
Neurovývojová porucha s mentální retardací spojená s genem PPP2R5D – první případ v České republice
Autori:
K. Slabá 1; H. Pálová 2; P. Veselá 2; Š. Aulická 2,3; P. Konečná 1; M. Štěrba 1; P. Jabandžiev 1,2; O. Slabý 2,4; D. Procházková 1,5
Pôsobisko autorov:
Pediatrická klinika, LF MU a FN Brno
1; CEITEC, MU, Brno
2; Klinika dětské neurologie, LF MU a FN Brno
3; Biologický ústav, LF MU, Brno
4; Ústav lékařské genetiky a genomiky, LF MU a FN Brno
5
Vyšlo v časopise:
Cesk Slov Neurol N 2021; 84/117(2): 205-207
Kategória:
Dopis redakci
doi:
https://doi.org/10.48095/cccsnn2021205
Vážená redakce,
diferenciální diagnostika opožděného psychického a motorického vývoje u dětí je velmi široká a náročná. Příčinami psychomotorické retardace mohou být perinatální asfyxie vyúsťující do hypoxické encefalopatie, endokrinní poruchy, poruchy výživy, dědičné poruchy metabolizmu či degenerativní poruchy CNS a v neposlední řadě také řada genetických syndromů. Diferenciální diagnostika u genetických syndromů je často velmi obtížná vzhledem ke skutečnostem, že řada onemocnění s postižením intelektu nemá vždy specifický fenotyp a pro přesnou diagnózu je nezbytné genetické laboratorní testování. Naše pacientka byla vzhledem k neobjasněné příčině psychomotorické retardace a již vyčerpaným diagnostickým možnostem zařazena do studie k provedení celoexomového sekvenování. Teprve díky této komplexní genetické analýze se podařilo nalézt kauzální mutaci v genu PPP2R5D (PPP2R5D-related intellectual disability and neurodevelopmental delay, MIM 601646) a stanovit diagnózu. V současnosti je celosvětově popsáno přibližně 100 pacientů s tímto onemocněním [1]. V naší kazuistice představujeme první pacientku s touto diagnózou v ČR.
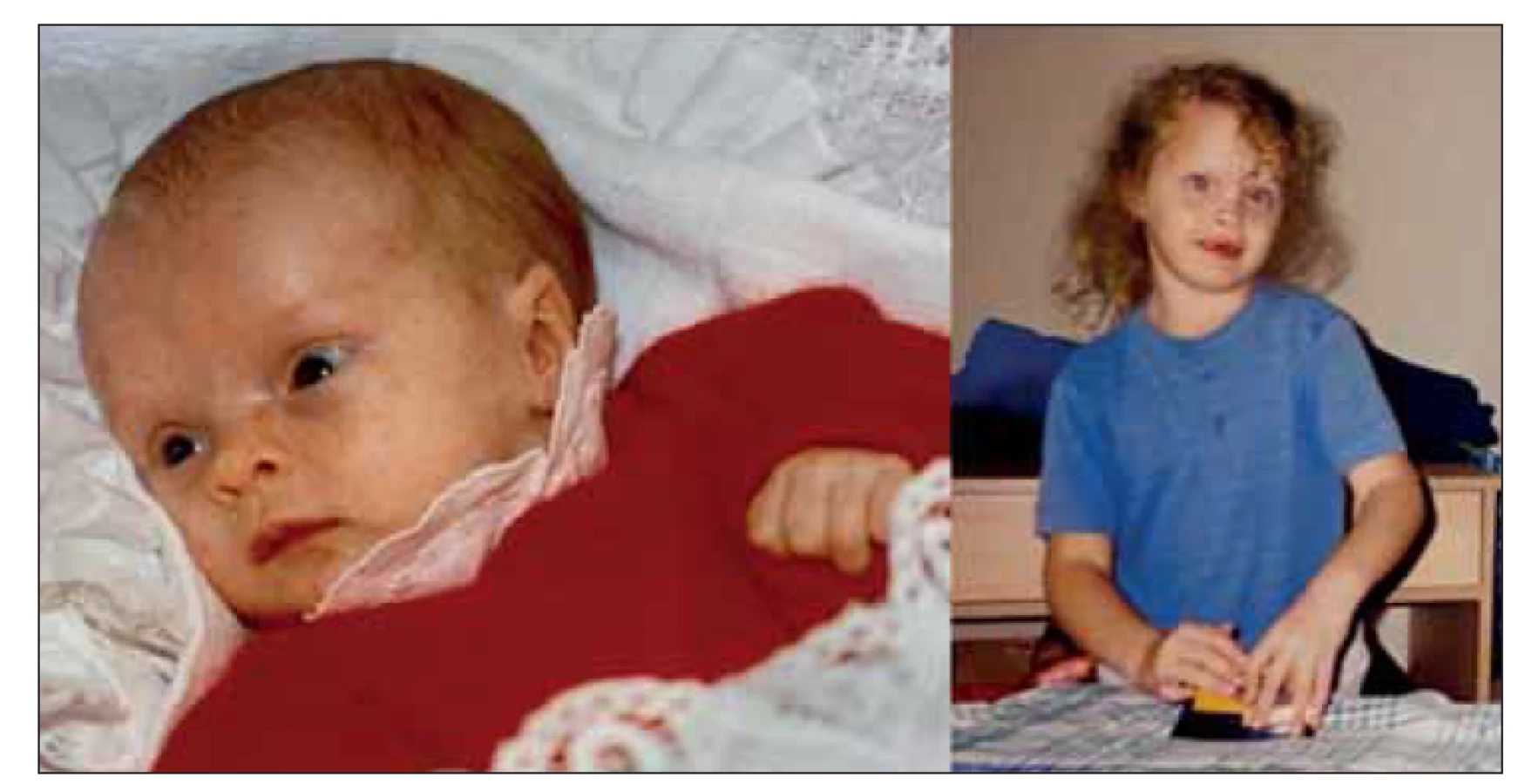
Dnes již 27letá žena se narodila v termínu jako eutrofický novorozenec. V 7. měsíci těhotenství byla matce provedena cerkláž děložního hrdla. Po porodu byla pro zkalenou plodovou vodu novorozenci nasazena antibiotická léčba. Rodinná anamnéza nevykazovala žádné pozoruhodnosti. Má jednoho staršího, nevlastního, zdravého sourozence. Od prvního týdne po narození byla sledována pro centrální hypotonický syndrom a novorozenecké křeče s dosažením plné kompenzace v 6 týdnech věku. Vzhledem k opakovanému výskytu hypoglykémie v kojeneckém věku bylo pomýšleno na dědičnou poruchu metabolizmu, avšak opakovaná biochemická, hematologická i specifická laboratorní vyšetření byla bez významných patologických nálezů. Na CT mozku byla v kojeneckém věku popsána lehká atrofie mozku, pozdější vyšetření MR tento nález nepotvrdilo. U pacientky byla od novorozeneckého věku pozorována kraniofaciální dysmorfie – široké vysoké čelo, prominence frontálních hrbolů, makrocefalie, hypertelorizmus, strabizmus, nízce posazené uši a gotické patro (obr. 1). Byla sledována na ortopedii pro hyperkyfózu hrudní páteře a pedes planovalgi. Psychomotorický vývoj byl od počátku velmi zpomalený. I přes intenzivní rehabilitační péči byla schopna samostatné chůze až ve 3 letech věku. Stagnovaly mentální vývoj i vývoj řeči. Dle psychologického vyšetření v dospělém věku se výše intelektu pacientky pohybuje v pásmu těžké až hluboké mentální retardace (IQ < 35), pacientka není schopna verbální ani neverbální komunikace. Je zcela nesoběstačná a závislá na péči své matky. Od časné dospělosti rodiče pozorují prvky agresivního chování, poruchu spánku, bruxizmus a známky sociální fobie, např. obavu z doteku cizích lidí. Antropometrické parametry naší probandky byly v dětství v normě, finální výška postavy činí 163 cm, index tělesné hmotnosti 25,21, tj. je na hranici nadváhy. Vzhledem k neobjasněné příčině psychomotorické retardace byla pacientka zařazena do studie k provedení celoexomového sekvenování.
Fig. 1. Photographs of the patient taken when she was 4 months and 8 years old
provided by her parents. Craniofacial dysmorphic features include macrocephaly,
bossing forehead, hypertelorism, low set ears and strabism.
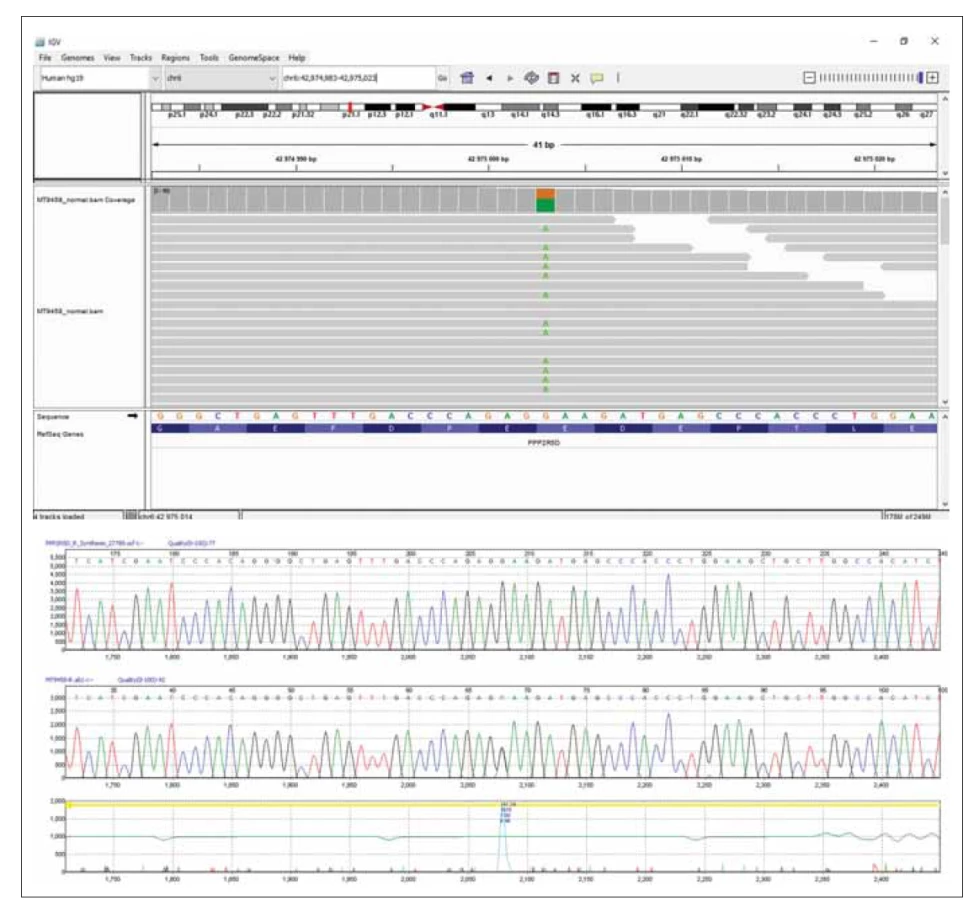
DNA pacientky byla izolována z periferní krve. Celoexomové sekvenování (Truseq Exome kit) bylo provedeno na plaftormě NextSeq 500 s využitím NextSeq 500/ 550 Mid Output Kit v2.5 (150 cycles) (vše Illumina, CA, USA). Minimálně 90 % cílových oblastí vykazovalo alespoň dvacetinásobné pokrytí. Identifikovali jsme heterozygotní variantu c.592G>A/ p.E198K v genu PPP2R5D (obr. 2A). Tato varianta byla již dříve popsána jako patogenní (třída 5) a způsobující neurovývojovou poruchu asociovanou s genem PPP2RD5, některými autory označovanou jako Jordanin syndrom (MIM 616355) [1,2]. U DNA pacientky a DNA jejích rodičů byla následně provedena přímá DNA analýza kodonu 198 exonu 5 genuPPP2R5D – polymerárová řetězová reakce následovaná Sangerovým sekvenováním (obr. 2B). V DNA ani jednoho z rodičů se patogenní varianta nevyskytovala. U pacientky byl potvrzen výskyt de novo varianty c.592G>A/ p.E198K v genu PPP2R5D v heterozygotním stavu. Tento nález umožnil stanovení diagnózy.
Fig. 2. Results of the sequencing analysis. (A) Exome sequencing of the patient’s DNA presenting pathogenic variant c.592G>A/p.
E198K in the PPP2R5D gene visualized using the Integrative Genomics Viewer software (Broad Institute and the Regents of the
University of California, USA). (B) Part of the PPP2R5D gene sequence showing heterozygous variant c.592G>A/p.E198K confi rmed
in the patient by Sanger sequencing.
Neurovývojová porucha s mentální retardací spojená s genem PPP2R5D je onemocnění s autozomálně dominantním typem dědičnosti vznikající na základě zárodečných de novo mutací v genu PPP2R5D a ovlivňující prenatální i postnatální vývoj CNS [3]. Některými autory je onemocnění nazýváno podle první diagnostikované pacientky Jordan Margaret Langové jako Jordanin syndrom (Jordan’s syndrome) [2]. Klinické příznaky zahrnují mentální retardaci různého stupně, centrální hypotonii, opoždění vývoje řeči a motorických dovedností. Vyskytovat se také mohou poruchy autistického spektra charakterizované zhoršenou komunikační zdatností a sociální interakcí [4]. Téměř 50 % pacientů trpí epileptickými záchvaty. Dále jsou u pacientů popisovány skeletální malformace, vývojové vady srdce a onemocnění očního aparátu. Typický je také nadměrný růst (overgrowth syndrome) a dysmorfické rysy – makrocefalie, hypertelorizmus, prominující čelo [5].
Gen PPP2R5D leží na chromozomu 6 a kóduje protein PPP2R5D složený z 602 amikokyselin, známý jako delta podjednotka B56 (B56δ), která je izoformou podjednotky B56 serintreoninové proteinfosfatázy 2A (PPA2). PPA2 je jedna ze čtyř hlavních serintreoninových fosfatáz a je zapojena do negativní kontroly buněčného růstu a dělení. Protein je tvořen společným heteromerním jaderným enzymem, který je složen z katalytické podjednotky a konstantní regulační podjednotky interagující s řadou regulačních podjednotek. Protein B56δ se nachází především v centrální nervové soustavě, kde hraje klíčovou roli při vývoji neuronů a regulaci neuronální signalizace [6]. Předpokládá se, že patogenní varianty genu PPP2R5D vedou ke tvorbě pozměněného proteinu B56δ, a tím snižují aktivitu PP2A. Abnormální aktivita PPA2 narušuje signální dráhy v neuronech a ovlivňuje jejich normální vývoj a funkci.
Varianta c.592G>A/ p.E198K v genu PPP2R5D (NM_006245.3, exon 5) je opakovaně popsána v databázích HGMD (CM153575) a ClinVar (RCV000202079.2) jako patogenní ve vztahu k vývojovým poruchám. Primárně byla popsána Fitzgeraldem et al v roce 2015 [7] a patogenita varianty byla potvrzena řadou dalších autorů, kteří ji popsali ve spojitosti s charakteristickými fenotypovými projevy (autizmus, overgrowth syndrome, opoždění řeči, hypotonie aj.) [1,3,8]. Prevalence tohoto onemocnění není zatím známá. Lze ji odhadnout ze studie Shanga et al, kteří identifikovali patogenní varianty v genu PPP2R5D pomocí celoexomového sekvenování u celkem 7 z 2 790 jedinců s neurovývojovou retardací, což v tomto případě činí 0,25 % [8].
Až u 50–75 % případů psychomotorické retardace se ani přes pokročilé diagnostické možnosti etiologie na individuální úrovni nepodaří objasnit [9]. U naší pacientky jsme stanovili diagnózu díky lepší dostupnosti necílených genomických metod, jako je celoexomové sekvenování. V současné době není u popsaného syndromu k dispozici žádná kauzální terapie. Stejně jako u jiných monogenně podmíněných chorob jsou však studovány možnosti genové terapie, která je do budoucna jedinou možnou kauzální léčbou [10].
Grantová podpora
Podpořeno MZ ČR – RVO (FNBr, 65269705).
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
doc. MUDr. Dagmar Procházková, Ph.D.
Pediatrická klinika
LF MU a FN Brno
Černopolní 9
625 00 Brno
e-mail: prochazkova.dagmar@fnbrno.cz
Přijato k recenzi: 9. 1. 2021
Přijato do tisku: 25. 2. 2021
Zdroje
1. Biswas D, Cary W, Nolta JA. PPP2R5D-related intellectual disability and neurodevelopmental delay: a review of the current understanding of the genetics and biochemical basis of the disorder. Int J Mol Sci 2020; 21(4): 1286. doi: 10.3390/ ijms21041286.
2. Jordan’s Guardian Angels. [online]. Available from URL: https:/ / jordansguardianangels.org/ the-history-of-jordans-syndrome/ .
3. Reynhout S, Jansen S, Haesen D et al. De novo mutations affecting the catalytic Ca subunit of PP2A, PPP2CA, cause syndromic intellectual disability resembling other PP2A-related neurodevelopmental disorders. Am J Hum Genet 2019; 104(1): 139–156. doi: 10.1016/ j.ajhg.2018.12.002.
4. Houge G, Haesen D, Vissers LE et al. B56d-related protein phosphatase 2A dysfunction identified in patients with intellectual disability. J Clin Invest 2015; 125(8): 3051–3062. doi: 10.1172/ JCI79860.
5. Loveday C, Tatton-Brown K, Clarke M et al. Mutations in the PP2A regulatory subunit B family genes PPP2R5B, PPP2R5C and PPP2R5D cause human overgrowth. HumMol Genet 2015; 24(17): 4775–4779. doi: 10.1093/ hmg/ ddv182.
6. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/ threonine phosphatases implicated in cell growth and signalling. Biochem J2001; 353(Pt 3): 417–439. doi: 10.1042/ 0264-6021:3530417.
7. Shang L, Henderson LB, Cho MT et al. De novo missense variants in PPP2R5D are associated with intellectual disability, macrocephaly, hypotonia, and autism. Neurogenetics 2016; 17(1): 43–49. doi: 10.1007/ s10048-015-0466-9.
8. Fitzgerald TW, Gerety SS, Jones WD et al. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015; 519(7542): 223–228. doi: 10.1038/ nature 14135.
9. Hladíková A, Grečmalová D, Černá D et al. Nové možnosti diagnostiky příčin mentální retardace u dětí. Pediatr praxi 2011; 12(6): 380–384.
10. Ilyas M, Mir A, Efthymiou S et al. The genetics of intellectual disability: advancing technology and gene editing. F1000Res 2020; 9: 22. doi: 10.12688/ f1000research.16315.1.
Štítky
Detská neurológia Neurochirurgia NeurológiaČlánok vyšiel v časopise
Česká a slovenská neurologie a neurochirurgie

2021 Číslo 2
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Tramadol a paracetamol v tlumení poextrakční bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
Najčítanejšie v tomto čísle
- Mortonova neuralgie, metatarzalgie
- Nemoc moyamoya
- Správná a chybná pojmenování obrázků pro náročnější test písemného Pojmenování obrázků a jejich vybavení (dveřní POBAV)
- Etiopatogeneze a diagnostika progresivní multifokální leukoencefalopatie u pacientů léčených natalizumabem