
Autofagické vakuolární myopatie – aneb co nás naučila diferenciální diagnostika vakuol ve svalové biopsii
Autophagic vacuolar myopathies: what we have learned from the differential diagnosis of vacuoles in muscle biopsy
Establishing the correct etiological diagnosis of autophagic vacuolar myopathies (AVM) is an imperative since the recent availability of the enzyme replacement therapy for the treatment of Pompe disease. Recent recognition of the complex functional pathology of these disorders dramatically changed the view on their pathogenesis and it may lead to identification of new approaches in the therapy. Muscle biopsy is a useful tool for the differential diagnosis of the AVM; however, in some cases it may fail.
Through a series of five short case studies we aim at demonstrating the histopathological findings and differential diagnosis of some AVM (Pompe disease, Danon disease and chloroquine myopathy) in muscle biopsy. We also want to address the fact that the way to the correct diagnosis of these disorders can be quite complicated.
Awareness of these diseases and the availability of a dry blood spot test for the non-invasive diagnosis of Pompe disease represent a good basis for detecting patients who are still kept under other diagnoses and thus escaping treatment.
Keywords:
autophagic myopathies – vacuoles – Pompe disease – Danon disease – chloroquine myopathy
Autori:
Josef Zámečník; Robert Artur Dahmen
Pôsobisko autorov:
Ústav patologie a molekulární medicíny, 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Čes.-slov. Patol., 49, 2013, No. 1, p. 39-45
Kategória:
Původní práce
Súhrn
Dostupnost efektivní enzymové substituční terapie pro léčbu Pompeho nemoci učinila ze stanovení správné etiologické diagnózy autofagických vakuolárních myopatií nutnost. Nedávný rozvoj poznání funkční patologie těchto poruch navíc zcela změnil pohled na jejich patogenezu a mohl by přinést nové přístupy v jejich léčbě. Svalová biopsie je dobrým nástrojem pro diferenciální diagnózu autofagických vakuolárních myopatií, v některých případech však může selhat.
V textu chceme ukázat na sérii pěti krátkých kazuistik histopatologický obraz a možnosti diferenciální diagnózy autofagických vakuolárních myopatií (Pompeho nemoc, Danonova choroba, chlorochinová myopatie) ve svalové biopsii. A také to, že cesta ke správné diagnóze těchto poruch může být poměrně komplikovaná.
Povědomí o těchto onemocněních a dostupnost neinvazivního testu suché krevní kapky pro diagnózu Pompeho choroby je dobrým předpokladem pro odhalení celé řady pacientů, kteří jsou zatím vedeni pod jinými diagnózami a přicházejí tak o možnost léčby.
Klíčová slova:
autofagické myopatie – vakuoly – Pompeho nemoc – Danonova nemoc – chlorochinová myopatie
Autofagie je vysoce regulovaným lysozomálním pochodem sloužícím k degradaci a obnově proteinů a poškozených nebo nepotřebných buněčných organel v cytoplazmě (1,2). Tento proces poskytuje buňce kromě “úklidu a obnovy” také dodatečnou energii (3). Je to jev zcela fyziologický a spontánní, ve zdravém svalovém vláknu je však morfologicky sotva zastižitelný. Vystupňovaný může být fyziologicky při nedostatku živin (3). Existuje však i dysfunkční autofagie, která doprovází řadu primárních lysozomálních poruch, stejně tak jako neurodegenerativní choroby, nádory, ale i různá zánětlivá onemocnění (2,4,5). Porucha lysozomálních funkcí vede v postižené buňce k tzv. autofagickému stresu (6), který doprovází patologická akumulace autofagických meziproduktů v cytoplazmě (7). Protože autofagie je fyziologicky vystupňovaná ve svalové (kosterní i srdeční) a nervové tkáni (8), bývají klinické a morfologické projevy lysozomálních dysfunkcí výrazné právě tam.
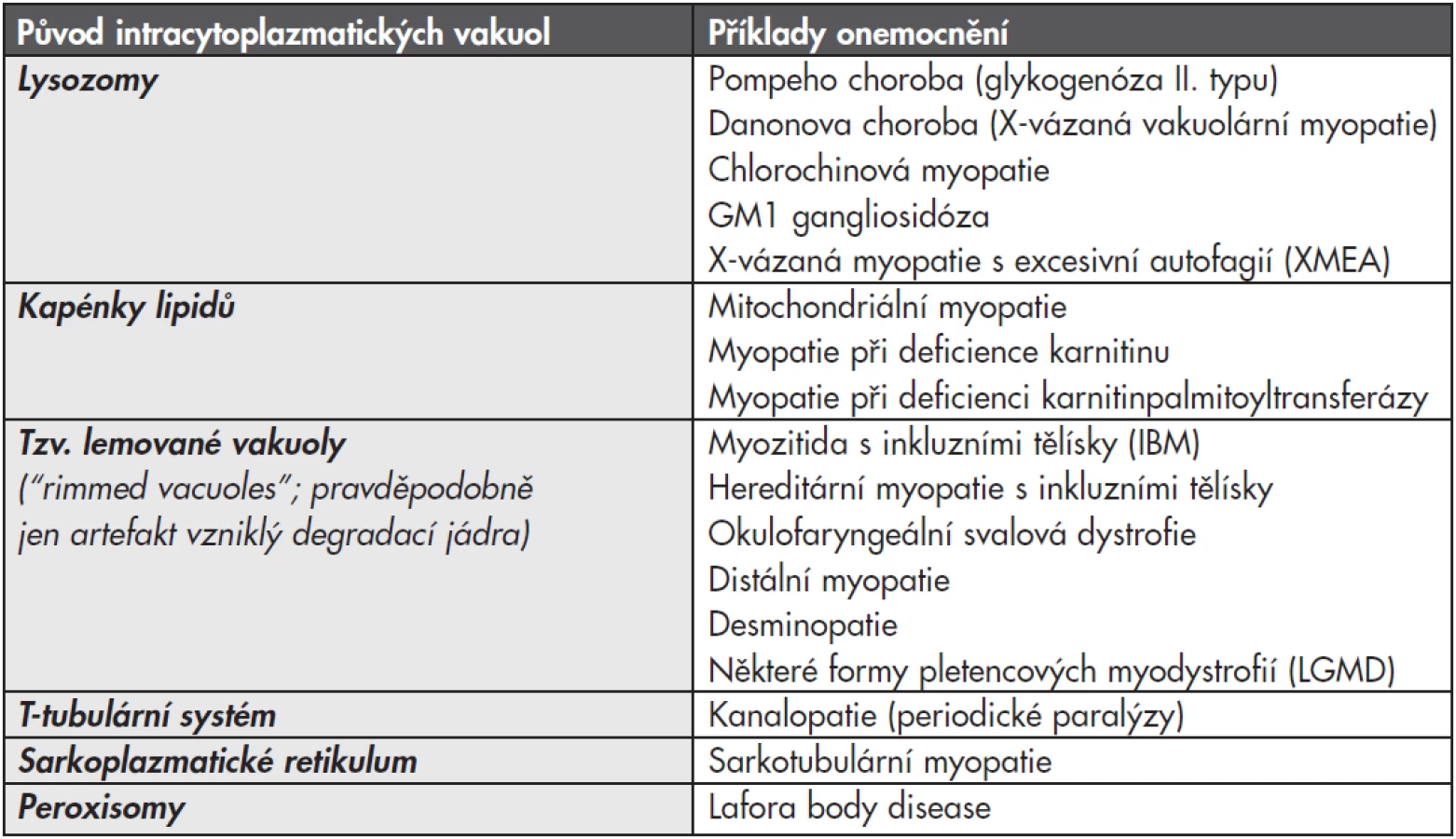
Myopatií, při kterých dochází k poruše autofagie z důvodu lysozomální dysfunkce, je celá řada a mají společný morfologický projev – vakuolizaci svalových vláken. Patří sem některé vrozené choroby (zejména Pompeho nemoc a Danonova choroba), ale podobný obraz může být způsobený i získanou poruchou, nejčastěji toxickým efektem některých léčiv, zejména antimalarik (tzv. chlorochinová myopatie). Vakuolizace svalových vláken však nemusí zdaleka vždy ukazovat jen na lysozomální poruchu (viz tabulka č. 1). Proto vyžaduje diagnostika vakuolárních myopatií ve svalové biopsii komplexní přístup s využitím enzymové histochemie, imunohistologie a elektronové mikroskopie; navíc jsou většinou nutná další vyšetření – enzymologická a molekulárně genetická.
Stanovení přesné etiologické diagnózy vakuolárních myopatií má dnes velký význam, který se poměrně nedávno přesunul z roviny spíše akademické do polohy čistě praktické, a to zejména kvůli recentně dostupné efektivní enzymové substituční léčbě (ERT, enzyme replacement therapy) Pompeho choroby (9). Navíc, i toxický efekt chlorochinů lze zastavit úpravou terapie.
V následujícím textu chceme ukázat na sérii krátkých kazuistik histopatologický obraz a možnosti diferenciální diagnózy autofagických vakuolárních myopatií ve svalové biopsii. A také to, že cesta ke správné diagnóze těchto poruch může být poměrně křivolaká.
KAZUISTIKY
Případ č. 1 (aneb typické případy Pompeho nemoci problémy nečiní)
17měsíčnímu batoleti mužského pohlaví, bez závažnější rodinné anamnézy stran neurologických a metabolických onemocnění, byla v šesti měsících věku náhodně zjištěna elevace jaterních transamináz. Po vyloučení jaterního onemocnění byla dále zjištěna elevace sérové kreatinkinázy (CK – 7,9 μkat/l, norma u batolat do 2,3 μkat/l ) a neurologické vyšetření odhalilo periferní hypotonický syndrom s mírně opožděným psychomotorickým vývojem. Elektromyografie (EMG) prokázala mírnou myogenní lézi, bez známek periferní neuropatie. S podezřením na kongenitální myopatii byla indikována svalová biopsie z m. tibialis anterior.
Svalová biopsie – velká část svalových vláken byla vyplněna různě velikými vakuolami, které byly buď opticky prázdné, nebo obsahovaly jemně bazofilní granula (obr. 1). Vakuoly reagovaly intenzivně v reakci na průkaz kyselé fosfatázy (ACP), což svědčilo pro jejich původ z lysozomů, a v reakci PAS na průkaz glykogenu. Na elektronmikroskopické úrovni odpovídaly vakuoly membránou obklopeným autofagickým vakuolám s heterogenním obsahem – některé obsahovaly jen glykogen, v jiných byly přítomny různé cytoplazmatické degradační produkty. Zmnožený glykogen jsme pozorovali i intracytoplazmaticky. Panel provedených enzymově histochemických vyšetření ani imunohistologie sarkolemálních proteinů neozřejmily jinou poruchu.




Na podkladě výše uvedeného jsme případ uzavřeli jakou autofagickou vakuolární myopatii a vyslovili podezření na deficit α-glukosidázy. Následně provedené enzymologické vyšetření diagnózu Pompeho choroby (glykogenózy II. typu) potvrdilo. Pacient je v současné době léčen ERT rekombinantní lidskou α-glukosidázou (rhGAA).
Případ č. 2 (aneb Danonova a Pompeho nemoc mají mnoho společných rysů, i když se jedná o zcela jiné poruchy)
28letý muž měl ve 13 letech věku diagnostikovánu hypertrofickou kardiomyopatii s významnou rodinnou zátěží (matka zemřela na srdeční selhání, u sestry již byla pro hypertrofickou kardiomyopatii provedena transplantace srdce). Pro postupný rozvoj srdečního selhávání byl indikován k transplantaci srdce. V rámci vyšetřování byla zjištěna elevace transamináz a CK. Ačkoli nebyl klinicky vyjádřený myopatický syndrom, EMG vyšetření odhalilo mírnou myogenní lézi. Pro upřesnění diagnózy tedy byla před transplantací provedena biopsie m. vastus lateralis.
Svalová biopsie – přítomen byl velmi mírný myopatický vzorec změn, zastiženo však bylo několik vláken s autofagickou vakuolizací, která elektronmikroskopicky odpovídala intralysozomálnímu střádání glykogenu a degradačních lysozomálních produktů. Vakuoly byly lemovány membránami, které reagovaly při imunohistochemickém průkazu sarkolemálních proteinů (dystrofin, dysferlin, sarkoglykany, merosin atp.) (obr. 2).





Na podkladě výše uvedeného jsme i tento případ uzavřeli jakou autofagickou vakuolární myopatii a opět vyslovili podezření na glykogenózu II. typu (m. Pompe). Provedená enzymologická vyšetření však deficit α-glukosidázy vyloučila. Laskavostí prof. Elledera bylo doplněno imunohistochemické vyšetření membránových proteinů lysozomálního systému a byla tak objevena absence proteinu LAMP-2 (lysosome-associated membrane protein-2) a paralelní molekulárně genetické vyšetření genu LAMP2 prokázalo patogenní mutaci. Byla tak stanovena diagnóza prvního českého případu X-vázané vakuolární myopatie (Danonova choroba).
Případ č. 3 (aneb normální nález ve svalové biopsii Pompeho nemoc nevylučuje)
30letý muž s anamnézou chronické hepatopatie byl před 6 lety vyšetřován v neurologické ambulanci pro chronické bolesti páteře a občasné vertigo. Později se objevily bolesti kloubů a svalů, pocit těžkých dolních končetin a zvýšená únavnost. Ačkoli byla zjištěna postupně se zvyšující hladina CK (v hladinách 8 – 9 μkat/l, norma do 3,24 μkat/l), vyšetření EMG myogenní lézi neprokázalo. Pro přetrvávání obtíží a elevaci svalových markerů byla provedena biopsie m. vastus lateralis.
Svalová biopsie však jednoznačné myopatické změny neozřejmila. Byla přítomna jen ojedinělá denervovaná angulární atrofická vlákna (svědčící pro možnou účast neurogenních vlivů na obtíže pacienta), panel enzymově histochemických i imunohistologických vyšetření neodhalil žádnou poruchu (obr. 3A,B). Ani pomocí elektronové mikroskopie jsme neidentifikovali žádné pozoruhodnosti.



Potíže pacienta však neustávaly a vyšetřování pokračovalo. Velikým překvapením pro nás bylo, že test suché krevní kapky na vyloučení Pompeho nemoci (provedený o rok později) přinesl diagnózu – enzymologicky byla potvrzena Pompeho choroba (glykogenóza II. typu) a pacient se dnes léčí ERT.
I přes relativně povzbudivý konec příběhu jsme se jen obtížně vyrovnávali se skutečností, že naše vyšetření neodhalila žádné známky této klinicky zřetelně manifestní choroby. Rozhodli jsme se tedy zpracovat v sériových řezech celý zbytek izopentanem zmraženého vzorku svalu, který byl archivován v hlubokomrazicím boxu. Po prokrájení jsme (ve více než 200 řezech) nakonec zastihli jediné vakuolizované vlákno (obr. 3C).
Případ č. 4 (aneb chlorochiny dokážou vyvolat obraz podobný Pompeho nemoci)
45leté ženě byl 3 roky před svalovou biopsií diagnostikovaný systémový lupus erythematodes (SLE) s pozitivitou antinukleárních protilátek, s kloubní a kožní symptomatologií, fotosenzitivitou, trombocytopenií a organickým psychosyndromem. Od té doby byla léčena glukokortikoidy a antimalariken hydroxychlorochinem. Poslední 3 měsíce se u ní rozvinula svalová slabost s bolestmi svalů horních i dolních končetin. EMG vyšetření myogenní lézi neprokázalo, byla však mírně zvýšená hladina CK (4,83 μkat/l, norma do 2,85 μkat/l). Pro vyloučení myotoxicity některého z léčiv či konkomitantní myozitidy byla indikována svalová biopsie z m. quadriceps femoris.
Svalová biopsie – přítomen byl mírný myopatický vzorec změn s kolísáním velikosti svalových vláken. Ve svalových vláknech jsme pozorovali zvýrazněnou granulární cytoplazmatickou pozitivitu kyselé fosfatázy (marker lysozomů), spíše fokálně jsme zastihli autofagické vakuoly (obr. 4). Nález jsme uzavřeli jako autofagickou vakuolární myopatii, etiologicky nejspíše toxickou - vlivem chlorochinového preparátu. Selektivní atrofii vláken II. typu, která by svědčila pro steroidní myopatii, ani známky autoimunitní zánětlivé myopatie, jsme nepozorovali.



Pacientce byl po stanovení diagnózy chlorochinové myopatie příslušný lék vysazen, krátce na to došlo k rychlé úpravě laboratorních parametrů a postupnému zlepšování svalové síly.
Případ č. 5 (aneb chlorochiny umějí vyvolat obraz podobný Pompeho nemoci… platí to ale bohužel i obráceně!)
48letá žena trpěla sedm měsíců před svalovou biopsií bolestmi zad v bederní páteři, bolestí kloubů a méně výraznou svalovou slabostí. Byla klinicky vedena jako případ seronegativní revmatoidní artritidy, pro kterou byla 5 měsíců léčena antimalarikem chlorochinem. Během toho však bylo zaznamenáno zvýšení sérové CK (kolem 12 μkat/l) a myoglobinu. Pro podezření z toxického efektu antimalarika, byl chlorochinový preparát dva měsíce před biopsií vysazen a nahrazen methylprednisolonem. Klinické obtíže však neustávaly a MRI vyšetření svalů navíc přineslo podezření na polymyozitidu. S klinickou diferenciální diagnózou polymyozitidy nebo chlorochinové myopatie byla provedena biopsie m. vastus lateralis.
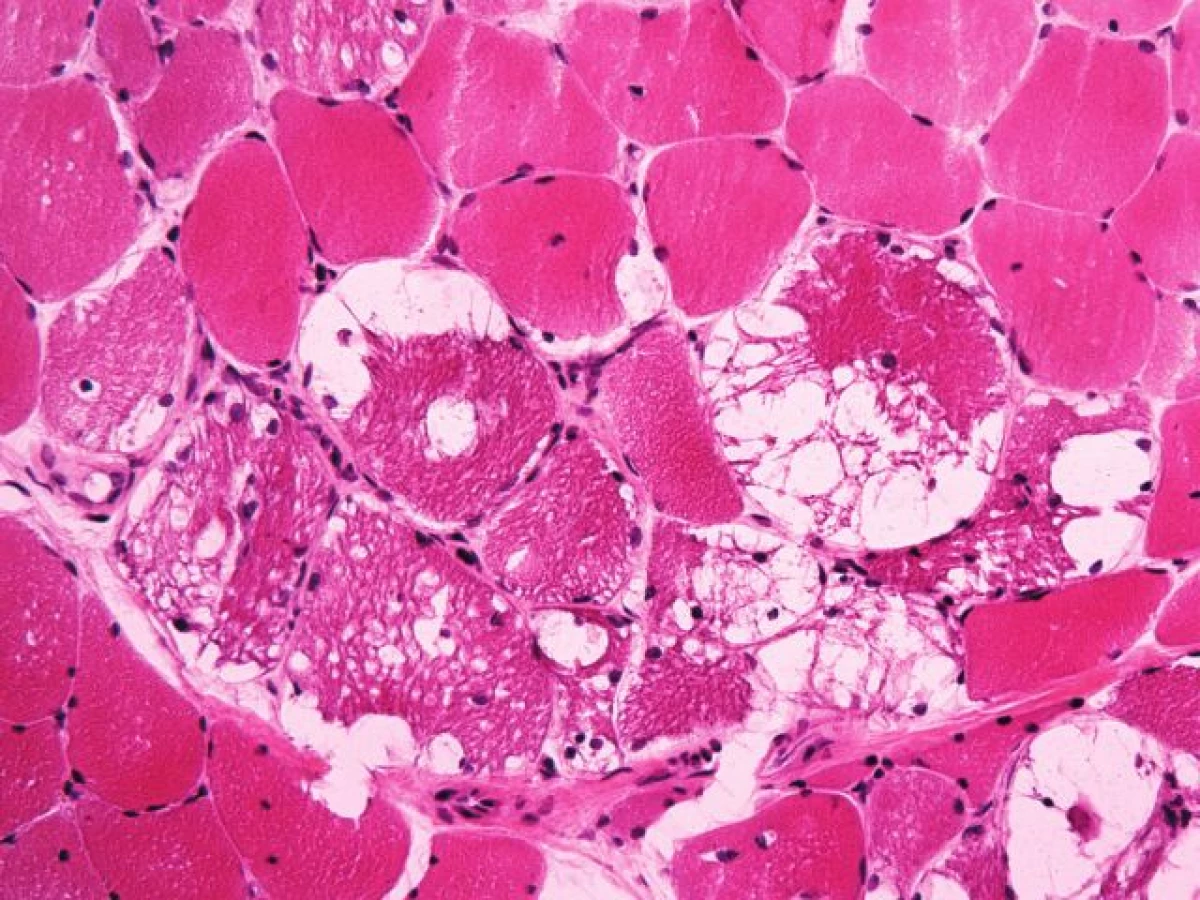
První svalová biopsie – známky zánětlivé myopatie nebyly nalezeny. Zato dominovala vakuolární myopatie s přítomností autofagických vakuol na úrovni světelného i elektronového mikroskopu (obr. 5). Vzhledem k nedávné anamnéze léčby chlorochinovým preparátem jsme nález uzavírali jako suspektní toxickou autofagickou vakuolární myopatii – chlorochinovou myopatii.
I přes vysazení antimalarik však k úpravě klinického stavu ani laboratorních parametrů nedošlo po dobu dalších šesti let. Pro trvající klinické podezření na myozitidu byla pacientka nadále léčena steroidy, dále pak také metotrexatem, avšak bez výraznějšího efektu. Pro nejasnost diagnózy tedy byla indikována druhá svalová biopsie. Zde byl obraz prakticky totožný s obrazem v první biopsii – autofagická vakuolární myopatie. Podrobným klinickým došetřením byla doplněna anamnéza, která nově ukázala, že svalové obtíže nejspíše předcházely začátku terapie antimalariky. Vzhledem k tomu, že teď bylo možné chlorochinovou myopatii z etiologie poruchy vyloučit, diagnózu jsme revidovali a indikovali provedení testu suché krevní kapky na průkaz Pompeho nemoci. Následná enzymologická konfirmace deficitu α-glukosidázy diagnózu glykogenózy II. typu (m. Pompe) potvrdila. Pacientka je nyní indikována k ERT.
DISKUZE A PŘEHLED LITERATURY
Pompeho nemoc
Pompeho nemoc (PN, glykogenóza II. typu), popsaná v roce 1932 holandským patologem J. C. Pompem (10) patří v současnosti mezi nejvýznamnější autofagické vakuolární myopatie. A to zejména proto, že od roku 2008 je i v ČR dostupná efektivní enzymová substituční léčba (ERT) rekombinantní lidskou α-glukosidázou (rhGAA) (9). Je to autozomálně recesivní střádavé lysozomální onemocnění způsobené primární deficiencí kyselé α-1,4-glukosidázy (GAA; syn.: kyselá maltáza). GAA je lysozomální hydrolázou, která hydrolyzuje glykogen na glukózu. Z enzymů, jejichž deficity jsou zodpovědné za rozvoj glykogenóz, je to jediný enzym, který je lokalizovaný v lysozomech. Ostatní glykogenózy jsou způsobeny deficitem cytoplazmatických enzymů. Ačkoli dochází při deficienci GAA k intralysozomální akumulaci glykogenu prakticky ve všech buňkách, nejvýrazněji je díky vystupňované autofagii postižena srdeční a kosterní svalovina (8).
Klinické formy PN. PN má velmi široké spektrum klinické prezentace, od nejtěžší infantilní formy, po méně progresivní „late-onset“ formy, rozčleněné ještě na juvenilní (případ č. 1) a adultní typ (případy č. 3 a 5) (11). Infantilní forma PN se může manifestovat už in utero, většinou je však diagnostikována v novorozeneckém období, kdy se prezentuje kardiomyopatií, makroglosií, hypotonií a respirační insuficiencí. Bez léčby je onemocnění letální, většinou do konce prvního roku života v důsledku kardiorespiračního selhání. U late-onset forem PN dominuje spíše slabost kosterního svalstva s mírnou elevací sérové kreatinkinázy (postiženy bývají hlavně proximální svalové skupiny a bránice, což později vede k rozvoji respirační insuficience), kardiální příznaky většinou tak výrazné nebývají. Obecně je známá inverzní korelace mezi tíží PN a hladinou reziduální enzymové aktivity GAA. Ačkoli je zřejmé, že různé mutace genu vedou k různému stupni reziduální aktivity GAA a tíži onemocnění, stanovení korelace mezi genotypem a fenotypem zatím možné není (12). Ukázalo se totiž, že kromě konkrétní mutace mají na fenotyp choroby výrazný vliv pravděpodobně i epigenetické faktory, neboť ve velké kohortě pacientů s totožnou mutací genu pro GAA bylo spektrum klinického postižení velmi široké (13). Tyto faktory bude nutné identifikovat, neboť by mohly být důležité pro rozvoj ještě efektivnější terapie PN.
Diagnostika. S tíží onemocnění koreluje i histopatologický obraz ve svalové biopsii. U těžších infantilních a juvenilních forem (případ č. 1) je obraz dramatický s intracytoplazmatickou přítomností vakuol, které jsou extrémně veliké a mohou zabírat i většinu vlákna. Jak jsme ukázali na případu č. 3, u late-onset formy může být obraz necharakteristický až minimální. Intracytoplazmatická vakuolizace prakticky nemusí být patrná a v těchto případech téměř normální nález ve svalové biopsii diagnózu PN nevylučuje. Navíc, metodika současné enzymové histochemie neumožňuje prokázat α-glukosidázu v tkáňovém řezu. Nemůžeme tak, na rozdíl od např. glykogenózy V. typu (m. McArdle, deficientní enzym: myofosforyláza) enzymatickou poruchu prokázat už v biopsii. Pro diagnózu PN je ale dostupný poměrně jednoduchý a neinvazivní test suché krevní kapky (DBS, dry blood spot test), který by měl podle našeho názoru a po zkušenostech s případy č. 3 a 5 vždy předcházet indikaci svalové biopsie. Testem, jehož principu se užívá i při skríningu jiných metabolických onemocnění, je stanovena aktivita příslušného enzymu. Pro konfirmaci diagnózy je nutno potvrdit tuto sníženou aktivitu ještě v kultivovaných fibroblastech, izolovaných leukocytech či lymfocytech z periferní krve, eventuálně verifikovat specifickou mutaci (je jich popsáno více než 150, gen leží na dlouhém raménku 17. chromozomu a má cca 20kb a 20 exonů) (14). DNA diagnostika PN je důležitá i pro prenatální diagnostiku a detekci heterozygotů.
Epidemiologie. Jak ukazují data ze zahraničních epidemiologických studií, celková incidence PN je asi 1:40.000 (15,16). Což znamená, že by mělo být v ČR diagnostikováno více než 200 případů. Tato léčitelná choroba je však u nás zřetelně poddiagnostikovaná, neboť diagnostikovaných a léčených případů v ČR není v současnosti ani patnáct. I proto jsme recentně dokončili projekt, ve kterém jsme se snažili identifikovat v našem registru ty případy svalových biopsií, u kterých jsme i přes klinické příznaky jednoznačnou diagnózu nestanovili. A u těch jsme ve spolupráci s klinickými kolegy indikovali provedení enzymologických vyšetření. Projekt už přinesl své výsledky a jen podtrhuje skutečnost, že normální nález ve svalové biopsii diagnózu late-onset Pompeho nemoci nevylučuje. Cesta k narovnání situace by mohla vést přes plošný skríning této choroby v populaci, který byl některými autory navržen a na Taiwanu už pilotně probíhá (17,18).
Patogeneze a kauzální léčba. Ačkoli je dostupná léčba PN, není ještě v oblasti patogeneze a mechanismu rozvoje tohoto onemocnění vyřešena celá řada otázek. Není jasný mechanismus, kterým by měla akumulace glykogenu vést k rozvoji svalové slabosti. Tradičně se mělo za to, že díky nedostatku glykogenu dochází k “energetické krizi” a to že je důvodem dysfunkce svaloviny. Řada prací však ukázala, že tomu tak zdaleka není a že mnohem důležitější než nedostatek energie (kterou si umí vlákno pořídit i jinak) je hromadění autofagických vakuol. To způsobuje poruchy intracelulární signalizace a změny v uspořádání cystoskeletu svalového vlákna, čehož důsledkem je omezení kontraktility (19). Dalším nevyřešeným momentem je skutečnost, že ERT (ačkoli výrazně prodlužuje život pacientů s infantilní formou PN (20) a vede ke stabilizaci onemocnění u late-onset forem (21)) nefunguje ani zdaleka tak stoprocentně, jak by se u kauzální léčby očekávalo (22). V srdečním svalu dochází při ERT k prakticky kompletnímu ústupu lysozomálního střádání a návratu funkce. U kosterního svalu ale zůstávají i při normalizaci hladiny aktivity enzymu morfologické známky dysfunkční autofagie (a do různé míry také myopatické příznaky) přítomny i nadále (20,23,24). Co za tímto stojí, není zatím jasno. Co však jasné je, že patologický mechanismus rozvoje PN ještě neznáme dokonale. Výzkum už přinesl první výsledky, když se ukázalo, že kromě lysozomů hrají u PN důležitou roli i mitochondriální abnormality (25). Navíc se objevilo několik dalších problémů. Například u malých dětí s infantilní formou PN s mutací způsobující úplné chybění GAA vzniká při léčbě rhGAA výrazná imunitní reakce (tzv. CRIM – cross-reactive immunological material) (26).
Budoucnost PN. Hlubší pochopení mechanismu rozvoje různých forem PN bude nutné pro vývoj nových generací ještě efektivnější léčby. Dostatečnému průniku rhGAA do lysozomů pacienta pravděpodobně brání komplexní autofagická dysfunkce, proto bylo navrženo několik nových strategií, jak terapeutický enzym do lysozomů dopravit – např. konjugací rhGAA se syntetickým oligosacharidem s navázaným manoso-6-fosfátem (27) nebo kombinací ERT a chaperony (28). Tyto malé molekuly se naváží na aktivní místo defektního enzymu a upraví jeho terciární strukturu tak, aby byl schopen transportu z endoplazmatického retikula do lysozomu.
Spektrum symptomatologie PN bude ale možná mnohem širší, než se nyní zdá. Existuje několik prací, které na podkladě pozorování mozkových arteriopatií tvrdí, že u PN je postižena i hladká svalovina (29,30)! Tím by se nutnost udržovat tuto léčitelnou glykogenózu v základní paletě diferenciální diagnostiky rozšířila daleko za hranice neuromuskulární diagnostiky či kardiopatologie.
Danonova choroba
Danonova choroba (DD), se od Pompeho nemoci liší, přestože mají v histopatologickém obraze i v klinické symptomatice mnoho společných rysů. Ačkoli byla DD dříve klasifikována jako glykogenóza typu IIb, dnes je jasné, že se o glykogenózu nejedná. DD je způsobena primární deficiencí glykoproteinu vnitřní strany lysozomální membrány – LAMP-2 (lysosome-associated membrane protein-2) (31). Tento protein hraje důležitou funkci při maturaci časných autofagických vakuol při fúzi endozomů s lysozomy, jeho nedostatek tedy způsobí poruchu normální autofagie se všemi důsledky této dysfunkce (32). Gen pro LAMP-2 je lokalizovaný na X chromozomu, a jako u jiných X-vázaných dominantních poruch, jsou i u DD muži postiženi výrazněji než ženské přenašečky (ale i ony mírnější a později nastupující klinickou symptomatiku rozvinou) (33). Pacienti s DD mají klasicky triádu obtíží: dominuje hypertrofická kardiomyopatie, méně výrazná je svalová slabost a spíše variantní bývá mentální retardace, která je překvapivě častější u postižených žen (34). Vzácněji bývají také postižena játra (35) nebo retina (36).
Ve svalové biopsii je podobně jako při PN pozorována vakuolizace svalových vláken, ta se však v některých detailech od vakuol u PN liší. Jednak, jen část vakuol je vodojasných s PAS pozitivním materiálem v lumen, častěji jsou pozorovány vakuoly s jemně bazofilním granulárním materiálem. Další zvláštností je skutečnost, že skupinky drobnějších lysozomálních granul bývají obklopeny membránou, ve které lze prokázat řadu proteinů, které jsou normálně součástí komplexu proteinů sarkoplazmatické membrány a extracelulární matrix (dystrofin, sarkoglykany, α2-laminin apod.) (37), jak jsme ukázali i na našem případu č. 2. Tyto vakuoly bývají označovány jako tzv. AVSF (autophagic vacuoles with sarcolemmal features) a pravděpodobně nevznikají vchlípením nebo endocytózou sarkolemy, jak se dříve předpokládalo, ale de novo při poruše transportu sarkolemálních proteinů do intracelulárních vakuol v cytoplazmě (37).
Konečná diagnostika Danonovy choroby je molekulárně genetická (i když i imunohistochemicky lze prokázat absenci LAMP-2 ve tkáni) a kvůli dominantně kardiologickému postižení by se na toto onemocnění mělo myslet (a provést skríning mutací genu pro LAMP-2) u všech pacientů s nevyjasněnou etiologií hypertrofické kardiomyopatie (31).
Chlorochinová myopatie
Další důležitou poruchou, která stojí v diferenciální diagnóze etiologie vakuolárních autofagických myopatií ve svalové biopsii, je tzv. chlorochinová myopatie. Hydroxychlorochin (a dříve používaný chlorochin) je antimalarikum, které se stále častěji používá při léčbě autoimunitních onemocnění (systémový lupus erythematodes, revmatoidní artritida), ale i při chronické reakci štěpu proti hostiteli (cGVHD) u pacientů po transplantaci kostní dřeně (38). Chlorochinové preparáty mají velmi komplexní vliv na imunitní systém – inhibují cytotoxicitu T lymfocytů, indukují jejich apoptózu a blokují jejich aktivaci (39), modulují zpracování a prezentaci antigenů pomocí HLA-II a ovlivňují produkci řady cytokinů (40-42). Jsou to velké amfifilní molekuly, které se rychle distribuují v celém těle (včetně kosterní a srdeční svaloviny) (43) a vstupují do lysozomů, kde se hromadí. Svou interakcí s lysozomy mění intralysozomální pH, čímž dochází k inhibici lysozomálních enzymů. Následkem toho vzniká lysozomální dysfunkce a tvorba autofagických vakuol (44). Nejčastějšími vedlejšími účinky terapie chlorochiny jsou však gastrointestinální poruchy, méně často bývá pozorována retinální toxicita, exantémy či útlum kostní dřeně (38); rozvoj myopatie je relativně vzácný (45).
Pacienti s autoimunitními onemocněními (včetně pacientů s cGVHD) jsou ale často léčeni více myotoxickými látkami najednou – kromě chlorochinových preparátů jsou to zejména kortikosteroidy (viz náš případ č. 4 a 5). Navíc, tyto systémové choroby (zejména revmatoidní artritida nebo SLE) mohou být asociovány s rozvojem autoimunitní myozitidy. Proto, pokud se u pacienta s některým z těchto onemocnění rozvine svalová slabost s elevací CK, je indikována svalová biopsie, která jediná dokáže rozlišit mezi chlorochinovou myopatií (vakuoly), steroidní myopatií (atrofie vláken typu IIB) (46) a dermatomyozitidou či polymyozitidou (47). Řešením nastalého stavu je pak buď zintenzivnění imunosuprese u zánětlivých myopatií, nebo naopak vysazení některého z myotoxických imunosupresivních preparátů, což vede (tak jako v našem případu č. 4) k dramatickému zlepšení svalových obtíží. Skutečným diagnostickým rébusem se ale stávají situace, kdy je nepoznaná hereditární autofagická vakuolární myopatie léčena myotoxickým preparátem, který rovněž vyvolává obraz autofagické vakuolární myopatie, tak jak tomu bylo u našeho posledního případu. Ponaučením je, že pokud po vysazení myotoxického léku k očekávanému zlepšení nedojde, je třeba situaci nevzdávat a pátrat dál. Celou situaci navíc komplikuje fakt, že efekt některých myotoxických léčiv může přetrvat i poměrně dlouhou dobu po jejich vysazení (48).
ZÁVĚR
Dostupnost efektivní ERT pro léčbu Pompeho nemoci učinila ze stanovení správné etiologické diagnózy autofagických vakuolárních myopatií nutnost. Svalová biopsie je dobrým nástrojem pro diferenciální diagnózu autofagických vakuolárních myopatií, v některých případech však selhává. Proto je podle našeho názoru vhodné indikovat provedení testu suché krevní kapky na vyloučení Pompeho nemoci před indikací svalové biopsie. Povědomí o tomto onemocnění a dostupnost neinvazivního testu je dobrým předpokladem pro odhalení celé řady pacientů, kteří jsou zatím vedeni pod jinými diagnózami a unikají tak léčbě.
Nedávný rozvoj poznání funkční patologie autofagických vakuolárních myopatií zcela změnil pohled na jejich patogenezu. Na Pompeho chorobu už nelze nahlížet jako na problém se střádáním glykogenu, stejně tak u Danonovy choroby není hlavní problém v lysozomální dysfunkci. U obou poruch hraje dominantní roli komplexní dysfunkce autofagického procesu, která hromaděním autofagických vakuol vede k disrupci myofibrilárního uspořádání a tím k poruše funkce. Identifikace celé komplexní patogeneze autofagických vakuolárních myopatií snad v budoucnu přinese ještě efektivnější terapii těchto onemocnění.
SEZNAM POUŽITÝCH ZKRATEK
ACP – kyselá fosfatáza;
AVSF – autophagic vacuoles with sarcolemal features;
cGVHD – chronická reakce štěpu proti hostiteli;
CK – sérová kreatinkináza;
CRIM – cross-reactive immunological material,
DBS – test suché krevní kapky (dry blood spot test);
DD – Danonova choroba;
EMG – elektromyografie;
ERT – enzymová substituční terapie (enzyme replacement therapy);
GAA – kyselá α-1,4-glukosidáza (synonymum: kyselá maltáza);
LAMP-2 – lysosome-associated membrane protein-2;
MRI – magnetická rezonance;
PN – Pompeho nemoc (glykogenóza II. typu, glycogen storage disease II, GSD II);
rhGAA – rekombinantní lidská α-glukosidáza
PODĚKOVÁNÍ
Podpora: Projekt (Ministerstva zdravotnictví ČR) koncepčního rozvoje výzkumné organizace 00064203 (FN Motol).
Autoři děkují za laskavou spolupráci a poskytnutí klinických údajů těmto kolegům: MUDr. J. Haberlová, Ph.D., MUDr. R. Mazanec, Ph.D. (FN Motol, Praha), MUDr. P. Ridzoň (FTNsP, Praha), MUDr. M. Kotrč (IKEM, Praha), MUDr. I. Patáková, CSc. (nemocnice Hořovice), MUDr. S. Skácelová, MUDr. H. Mann, prof. MUDr. J. Vencovský, DrSc. (Revmatologický ústav, Praha), MUDr. V. Malinová (VFN, Praha).
Autoři děkují za kritické poznámky k rukopisu prof. MUDr. Markétě Hermanové, Ph.D.
VĚNOVÁNÍ
Autoři věnují tento text památce významného českého lékaře, vědce a pedagoga prof. MUDr. Milana Elledera, DrSc.
(4. 12. 1938 – 25. 9. 2011).
Adresa pro korespondenci:
Doc. MUDr. Josef Zámečník, Ph.D.
Ústav patologie a molekulární medicíny
2. LF UK a FN v Motole
V Úvalu 84, 150 06 Praha 5
tel.: 224 435 635, fax: 224 435 620
e-mail: josef.zamecnik@lfmotol.cuni.cz
Zdroje
1. Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med 2003; 9(3–4): 65–76.
2. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306(5698): 990–995.
3. Mizushima N. Autophagy: process and function. Genes Dev 2007; 21(22): 2861-2873.
4. Raben N, Shea L, Hill V, Plotz P. Monitoring autophagy in lysosomal storage disorders. Methods Enzymol 2009; 453: 417–449.
5. Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 2009; 1793(4): 684–696.
6. Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol 2006; 65(5): 423–432.
7. Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes and oxidative stress in aging and apoptosis. Biochim Biophys Acta 2008; 1780(11): 1291–1303.
8. Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15(3): 1101–1111.
9. Amalfitano A, Bengur AR, Morse RP et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 2001; 3(2): 132–138.
10. Pompe J. Over idiopatische hypertrophie van het hart. Ned Tijdshr Geneeskd 1932; 76: 304-312.
11. Di RM, Buzzi D, Taro M. Glycogen storage disease type II: clinical overview. Acta Myol 2007; 26(1): 42–44.
12. Kroos M, Hoogeveen-Westerveld M, van der PA, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet 2012; 160(1): 59–68.
13. Kroos MA, Pomponio RJ, Hagemans ML et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007; 68(2): 110–115.
14. Malinová V. Glykogenóza II. typu (GSD II, m. Pompe). Současné možnosti diagnostiky a terapie. Klinická kazuistika. Neurol prax 2010; 11(5): 326–330.
15. Ausems MG, Verbiest J, Hermans MP et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999; 7(6): 713–716.
16. Martiniuk F, Chen A, Mack A et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998; 79(1): 69–72.
17. Burton BK. Newborn screening for Pompe disease: an update, 2011. Am J Med Genet C Semin Med Genet 2012; 160(1): 8–12.
18. Chien YH, Lee NC, Huang HJ, Thurberg BL, Tsai FJ, Hwu WL. Later-onset Pompe disease: early detection and early treatment initiation enabled by newborn screening. J Pediatr 2011; 158(6): 1023–1027.
19. Fukuda T, Ewan L, Bauer M et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol 2006; 59(4): 700–708.
20. Kishnani PS, Corzo D, Nicolino M et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007; 68(2): 99–109.
21. Van der Ploeg AT, Clemens PR, Corzo D et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010; 362(15): 1396–1406.
22. Reuser AJ. Enzyme therapy in Pompe disease: questions remain. Mol Genet Metab 2012; 107 (3): 485-489.
23. Nicolino M, Byrne B, Wraith JE et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med 2009; 11(3): 210–219.
24. Raben N, Jatkar T, Lee A et al. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther 2002; 6(5): 601–608.
25. Raben N, Wong A, Ralston E, Myerowitz R. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med Genet 2012; 160(1): 13–21.
26. Kishnani PS, Goldenberg PC, DeArmey SL et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 2010; 99(1): 26–33.
27. Zhu Y, Jiang JL, Gumlaw NK et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther 2009; 17(6): 954–963.
28. Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med 2009; 1(5): 268– 279.
29. Laforet P, Petiot P, Nicolino M et al. Dilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology 2008; 70(22): 2063–2066.
30. Sacconi S, Bocquet JD, Chanalet S, Tanant V, Salviati L, Desnuelle C. Abnormalities of cerebral arteries are frequent in patients with late-onset Pompe disease. J Neurol 2010; 257(10): 1730–1733.
31. Nishino I, Fu J, Tanji K et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000; 406(6798): 906–910.
32. Tanaka Y, Guhde G, Suter A et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000; 406(6798): 902–906.
33. Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep 2003; 3(1): 64–69.
34. Danon MJ, Oh SJ, DiMauro S et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981; 31(1): 51–57.
35. Sugie K, Yamamoto A, Murayama K et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002; 58(12): 1773–1778.
36. Schorderet DF, Cottet S, Lobrinus JA, Borruat FX, Balmer A, Munier FL. Retinopathy in Danon disease. Arch Ophthalmol 2007; 125(2): 231–236.
37. Sugie K, Noguchi S, Kozuka Y et al. Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J Neuropathol Exp Neurol 2005; 64(6): 513 –522.
38. Gilman AL, Chan KW, Mogul A et al. Hydroxychloroquine for the treatment of chronic graft-versus-host disease. Biol Blood Marrow Transplant 2000; 6(3A): 327–334.
39. Goldman FD, Gilman AL, Hollenback C, Kato RM, Premack BA, Rawlings DJ. Hydroxychloroquine inhibits calcium signals in T cells: a new mechanism to explain its immunomodulatory properties. Blood 2000; 95(11): 3460–3466.
40. Fox RI, Kang HI. Mechanism of action of antimalarial drugs: inhibition of antigen processing and presentation. Lupus 1993; 2(Suppl 1): 9–12.
41. Schultz KR, Su WN, Hsiao CC et al. Chloroquine prevention of murine MHC-disparate acute graft-versus-host disease correlates with inhibition of splenic response to CpG oligodeoxynucleotides and alterations in T-cell cytokine production. Biol Blood Marrow Transplant 2002; 8(12): 648–655.
42. Bondeson J, Sundler R. Antimalarial drugs inhibit phospholipase A2 activation and induction of interleukin 1beta and tumor necrosis factor alpha in macrophages: implications for their mode of action in rheumatoid arthritis. Gen Pharmacol 1998; 30(3): 357–366.
43. McChesney EW. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med 1983; 75(1A): 11–18.
44. Sundelin SP, Terman A. Different effects of chloroquine and hydroxychloroquine on lysosomal function in cultured retinal pigment epithelial cells. APMIS 2002; 110(6): 481–489.
45. Bolanos-Meade J, Zhou L, Hoke A, Corse A, Vogelsang G, Wagner KR. Hydroxychloroquine causes severe vacuolar myopathy in a patient with chronic graft-versus-host disease. Am J Hematol 2005; 78(4): 306–309.
46. Zámečník J, Veselý D, Jakubička B et al. Atrophy of type II fibres in myasthenia gravis muscle in thymectomized patients: steroid-induced change with prognostic impact. J Cell Mol Med 2009; 13(8B): 2008–2018.
47. Zámečník J, Veselý D, Jakubička B et al. Muscle lymphocytic infiltrates in thymoma-associated myasthenia gravis are phenotypically different from those in polymyositis. Neuromuscul Disord 2007; 17(11–12): 935–942.
48. Kuncová K, Sedláčková M, Vencovský J, Mann H, Wenchich L, Zámečník J. Inflammatory myopathy associated with statins: Report of 3 Cases. Mod Rheumatol 2013; in press.
Štítky
Patológia Súdne lekárstvo ToxikológiaČlánok vyšiel v časopise
Česko-slovenská patologie

2013 Číslo 1
Najčítanejšie v tomto čísle
- Česká eponyma v patologii
-
Dysplázie sliznice žaludku.
Klinickopatologická studie 35 případů - Autofagické vakuolární myopatie – aneb co nás naučila diferenciální diagnostika vakuol ve svalové biopsii
-
Nanopatologie - nový vědecký obor
Minireview