalizovaných hemangioblastomů a zhodnotit její aplikaci na souboru našich pacientů. Nejprve bylo nutné odlišit pacienty se sporadickými hemangioblastomy od pacientů s von Hippel‑ Lindauovou (VHL) chorobou. Tato geneticky podmíněná choroba se dříve diagnostikovala pouze na základě klinického nálezu (výskyt kombinace hemangioblastomů a extraneurálních nádorů spojených s VHL, familiární výskyt hemangioblastomů, existence mnohočetných či recidivujících hemangioblastomů v CNS), což vedlo k mnoha falešně negativním i pozitivním závěrům. Zavedení DNA analýzy VHL genu do klinické praxe umožnilo jednoznačnou identifikaci pacientů s VHL chorobou. Následně bylo nutné zajistit skríning pacientů. V případě, že VHL nebyla potvrzena a pooperační MR byla negativní, další vyšetření již nebyla indikována. Při průkazu zárodečné mutace VHL genu a negativním nálezu v CNS opakujeme MR 1krát ročně. U nově objevených, k operaci neindikovaných hemangioblastomů v CNS provádíme MR každých šest měsíců s opakovaným zvažováním chirurgického výkonu. Vše závisí na klinickém stavu pacienta, velikosti cysty i solidní části a především na lokalizaci nádorů. Klinický projev je ovlivněn také rychlostí a charakterem růstu hemangioblastomů. Uvedené postupy byly extrapolovány také na extraneuronální nádory spojené s VHL chorobou. U většiny pacientů v našem souboru, u nichž se potvrdila mutace VHL genu, byl hemangioblastom v CNS jediným projevem VHL choroby. Pro chirurgické postupy byla podstatná lokalizace a charakter nádoru. S využitím DNA analýzy jsme vytvořili strategii dispenzarizace, kterou využíváme u všech pacientů po operaci histologicky verifikovaného hemangioblastomu. Cílem je zachytit vznik dalších, klinicky němých projevů VHL choroby ještě před jejich manifestací.
Klíčová slova:
hemangioblastom – von Hippel‑ Lindauova choroba – genetické vyšetření
Úvod
Hemangioblastom je pomalu rostoucí, benigní, intra‑ axiální nádor (WHO gr. I), jenž postihuje stejným poměrem obě pohlaví. Tvoří 0,9 až 2,1 % všech nádorů CNS; z toho 7– 12 % nádorů zadní jámy lební a 1,8– 3 % intraspinálních nádorů. Přibližně 35 % hemangioblastomů je solidních a 65 % cystických, s typicky excentricky uloženým, solidním tumorózním uzlem [1,2].
Hemangioblastomy se v 75– 80 % případů vyskytují jako sporadické a v 20– 25 % jako hereditární, v rámci von Hippel‑ Lindauovy (VHL) choroby. VHL choroba je autozomálně dominantně geneticky podmíněné onemocnění popsané v roce 1904 E. von Hippelem a v roce 1924 A. Lindauem [1,2]. Incidence v populaci se pohybuje v rozmezí 1 : 35 000 až 1 : 45 000 a choroba se manifestuje prakticky v 100 % do 65 let. Oba typy hemangioblastomů se liší i věkem manifestace; sporadický typ se obvykle vyskytuje mezi 45. a 50. rokem, zatímco hemangioblastomy pojící se s mVHL už mezi 29. a 30. rokem života. Prvním klinickým projevem mVHL bývá v 40– 68 % hemangioblastom retiny a očekávaná délka života nemocných se uvádí mezi 40 a 50 lety [2– 4].
Cílem naší práce bylo stanovit postup identifikace nemocných s mutací VHL genu a také vytvořit strategii jejich dalšího sledování a léčby.
Materiál a metodika
Pacienty po operaci hemangioblastomů v CNS jsme sledovali od srpna 2007 do prosince 2009 retrospektivně a od ledna 2009 do prosince 2012 prospektivně. Doba jejich pooperačního sledování byla nejméně 12 měsíců.
Všem pacientům po operaci byl proveden základní skríning k odhalení projevů VHL choroby. Nemocní podstoupili vyšetření očního pozadí, ultrazvukové vyšetření (UZ) dutiny břišní a MR zbývajících částí CNS. V případě pozitivního UZ nálezu v abdominální dutině bylo indikováno CT či MR s kontrastem, s následným urologickým, chirurgickým, popř. endokrinologickým vyšetřením. Všem nemocným bylo také provedeno genetické vyšetření. Pomocí PCR (Polymerase Chain Reaction) s přímým sekvenováním (MLPA, kit P016, MRC Holland) byla zjišťována přítomnost somatické mutace genu VHL. U pacientů s pozitivní genetickým nálezem bylo, vzhledem k 50% riziku penetrace do další generace, doporučeno vyšetření příbuzných. Ti, kteří vyšetření podstoupili, byli do studie zahrnuti.
K operační revizi jsme indikovali všechny klinicky manifestní hemangioblastomy. Ve dvou případech jsme se rozhodli k operaci klinicky němých, intramedulárních hemangioblastomů o velikosti 10 mm, resp. 15 mm. Obecně jsme opakovaně zvažovali exstirpaci nádoru velikosti nad 5 mm. Důležitým kritériem byla i lokalizace tumoru, charakter růstu, perifokální edém a poměr cysty i solidní části nádoru. Existovaly dva póly rozhodovacího procesu. Více než 10 mm velký, klinicky manifestní hemangioblastom s velkou doprovodnou cystou, s významným perifokálním edémem uložený periferně v mozečkové hemisféře byl jednoznačně indikován k operaci. Naopak solidní, klinicky němý hemangioblastom, do 5– 7 mm velikosti, s minimálním či žádným perifokálním edémem uložený v mozkovém kmeni byl indikován k dispenzarizaci (pacient 3 a 6). Mezi oběma hemangioblastomy však existoval široký přechod nálezů vyžadující individuální přístup. Jak již bylo řečeno, hemangioblastomy nemají pravidelný růst a v případě potvrzení skokového zvětšení u sledovaného hemangioblastomu jsme se rozhodli pro operaci ložisek i v kritických strukturách jako u pacienta 6, který podstoupil exstirpaci solidního, intraspinálního hemangioblastomu. Autoři prohlašují, že studie na lidských subjektech popsaná v manuskriptu byla provedena v souladu s etickými standardy příslušné komise (institucionální a národní) odpovědné za provádění klinických studií a Helsinskou deklarací z roku 1975, revidovanou v r. 2000.
Výsledky
Na základě našich kritérií vznikla skupina 17 pacientů. Operováno bylo 15 pacientů, z toho 14 pro hemangioblastomy v CNS (tab. 1). Všichni naši pacienti (až na dva, kteří již zemřeli) podstoupili DNA analýzu. VHL choroba se potvrdila u devíti z nich. Genetická analýza odhalila u čtyř nemocných úplnou deleci VHL genu (za podmínky ztráty heterogozity je gen zcela nefunkční), u čtyř deleci exonu 1 (poškození čtení genu, kdy výsledný protein je defektní, neaktivní, méně aktivní, neodbouratelný, s poškozeným rozeznáváním vazebných oblastí apod.) a u jedné pacientky bodovou mutaci (vymizení nebo záměna jednoho nukleotidu v sekvenci DNA s následnou změnou nebo posunem „čtecího rámce“; dle uložení změny je výsledkem buď úplné chybění proteinu, nebo různě funkčně postižený protein).
V tab. 1 mají probandi z jedné rodiny vždy stejné pořadové číslo. Byly vysledovány čtyři generace rozvětvené rodiny zatížené výskytem hemangioblastomů v CNS (tab. 1). Na počátku stála pacientka 1a její syn 1a, kteří byli opakovaně operováni pro v CNS lokalizované hemangioblastomy. Oba zemřeli na komplikace této choroby. Protože je při hodnocení přenosu mutace VHL genu v této rodině považujeme za klíčové, byli i oni zařazeni do studie. Vyšetření VHL genu u nich nebylo provedeno a diagnóza VHL choroby byla u nich stanovena na základě klinického nálezu. DNA analýza u ostatních členů rodiny potvrdila mutaci VHL genu. Pacientka 1b (je dcerou pacientky 1 a sestrou pacienta 1a) jako jediná nemá CNS projevy VHL choroby, ale je po nefrektomii pro renální karcinom. Synové (1baa, 1bab) pacientky 1ba podstoupili genetické vyšetření mutace VHL genu, oba s pozitivním závěrem, ale zatím jsou bez klinických i radiologických projevů této nemoci.
Pouze pacienti 3 a 6 jsou sledováni pro výskyt mnohočetných extraneurálních nádorů pojících se s VHL chorobou. U nemocného 1c jsme během skríningu zachytili vývoj dvou nových, solidních supratentoriálních tumorózních ložisek, zatím do 4 mm velikosti. Nemocný 6 podstoupil tři operace hemangioblastomů v různých částech CNS a skríning objevil další, v současnosti klinicky němé, intra‑ i extraneurální nádory, které jsou předmětem dalšího sledování. Pouze u nemocné 3 se nemoc nejprve manifestovala retinálním hemangioblastomem a poté i vývojem dalších nádorů. Retinální ložisko bylo léčeno gama nožem, přesto pacientka postupně oslepla. Na našem pracovišti podstoupila operaci cystického cerebelárního hemangioblastomu.
V našem souboru byl průměrný věk manifestace VHL hemangioblastomů 28 let a sporadických hemangioblastomů 42,7 let, což odpovídá deklarovanému věkovému rozmezí výskytu u obou podtypů.
U sedmi pacientů s potvrzenou mutací VHL genu byly intraneurální hemangioblastomy prozatím jedinými projevy této choroby. Naopak u žádného pacienta bez prokázané mutace VHL genu nebyly nalezeny další intra‑ či extraneuronální léze vázané na VHL chorobu.
Oba typy hemangioblastomů se nelišily ve výsledné chirurgické morbiditě a mortalitě. Žádný pacient nezemřel v souvislosti s operačním výkonem. Pacient 5 má od operace intramedulárního hemangioblastomu stacionární frustní hemiparézu. Nebyla u něj potvrzena VHL choroba. Opakované kontrolní MR po dobu čtyř let ukazují neměnný nález drobného rezidua nádoru. Dva pacienti 3 a 6 s prokázanou mutací VHL genu byli operováni pro solidní hemangioblastomy lokalizované v mozkovém kmeni a intramedulárně. U obou jsme zaznamenali pouze přechodný pooperační neurologický deficit: dystaxe horní končetiny, resp. obtíže s mikcí. Ostatní pacienti byli bez pooperační neurologického deficitu a výkony byly, dle kontrolních MR, chirurgicky radikální.
Diskuze
Sporadická forma hemangioblastomů je většinou solitární, vyskytuje se ve vyšším věku a je lokalizována v zadní jámě lební. S VHL chorobu spojené hemangioblastomy jsou spíše mnohočetné, lokalizované supratentoriálně, v retině, v mozkovém kmeni, míše a v periferních nervech [1]. Původně byla diagnóza VHL choroby stanovována pouze na základě klinického nálezu. Šlo o kombinaci hemangioblastomu v CNS, včetně retiny, a alespoň jednoho extraneurálního, na VHL chorobu vázaného nádoru. Taktéž sem byli řazeni nemocní s mnohočetným a familiárním výskytem hemangioblastomů [4]. Jak bylo v literatuře prokázáno, výskyt mnohočetných hemangioblastomů v CNS však není spolehlivým průkazem VHL choroby [5]. A jak ukázala naše pacientka 9, i kombinace věku a lokalizace nádoru není zcela signifikantní. Tato 44letá nemocná byla postižena kortikálně, supratentoriálně uloženým hemangioblastomem, a přesto mutace VHL genu nebyla prokázána. Vše se změnilo po zavedení genetické analýzy VHL genu do klinické praxe.
Autoři si jsou vědomi limitovaného počtu pacientů ve vlastním souboru. Ve vymezeném období bylo operováno na našem pracovišti 12 pacientů s hemangioblastomy v CNS. Zbývajících pět pacientů jsou příbuzní pacientů s pořadovým číslem 1.
Celkově u šesti operovaných pacientů (50 %) byla potvrzena mutace VHL genu, což je dvojnásobek deklarovaného výskytu VHL choroby. Za jeden z důvodů považujeme malý počet pacientů v souboru a existence dvou rodin (pořadové číslo 1 a 7) s VHL chorobou, u jejichž členů stejné generace (bratranec 1aa + sestřenice 1ba, sestra 7a + bratr 7b) se hemangioblastomy manifestovaly ve sledovaném období.
Dalším důvodem může být podhodnocení incidence VHL choroby. Někteří autoři [6] uvádějí, že mutace VHL genu u pacientů s hemangioblastomy v CNS se vyskytuje až v 40 %. Avšak recentní práce, které by se tímto na větších souborech zabývaly, chybí.
Problém je i nedůsledné provádění DNA analýzy u pacientů s nádory vyskytujícími se v rámci VHL choroby. Týká se to především pacientů s renálními karcinomy (viz naše pacientka 1b), nádory pankreatu apod. Z toho vyplývá potřeba vyšetřit pacienty i s extraneuronálními nádory patřícími k VHL chorobě, k čemuž by měla směřovat diskuze s kolegy dalších odborností.
Na straně druhé byla potvrzena vhodnost vyšetření mutace VHL genu u příbuzných pacienta s potvrzenou VHL chorobou. Jde však o složitý medicínsko‑ etický problém, a proto ne všichni příbuzní našich pacientů byli toto vyšetření ochotni podstoupit.
Za pozoruhodný považujeme také fakt výskytu hemangioblastomu jako jediného projevu VHL choroby u velkého počtu našich pacientů. Dokonce pacienti 1 a 1a zemřeli na komplikace recidivujících hemangioblastomů bez vzniku jiné, na VHL chorobu vázané patologie.
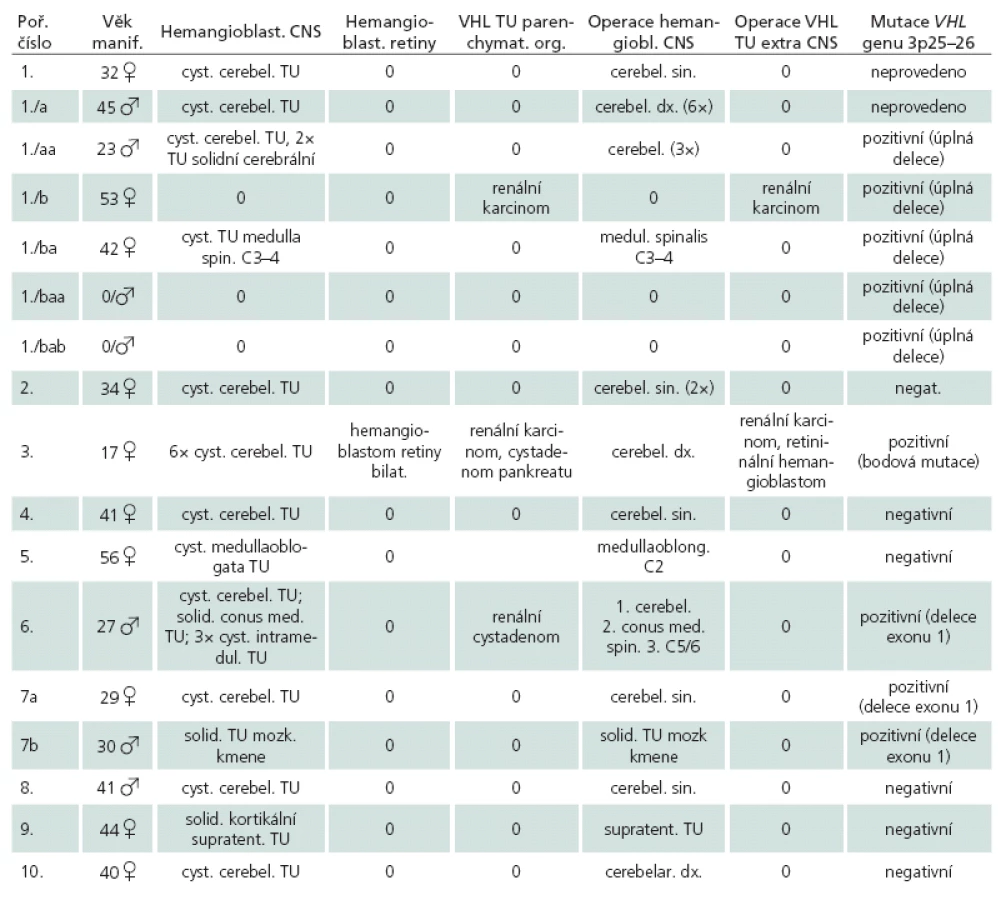
Ve vlastním histologickém obraze hemangioblastomu dominuje bohatá cévní tkáň, doplněná o vlastní polygonální, stromální, tumorózní buňky (obr. 1). Jejich původ je dosti kontroverzní; uvažuje se o vztahu ke gliálním, endoteliálním a arachnoideálním buňkám, k embryonálnímu choroideálnímu plexu, ale taktéž k neuroendokrinním, fibrohistiocytárním buňkám apod. Nicméně schopnost nádorových buněk produkovat proteiny typické pro hemoblastické zárodečné buňky, či potenciál nádoru vytvářet extramedulární ložiska hematopoézy a exprimovat erytropoetinové receptory spíše ukazují na příbuznost s hemoblastickými, progenitorovými buňkami [2].
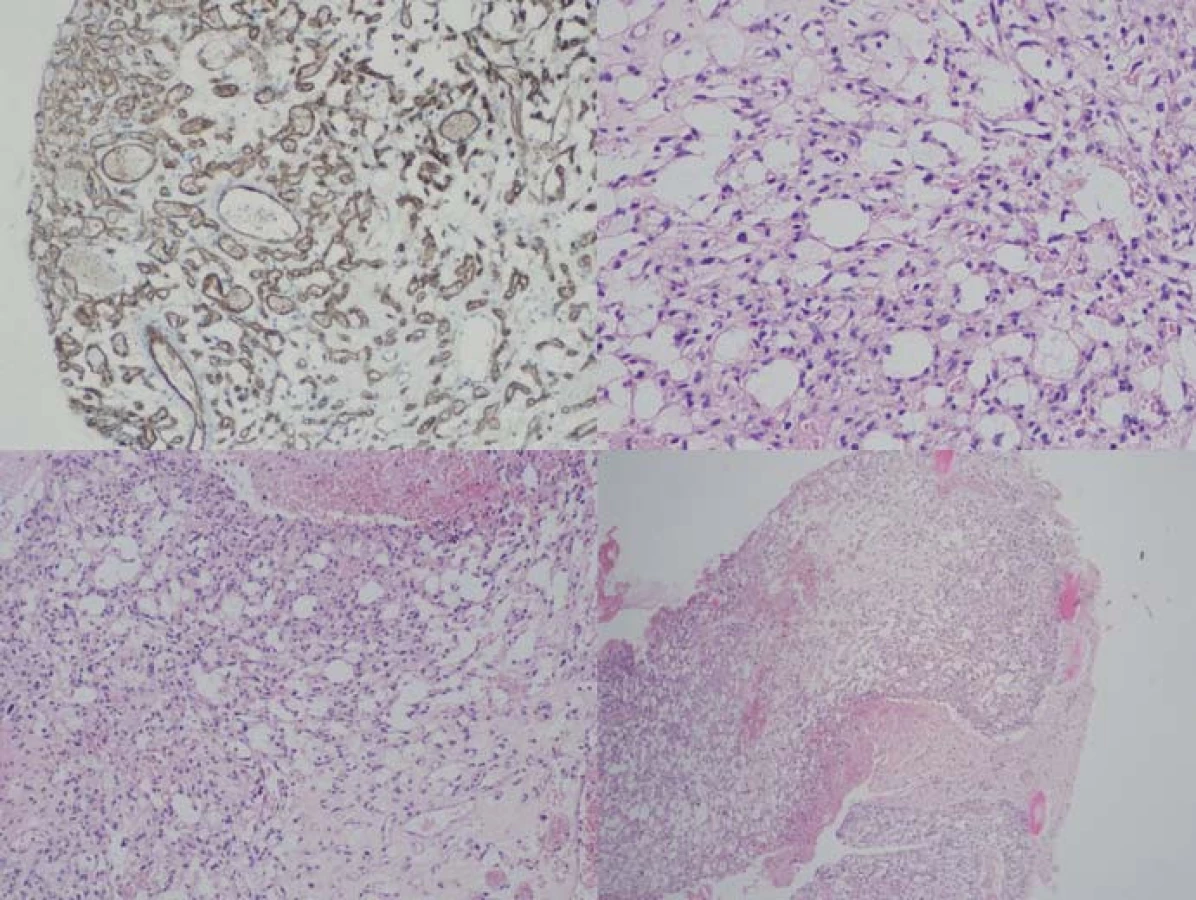
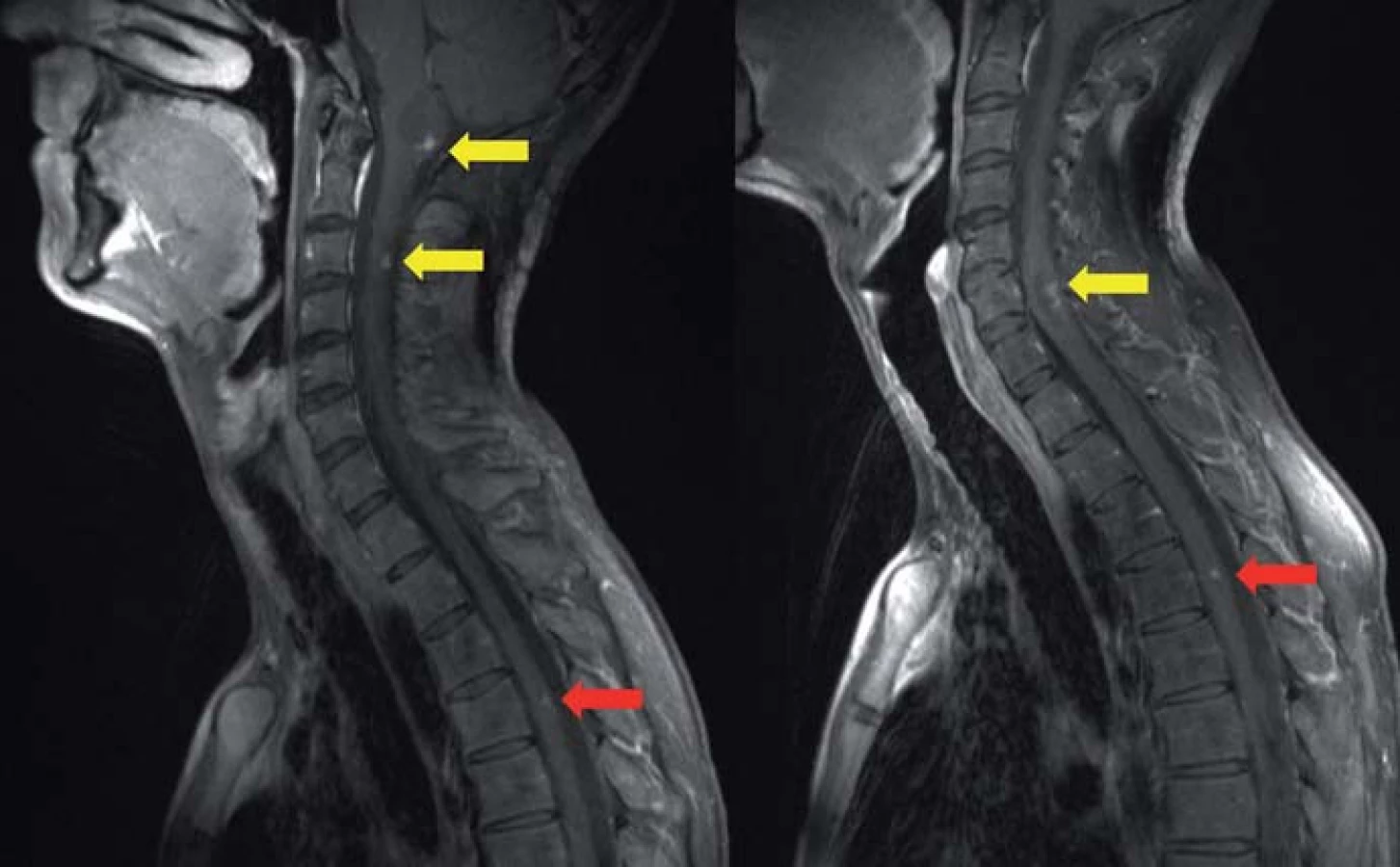
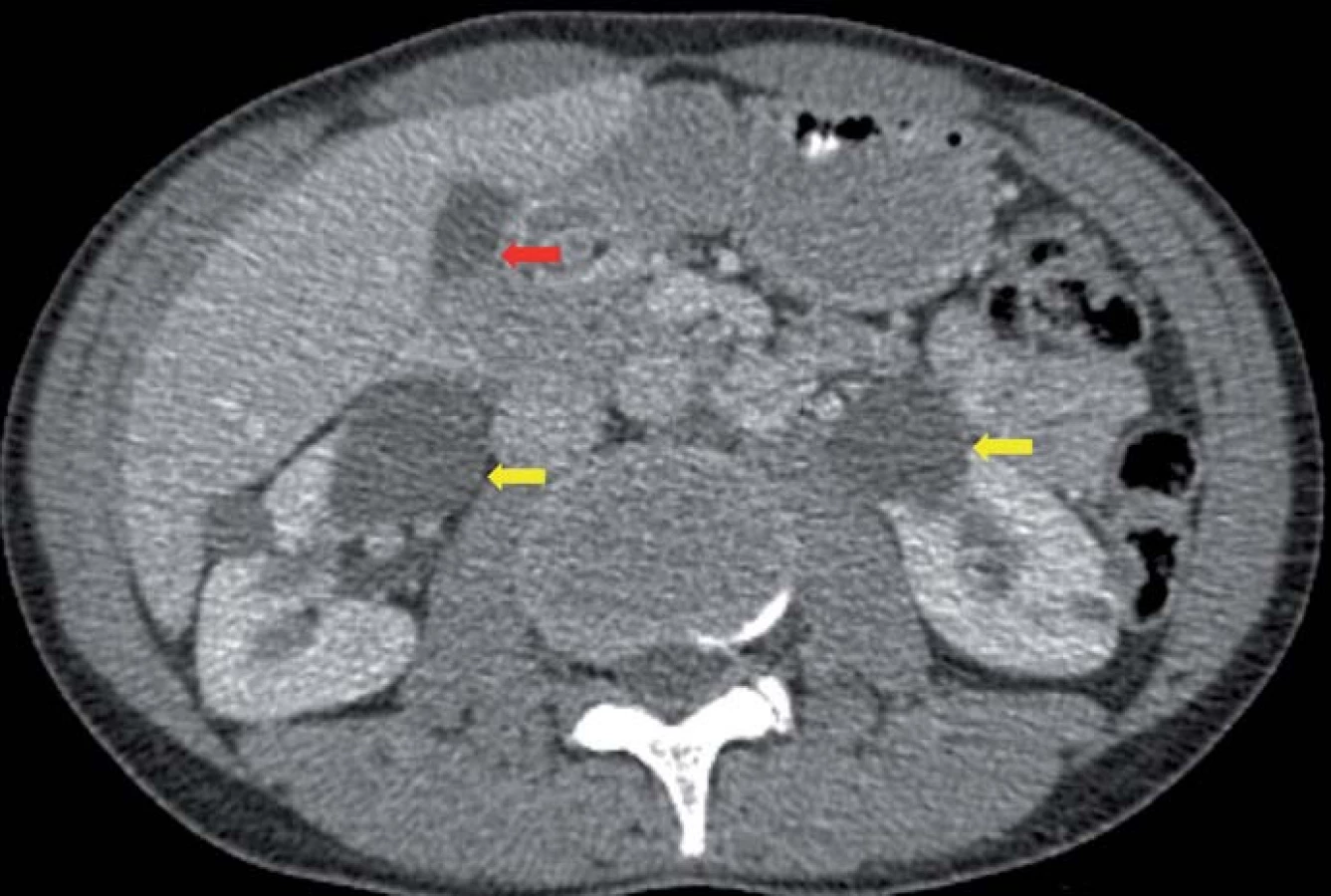
Genetickým podkladem vývoje onemocnění je mutace tumor‑ supresorového genu, lokalizovaného na krátkém ramenu chromozomu 3, v lokusu 25– 26. VHL gen je exprimován pouze v CNS, retině, periferních nervech, ledvinách, nadledvinách, pankreatu, v saccus a ductus endolymfaticus, v nadvarleti a adnexách. Nemocní jsou proto ohroženi jak vývojem recidiv a nových nádorových ložisek v CNS (obr. 2), popř. v retině, tak i vznikem specifických extraneurálních nádorů (obr. 3), jako jsou hemangioblastomy periferních nervů, karcinomy a cystadenomy ledviny, pankreatu, feochromocytomy nadledvin, paragangliomy, nádory saccus a ductus endolymfaticus, cystadenomy nadvarlete a adnex [1].
VHL gen ovlivňuje několik signálních drah buněčného cyklu. Především jde o regulaci exprese a degradace angiogenezi ovlivňujících proteinů, jako jsou např. hypoxií indukovaný faktor (HIF), VEGF, VEGFR, transformující růstový faktor (TGF). VHL gen také kontroluje transmembranózní pohyb oxidů uhlíku, a tím i extracelulární pH. Napodobuje stav tkáňové hypoxie, a tak indukuje novotvorbu cév. Toto je důvodem bohaté kapilární komponenty u všech tumorů spojených s VHL chorobou. Gen udržuje také hladinu inhibitorů metaloproteináz, a tím usnadňuje šíření nádorových buněk mezibuněčným prostorem. V neposlední řadě gen reguluje blokádu buněčného cyklu v G2 fázi a akumulaci inhibitorů proteinu 27. Porucha těchto regulačních funkcí je podkladem vývoje maligního renálního karcinomu.
Námi prováděné genetické vyšetření odhalí somatické postižení VHL genu na 3p25– 26, a tím heterozygotního nosiče mutace jednoho z jeho tří exonů. Pro rozvoj VHL choroby je však nutná ztráta heterozygozyty, tedy bialelická inaktivace genu [1,2]. První, germinální mutace vzniká na podkladě vrozeného defektu. Změna u druhé alely vzniká během života a je detekovatelná pouze v nádorové tkáni. Odhalení bialelických změn je však technicky velmi obtížné, protože vlastní tumorózní tkáň tvoří jen velmi malou část nádoru oproti cévní složce, a dále podstatné příměsi arteficiální, peritumorózní tkáně (makrofágy, reaktivní astrocyty apod.). Pro evoluci této dosti komplexní choroby však nejspíše budou nutné změny genů v jiných lokusech třetího či dalších chromozomů.

Na základě genetických změn se VHL choroba dělí na čtyři klinické subtypy. Základem je fenotypově‑genotypová korelace mezi výskytem hemangioblastomů, renálního karcinomu a feochromocytomu na jedné straně a missense mutace (mutace měnící řazení aminokyselin v peptidovém řetězci) a nonsense mutace (mutace zapříčiňující tvorbu předčasného terminačního kodonu a produkci afunkčních proteinů) v „hot spot“ kodonu 167, úplná delece exonu, splice acceptor mutace (mutace sestřihu RNA při přípravě pro přepis do proteinových řetězců) a nakonec kompletní a parciální delece VHL genu na straně druhé. Typ 1 je charakterizován výskytem hemangioblastomu, renálního karcinomu a zcela minimálním výskytem feochromocytomu. Typ 2A je charakterizován výskytem hemangioblastomu, feochromocytomu a zcela minimálně renálního karcinomu. Typ 2B je charakterizován vysokým výskytem feochromocytomu a renálního karcinomu a typ 2C vysokým výskytem feochromocytomu. V budoucnosti bychom po zjištění typu genetické poruchy VHL genu mohli predikovat, kterým z nádorů se choroba manifestuje a tím směrem zaměřit svoji dispenzarizaci [1,2].
Otázkou zůstává genetický podklad vzniku sporadických hemangioblastomů. Protože mutace VHL genu byla v tomto případě nalezena jen asi v 18 % případů, uvažuje se o dalších genech lokalizovaných na krátkém ramenu třetího chromozomu [1].
Současný výzkum se zaměřuje obecně na odhalení genetického podkladu vzniku hemangioblastomů a vztahu mezi germinální mutací a vývojem VHL choroby.
Základní diagnostická metoda intraneuronálních hemangioblastomů je MR. V literatuře uváděnou angiografii, někdy doplněnou o embolizaci, jsme nebyli nuceni využít [6,7]. Postupy chirurgického řešení u sporadických i hereditárních hemangioblastomech odpovídají mikrochirurgickým standardům [3,4,8,9] v závislosti na lokalizaci nádoru. Podstatou je radikální odstranění solidní porce nádoru, bez resekce stěny cysty tvořené astroglií. V literatuře je popisována vyšší morbidita u solidních hemangioblastomů oproti cystické varietě nádoru. To odpovídá i naší zkušenosti. Důvodem je relativně jednodušší exstirpace nádoru při využití koridoru tvořeného nádorovou cystou [4,7–12]. Ošetření gama nožem jsme u našich pacientů neindikovali. Jedna pacientka podstoupila radiochirurgii retinálního hemangioblastomu ještě před příchodem na naše pracoviště. Opravdu dlouhodobá kontrola růstu takto ošetřeného nádoru, obzvláště při koexistující cystě, je dosti problematická [13,14]. O této terapii, dle našeho názoru, lze uvažovat pouze pro ošetření klinicky manifestních a opravdu chirurgicky nedostupných hemangioblastomů. Využití profylaktické radiochirurgie/ radioterapie u vícečetných hemangioblastomů v CNS, i přes některé opačné názory, nepovažujeme za indikované [15].
Na základě těchto zjištění jsme vytvořili strategii přístupu k pacientům po neurochirurgické operaci histologicky verifikovaných hemangioblastomů. Vzhledem k dobré dostupnosti každý nemocný podstoupí vyšetření očního pozadí a UZ abdominální krajiny. Z důvodu již výše uvedené věkové distribuce a manifestace hemangioblastomů vázaných na VHL chorobu (bylo potvrzeno i naším souborem) jsou indikováni k DNA analýze především pacienti mladší 65 let. Ale i u starších pacientů o vyšetření doporučujeme uvažovat. K indikaci přispívá lokalizace hemangioblastomu v retině, intrakraniálně, supratentoriálně, v mozkovém kmeni a intraspinálně. Taktéž přítomnost recidivujících a mnohočetných hemangioblastomů v CNS, extraneurálních nádorů spojených s VHL chorobou a nakonec pozitivní rodinné anamnézy všech těchto patologií. Nepřítomnost mutace VHL genu u pacienta staršího 65 let s infratentoriálním hemangioblastomem však nelze stoprocentně vyloučit.
U pacientů bez prokázané zárodečné VHL mutace provádíme kontrolní MR za tři a 12 měsíců od operace. Pokud není přítomno reziduum či recidiva nádoru, další vyšetření neindikujeme. V případě rezidua nádoru zvažujeme reoperaci nebo sledování pomocí klinických a MR kontrol 1krát ročně.
U pacientů s prokázanou VHL mutací doplňujeme MR zbývající kraniospinální osy. Průměrná doba nutná pro vznik nové, na MR detekovatelné léze v CNS, se pohybuje okolo jednoho roku. Proto při negativním MR nálezu opakujeme vyšetření v této frekvenci [4].
U nově zjištěných, prozatím neoperovaných CNS tumorů provádíme MR každých šest měsíců, s opakovaným zvažováním operace. K operační revizi jsou indikovány všechny klinicky manifestní hemangioblastomy. U klinicky němých nádorů doporučujeme uvažovat o exstirpaci nádoru velikosti nad 5 mm. Přihlíží se ke klinickému stavu pacienta, velikosti cysty a solidní části nádoru a také lokalizaci nádoru. V mozečku je jako symptomatický uváděn hemangioblastom o objemu přibližně 69 mm3, v míše již o objemu 22 mm3 a v mozkovém kmeni o objemu 24,5 mm3. Na klinickém projevu se podílí rychlost a charakter růstu, kdy hemangioblastomy mají většinou výrazně nepravidelný, spíše skokový charakter růstu. Obecně se klinicky manifestují hemangioblastomy s průměrem velikosti nad 5 mm, v míše již od 3,5 mm [4,8,9,11]. V rámci multioborové spolupráce je aplikován obdobný postup na extraneurální léze. Sem patří opakovaná oční vyšetření, UZ, CT, MR abdominální krajiny, vyšetření katecholaminů a jejich metabolitů (feochromocytom) [16,17].
Závěr
U hemangioblastomů považujeme chirurgii za terapeutickou metodu první volby. Operované CNS hemangioblastomy byly často jediným projevem VHL choroby a další vývoj choroby je jen omezeně predikovatelný. V našem souboru jsme zjistili překvapivě vysoký výskyt mutace exonů VHL genu, což potvrdilo naše doporučení provádět genetickou analýzu VHL genu u všech pacientů po operaci hemangioblastomů v CNS. Vzhledem k 50% riziku penetrace do další generace všem pacientům s potvrzenou germinální mutací doporučujeme provést genetické vyšetření u příbuzných.
Snahou bylo vytvořit strategii multioborového (neurochirurg, genetik, onkolog, neurolog, radiolog, oftalmolog, urolog, chirurg, internista) sledování pacientů, která umožní zachytit vznik nových, klinických němých projevů VHL choroby ještě před jejich manifestací, a opakovaně zhodnotit další postup.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Ondřej Kalita, Ph.D.
Neurochirurgická klinika
LF UP a FN Olomouc
I. P. Pavlova 6
775 20 Olomouc
e-mail: ondrej.kalita@fnol.cz
Přijato k recenzi: 4. 2. 2013
Přijato do tisku: 13. 5. 2013
Zdroje
1. Plate KH, Vortmeyer AO, Zagzag D, Neumann HP. Von Hippel‑ Lindau disease and haemangioblastoma. In: Louis DN, Ohgaki H, Otmar D et al (eds). WHO Classfication of Tumours of the Central Nervous System. Lyon: IARC Press 2007: 215– 217.
2. Sanberg AA, Stone JF. Haemangioblastoma of the Central Nervous system. In: Sanberg AA, Stone JF (eds). The Genetics and Molecular Biology of Neural Tumors. Totowa, NJ: Humana Press 2008: 145– 164.
3. Jagannathan J, Lonser RR, Smith R, deVroom HL, Oldfield EH. Surgical management of cerebellar hemangioblastomas in patients with von Hippel‑ Lindau disease. J Neurosurg 2008; 108(2): 210– 222.
4. Ammerman JM, Lonser RR, Dambrosia J, Butan JA, Oldfield EH. Long‑term natural history of hemangioblastomas in patients with von Hippel‑ Lindau disease: implications for treatment. J Neurosurgery 2006; 105(2): 248– 255.
5. Ortego‑ Martínez M, Cabezudo FM, Fernández‑ Portales I, Pineda‑ Palomo M, Rodrígues‑ Sánches JA, Bernal‑ García LM. Multiple filum terminale hemangioblastomas symptomatic dutiny pregnancy. Case report. J Neurosurg Spine 2007; 7(2): 254– 258.
6. Niemelä M, Lemeta S, Summanen P, Böhling T, Sainio M, Kere J et al. Long‑term prognosis of haemangioblastoma of the CNS: impact of von Hippel‑ Lindau disease. Acta Neurochir 1999; 141(11): 1147– 1156.
7. Přibáň V, Fiedler J, Řehoušek P, Štěrba L, Štěpánková H, Křepelová A. Kombinovaná mikrochirurgická a endovaskulární terapie intramedulárního hemangioblastomu: kasuistika. Cesk Slov Neurol N 2007; 70/ 103(5): 580– 583.
8. Roonprapunt C, Silvera VM, Setton A, Freed D, Epstein FJ, Jallo GI. Surgical management of isolated haemangioblastomas of the spinal cord. Neurosurgery 2001; 49(2): 321– 328.
9. Symon L, Murota T, Pell M, Bordi L. Surgical management of haemangioblastoma of the posterior fossa. Acta Neurochir 1993; 120(3– 4): 103– 110.
10. Shin DA, Kim SH, Kim KN, Shin HC, Yoon DH. Surgical management of spinal cord haemangioblastoma. Acta Neurochir 2008; 150(3): 215– 220.
11. Zhou LF, Du G, Mao Y, Zhang R. Diagnosis and surgical treatment of brainstem hemangioblastomas. Surg Neurol 2005; 63(4): 307– 315.
12. Pietilä TA, Stendel R, Schilling A, Krznaric I, Brock M. Surgical treatment of spinal hemangioblastomas. Acta Neurochir 2000; 142(8): 879– 886.
13. Asthagiri AR, Mehta GU, Zach L, Li X, Butman JA, Camphausen KA, Lonser RR. Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel‑ Lindau disease. Neuro Oncol 2010; 12(1): 80– 86.
14. Valchář J, Liščák R, Šimonová G, Vymazal J. Hemangioblastom a jeho léčba pomocí Leksellova gama nože. Cesk Slov Neurol N 2008; 71/ 104(2): 216– 222.
15. Simone CB jr, Lonser RR, Ondos J, Oldfield EH, Camphausen K, Simone NL. Infratentorial craniospinal irradiation for von Hippel‑ Lindau: a retrospective study supporting a new treatment for patients with CNS hemangioblastomas. Neuro Oncol 2011; 13(9): 1030– 1036.
16. Smrcka M, Smrcka V. CNS haemangioblastoma: its role in von Hippel‑ Lindau disease. Bratisl Lek Listy 2000; 101(9): 503– 506.
17. Plevová P, Novotný J, Křepelová A. Von Hippel‑ Lindauova choroba. Klin Onkol 2009; 22 (Suppl): 23– 24.
Štítky
Detská neurológia Neurochirurgia NeurológiaČlánok vyšiel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 5
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Tramadol a paracetamol v tlumení poextrakční bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
Najčítanejšie v tomto čísle
- Wilsonova nemoc
- Multiformní glioblastom – přehled nových poznatků o patogenezi, biomarkerech a perspektivách léčby
- Tumoriformní varianta roztroušené sklerózy – dvě kazuistiky
- Test 3F Dysartrický profil – normativní hodnoty řeči v češtině