
Klinický význam vyšetřování mutací v genu pro transkripční faktor PROP1 u dětí s vrozeným deficitem růstového hormonu
Clinical Importance of Identification of PROP1 Gene Mutations in Children with Inherited Growth Hormone Deficiency
Objective:
PROP1 gene is the transcription factor regulating the differentiation of anterior pituitary cells during embryologic development. Mutations found in this gene are the most frequent cause of inherited pituitary hormone deficiency in the Middle and Eastern Europe. The aim of this study was to identify the individuals with PROP1 gene mutations in the group of children with idiopathic combined pituitary hormone deficiency and with isolated growth hormone deficiency. Secondly, we evaluated clinical importance of PROP1 gene molecular genetic analysis.
Patients and methods:
We collected DNA samples and phenotypic data of 94 patients with isolated growth hormone deficiency and of 55 patients with combined pituitary hormone deficiency treated in six Czech and one foreign centre. In patients with isolated deficiency we performed a screening method to detect two most frequent PROP1 gene mutations; DNA samples of patients with combined deficiency were subjected to direct PROP1 gene sequencing.
Results:
In the group of 55 patients with combined pituitary hormone deficiency four were homozygous for 296delGA mutation; no other mutation was found in the rest of the group. In the group of 94 patients with isolated growth hormone deficiency one was heterozygous for 296delGA mutation.
Conclusions:
This study has not confirmed the clinical importance of routine identification of PROP1 gene mutations in the phenotypic heterogeneous group of patients with idiopathic isolated or combined growth hormone deficiency. It may play a role in subjects with precisely defined phenotype.
Key words:
PROP1, embryogenesis, differentiation, pituitary, transcription factor, growth hormone
Autoři:
B. Obermannová 1; J. Černá 2; V. Janštová 2; S. Koloušková 1; D. Neumann 3; D. Novotná 4; R. Pomahačová 5; J. Škvor 6; M. Šnajderová 1; Z. Šumník 1; J. Zapletalová 7; J. Lebl 1
Působiště autorů:
Pediatrická klinika UK 2. LF a FN Motol, Praha
přednosta prof. MUDr. J. Lebl, CSc.
1; Klinika dětského lékařství FN, Ostrava-Poruba
přednosta doc. MUDr. J. Slaný, CSc.
2; Dětská klinika FN, Hradec Králové
přednosta prof. MUDr. M. Bayer, CSc.
3; II. dětská klinika LF MU, FN Brno
přednosta prof. MUDr. Z. Doležel, CSc.
4; Dětská klinika FN, Plzeň
přednosta doc. MUDr. J. Kobr, PhD.
5; Dětská klinika, Masarykova nemocnice, Ústí nad Labem
přednosta MUDr. J. Škvor, CSc.
6; Dětská klinika FN, Olomouc
přednosta prof. MUDr. V. Mihál, CSc.
7
Vyšlo v časopise:
Čes-slov Pediat 2009; 64 (6): 296-304.
Kategorie:
Původní práce
Souhrn
Účel studie:
PROP1 je transkripční faktor, který reguluje diferenciaci buněčných linií adenohypofýzy v průběhu embryonálního vývoje. Jeho defekt je ve střední a východní Evropě nejčastější příčinou vrozeného deficitu hypofyzárních hormonů. Cílem studie bylo identifikovat jedince s mutací PROP1 genu ve skupině dětí s idiopatickým kombinovaným deficitem hypofyzárních hormonů a s izolovaným deficitem růstového hormonu; posoudit klinický přínos této diagnostiky a výtěžnost vyšetřování PROP1 genu jako rutinní diagnostiky u těchto pacientů.
Pacienti a metody:
Z šesti českých a jednoho zahraničního centra byly k dispozici vzorky DNA a fenotypická data 94 pacientů s idiopatickým izolovaným deficitem růstového hormonu a 55 pacientů s idiopatickým kombinovaným deficitem hypofyzárních hormonů. U pacientů s izolovaným deficitem byla provedena screeningová metoda zaměřená na záchyt dvou nejčastějších mutací PROP1 genu; vzorky DNA pacientů s kombinovaným deficitem byly podrobeny přímé sekvenaci PROP1 genu.
Výsledky:
Ve skupině 55 pacientů s kombinovaným deficitem byli zachyceni čtyři homozygotní nositelé mutace 296delGA, u ostatních vyšetřovaných pacientů mutace zjištěna nebyla. Z 94 pacientů s izolovaným deficitem růstového hormonu byl jeden heterozygot pro mutaci 296delGA.
Závěr:
Tato studie nepotvrdila výtěžnost rutinní diagnostiky PROP1 mutací u fenotypicky heterogenní skupiny pacientů s idiopatickým izolovaným nebo kombinovaným deficitem růstového hormonu. Svůj význam může mít u fenotypicky přesně charakterizovaných osob.
Klíčová slova:
PROP1, embryogeneze, diferenciace, hypofýza, transkripční faktor, růstový hormon
Úvod
Vrozené poruchy funkce hypofýzy zahrnují jednak izolované deficity jednoho hormonu, například izolovaný deficit růstového hormonu (IGHD, Isolated Growth Hormone Deficiency), nebo kombinovaný deficit hypofyzárních hormonů (CPHD, Combined Pituitary Hormone Deficiency). Tyto poruchy byly v minulosti obvykle hodnoceny jako idiopatické. Detailní poznání genové kontroly regulace vývoje a funkce hypofýzy umožňuje v současné době u mnoha z těchto pacientů prokázat geneticky podmíněný defekt. V případě CPHD se jedná zpravidla o poruchu některého z genů, které kódují transkripční faktory ovlivňující embryonální vývoj hypofýzy [1].
Embryonální vývoj hypofýzy je řízen kaskádou signálních molekul a transkripčních faktorů produkovaných jednak okolními tkáněmi, jednak buňkami zárodečných tkání samotné žlázy [2, 3, 4]. Proteinové produkty těchto genů jsou specifické transkripčními faktory, které aktivují nebo blokují transkripci dalších genů. Pouze jejich správná exprese v prostoru a čase vede ke vzniku morfologicky i funkčně plnohodnotné tkáně. Tyto geny vedou nejdříve k vzniku základu orgánu, poté k buněčné proliferaci, tkáňové specializaci buněk a konečně k diferenciaci jednotlivých buněčných linií adenohypofýzy [5, 6].
Přední a střední lalok žlázy má původ v orálním ektodermu (Rathkeho výchlipka), zadní lalok v neuroektodermu diencefala. Vývoj předního laloku má dvě fáze, ve kterých se uplatňují rozdílné transkripční faktory [1]. Schéma embryonálního vývoje hypofýzy a zúčastněných transkripčních faktorů shrnuje obrázek 1.
![Signální molekuly a transkripční faktory zapojené v embryonálním vývoji hypofýzy. Schéma ukazuje expresi jednotlivých regulačních molekul v průběhu embyogeneze, příslušné vývojové fáze hypofýzy a středočárových mozkových struktur a vznik specializovaných buněčných linií adenohypofýzy.
(Upraveno dle [8]).
Vysvětlivky:
e – embryonální den vývoje myši; ACTH – kortikotropní buňky (produkce ACTH); FSH/LH – gonadotropní buňky (produkce gonadotropinů); GH – somatotropní buňky (produkce růstového hormonu); PRL – laktotropní buňky (produkce prolaktinu);TSH – tyreotropní buňky (produkce TSH); jednotlivé regulační molekuly jsou podrobněji popsány v textu.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/06d5b61c202905016ae07473b3eee0a5.jpg)
V první fázi vzniká základ orgánu (morfogeneze adenohypofýzy). V časné organogenezi hrají zásadní roli extrinsické molekuly produkované okolními tkáněmi, zejména ventrálním diencefalem. Patří mezi ně nedávno popsané molekuly sonic hedgehog homologue (SHH), tyreoidální transkripční faktor 1 (TTF-1) [2, 7, 8] a orthodenticle homeobox 2 faktor (OTX2), který transaktivuje následující faktory HESX1 a POU1F1. Ztrátová mutace genu pro OTX2 vede zpravidla k anoftalmii, která může být spojena s kombinovaným deficitem hypofyzárních hormonů, malou hypofýzou, ektopickou neurohypofýzou a Chiariho malformací [9]. Do vývoje hypofýzy však již v této fázi zasahují i produkty genů exprimovaných přímo v buňkách základu hypofýzy (intrinsické transkripční faktory). Defekty jednotlivých transkripčních faktorů vedou k různému spektru hormonálních deficitů a současně k poruchám vývoje mozku a dalších struktur. Jedná se zejména o mutace faktorů HESX1 [8], PITX2 [5], LHX3 a LHX4 [10].
Ve druhé fázi vývoje dochází k diferenciaci jednotlivých buněčných linií adenohypofýzy. Vedle řady extrapituitárních signálních molekul řídí tento proces dva po sobě následující transkripční faktory – PROP1 a PIT1 (POU1F1) [1].
Pacienti s mutací genu pro faktor POU1F1 (dříve nazývaný PIT1) mají těžký deficit růstového hormonu a prolaktinu a progredující deficit tyreotropního hormonu. Hypofýza bývá brzy po narození normální velikosti, v průběhu života však většinou atrofuje. Dědičnost je autozomálně dominantní nebo recesivní [11].
PROP1 je zkratkou pro anglické „prophet of PIT1“, tedy „prorok (předchůdce) faktoru PIT1“. PROP1 gen je exprimován v prvních týdnech nitroděložního vývoje, nejprve v dorzální části Rathkeho výchlipky, následně pak v její kaudomediální oblasti. Poté jeho funkce vyhasíná. V buňkách zralé adenohypofýzy se neexprimuje [12].
Mutace PROP1 genu jsou u člověka nejčastější příčinou geneticky podmíněné formy CPHD. V naší populaci tvoří až čtvrtinu všech forem CPHD [13]. PROP1 gen leží na dlouhém raménku 5. chromozomu. Je dlouhý 3 kb a skládá se ze tří exonů a dvou intronů. Kóduje protein o 226 aminokyselinách. Druhý a třetí exon kódují oblast od 69. do 128. aminokyseliny, kde bylo u člověka popsáno nejvíce mutací. Je to oblast homeodomény s transaktivační kapacitou a oblast vazby na DNA, která se skládá ze tří alfa helixů. Ta díky vazbě na DNA aktivuje transkripci cílového genu, tedy genu POU1F1 (PIT1) [12]. Pokud dojde k mutaci PROP1 genu, selhává aktivace genu pro POU1F1 a tím i diferenciace hypofyzárních buněk na něm závislých (somatotropy, laktotropy a thyreotropy) [4].
Dědičnost známých mutací PROP1 genu je autozomálně recesivní. Až na jednu výjimku postihují mutace oblast homeodomény ve II. a III. exonu genu. Nejčastěji se vyskytují v oblasti tří tandemově se opakujících GA nukleotidů v exonu II (296-GAGAGAG-302), tzv. „hot-spot“ oblasti [14]. Mutace delA301,G302 (nyní známá spíše jako 296delGA) je vůbec nejčastěji diagnostikovaná mutace PROP1 genu a tvoří asi 55 % všech dosud nalezených mutací u kombinovaného deficitu hypofyzárních hormonů. Tato delece dvou párů bází vede k posunu čtecího rámce od 102. kodonu se vznikem předčasného stop kodonu na pozici 109. aminokyseliny (S109X) a k expresi zkráceného dysfunkčního proteinu. Ten ztrácí schopnost vázat DNA a aktivovat transkripci, jeho funkce je tedy nulová [14, 12]. Druhá nejčastější mutace je 150delA. Jedná se o deleci jednoho páru bází v kodonu 50. aminokyseliny, která také způsobuje posun čtecího rámce a vznik předčasného stop kodonu na pozici 53. aminokyseliny. Vede také ke vzniku zkráceného nefunkčního PROP1 proteinu [1, 14].
Pacienti s defekty PROP1 genu mají deficit hypofyzárních hormonů, ale nemají další abnormality mozku či jiných orgánů. Endokrinní fenotyp je různorodý. V časném dětství se zpravidla projevuje deficit růstového hormonu (growth hormone, GH), TSH a prolaktinu, později se manifestuje deficit gonadotropinů a případně i ACTH. Hormonální deficity bývají méně závažné než při mutaci genu POU1F1. Deficit GH se projeví růstovou retardací, spolu s deficitem TSH také opožděním kostního věku. Ačkoliv u většiny nastává deficit růstového hormonu již časně v životě, je možný i normální růst v časném dětství a dokonce i normální finální výška v dospělosti u neléčených pacientů. Deficit TSH se projevuje sekundární hypotyreózou s nízkými hladinami TSH i periferních tyreoideálních hormonů [8, 10].
Ačkoliv je transkripční faktor PROP1 nezbytný pro diferenciaci gonadotropních buněk během fetálního vývoje, je deficit gonadotropinů variabilní. Obvykle jsou pacienti hypogonadální a chybí jim puberta; někteří ale spontánně dospívají a dokonce jsou i fertilní. Přesný mechanismus tohoto různorodého fenotypu zůstává nejasný. Předpokládá se, že PROP1 je nezbytný pro diferenciaci gonadotropních buněk, ne však pro jejich determinaci [2].
U některých pacientů s defektem genu PROP1 se během života rozvine deficit ACTH a tedy sekundární hypokortikalismus („late-onset“ deficit). PROP1 se sice neexprimuje v kortikotropech, jejich funkce je však závislá na signálech z ostatních buněk adenohypofýzy, které jsou porušené [15]. Jiná teorie vysvětluje pozdní rozvoj deficitu gonadotropů a kortikotropů spoluúčastí ischemického postižení buněk hypofýzy při nesprávném embryonálním utváření žlázy a jejího cévního zásobení. Gonadotropní buňky jsou umístěny v kaudomediální oblasti žlázy a jsou náchylnější k ischemickému postižení než kortikotropy, které jsou převážně v lépe prokrvené centrální oblasti. Proto dochází k dřívější manifestaci deficitu gonadotropů než kortikotropů.
Mutace PROP1 obvykle mění velikost hypofýzy. Předpokládá se, že hypofýza u pacientů s mutacemi PROP1 prochází fází hyperplazie v dětství a následné regrese k hypoplastické hypofýze v pubertě nebo časné dospělosti až do stadia „empty sella“ [16]. Možným vysvětlením tohoto jevu je nepřiměřeně prodloužená exprese předcházejícího faktoru HESX1. Proto přežívají nediferenciované prekurzory hypofyzárních buněčných linií. Jiná hypotéza předpokládá, že hmota tkáně zvětšené hypofýzy pochází z intermediálního laloku. U jedinců s mutací PROP1 genu je oddálena involuce intermediálního laloku a migrace buněk v něm obsažených, která probíhá za normálních okolností ke konci fetálního období. Byl popsán případ biopsie hyperplastické hypofýzy u pacienta s PROP1 defektem, histologický výsledek byl nespecifický – amorfní materiál bez diferencovaných buněčných linií [2]. V časném dětství může toto benigní zvětšení hypofýzy připomínat hypofyzární adenom, kraniofaryngeom, germinom nebo cystu z Rathkeho výchlipky (obr. 2a, b).

Pokud je zjištěn defekt PROP1, není indikováno chirurgické řešení. Postačí dlouhodobé sledování zobrazovacími metodami, jelikož tento nález časem spontánně involuje [8].
Cíl studie
Cílem naší studie bylo identifikovat jedince s mutací PROP1 genu mezi dětmi s idiopatickým kombinovaným deficitem hypofyzárních hormonů i mezi dětmi s izolovaným deficitem růstového hormonu a posoudit klinický přínos této diagnostiky. Studie navazovala na práci z roku 2005, kdy byl PROP1 gen vyšetřen u 74 českých pacientů s kombinovaným deficitem hypofyzárních hormonů. Z nich bylo 24 % pozitivních pro homozygotní nebo složeně heterozygotní mutaci PROP1 genu [13].
Hlavním cílem nové studie bylo ověřit možnost zavedení vyšetřování mutací v PROP1 genu jako rutinní diagnostiky u všech dětských pacientů s deficitem růstového hormonu bez prokázané organické příčiny a posoudit možnosti a přínos genetického poradenství v postižené rodině.
Pacienti a metody
Pacienti
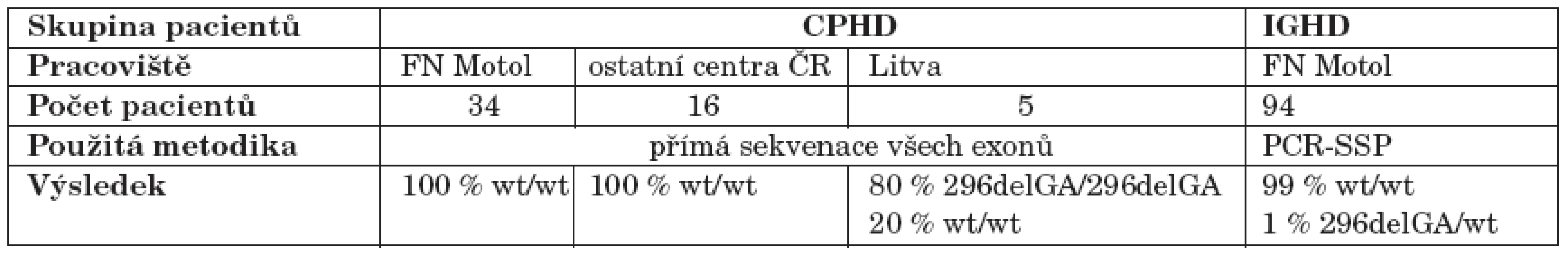
Do studie byli zařazeni dětští pacienti sledovaní na Pediatrické klinice FN Motol k 31. 3. 2007, u kterých byl prokázán vrozený („idiopatický“) deficit růstového hormonu (postimulační hladina GH nižší než 10 µg/l ve dvou stimulačních testech), buď izolovaný (IGHD) nebo spojený alespoň s jedním dalším deficitem hypofyzárního hormonu (CPHD), a kteří nebyli vyšetřeni v rámci předcházející české studie z roku 2005. Byly tak shromážděny vzorky DNA a klinická data 94 pacientů s IGHD a 34 pacientů s CPHD. Skupina s CPHD byla doplněna o 16 pacientů z dalších center v ČR (Brno, Ostrava, Olomouc, Hradec Králové, Ústí nad Labem, Plzeň a České Budějovice) a o pět pacientů z Kaunasu (Litva).
U pacientů s CPHD byl posouzen endokrinní fenotyp (spektrum hormonálních deficitů) a eventuální výskyt dalších genotypických projevů – morfologie hypofýzy (hypoplazie až „empty sella“, hyperplazie, výskyt cyst, morfologie neurohypofýzy) a celého mozku (výskyt ageneze středočárových struktur mozku, hypoplazie optických nervů) a poruchy embryonálního vývoje ostatních tělních struktur). Z 55 dětí s CPHD jich 16 splňovalo klinickou charakteristiku septo-optické dysplazie. Podle popisu MRI měli čtyři pacienti přerušenou stopku hypofýzy, u 29 pacientů byla popsána hypoplastická hypofýza a devět pacientů mělo nález „empty sella“. U žádného pacienta nebyla popsána hypertrofie hypofýzy.
Zákonní zástupci všech pacientů podepsali informovaný souhlas s odebráním vzorku periferní krve pro extrakci DNA, jehož znění bylo schváleno Etickou komisí 2. lékařské fakulty Univerzity Karlovy.
Molekulárně genetická analýza mutací PROP1 genu
Genomická DNA všech vyšetřovaných pacientů byla extrahována z leukocytů žilní krve nebo suché kapky krve použitím standardních metod (vysolování) nebo firemního kitu; QIAmp DNA Blood micro kit (Quiagen, Hilden, Germany).
U pacientů s IGHD jsme nejprve screeningovou metodou vyšetřovali dvě nejčastější mutace (296delGA a 150delA) pomocí PCR se specifickými primery (Polymerase chain reaction with sequence-specific primers, PCR-SSP). U jedinců pozitivních pouze na jednu mutaci jsme druhou mutaci hledali přímou sekvenací.
U pacientů s CPHD jsme nejprve amplifikovali tři kódující exony PROP1 genu a poté je sekvenovali, s cílem zachytit všechny mutace v kódujících oblastech genu. Pro amplifikaci všech tří exonů a navazujících intronických oblastí PROP1 genu byly použity tři páry dříve publikovaných primerů [12]. Vzniklé PCR produkty exonů byly přečištěny a poté podrobeny přímé sekvenaci za použití BigDye® Terminator 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) a následně analyzovány pomocí ABI PRISM® 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). K sekvenaci jsme použili totožné primery jako pro amplifikaci, pouze v nižších koncentracích. Přesné sekvence všech použitých primerů jsou uvedeny v tabulce 1.

Výsledky
U všech 55 probandů s CPHD jsme přímo sekvenovali tři kódující exony PROP1 genu. U žádného českého pacienta s CPHD nebyla nalezena mutace ve vyšetřovaných oblastech PROP1 genu. Z pěti litevských pacientů s CPHD byli čtyři homozygoti pro mutaci 296delGA, z nichž dva byli sourozenci se stejným endokrinním fenotypem, jeden pacient byl homozygot pro „wild-type“ alelu.
Z 94 probandů s IGHD byl screeningovou metodou PCR-SSP zachycen jeden proband pozitivní pro mutaci 296delGA. Přímá sekvenace potvrdila přítomnost mutace na jedné alele, ve druhé alele však neprokázala žádnou mutaci.
U žádného z vyšetřovaných pacientů nebyla nalezena mutace 150delA ani jiná, v populaci méně častá mutace.
Souhrn výsledků molekulárně genetické analýzy u všech pacientů shrnuje tabulka 2.
Diskuse
Jedna z nejrozsáhlejších studií o problematice genetických příčin poruch embryogeneze hypofýzy byla publikována v roce 2006. Tato multicentrická studie zkoumala genotyp 195 pacientů s idiopatickým CPHD a nalezla mezi 109 pacienty s CPHD bez dalšího sdruženého fenotypu (tedy s vyloučením pacientů se syndromem přerušené stopky hypofýzy nebo septo-optickou dysplazií) dvacet pacientů s defektem v PROP1 genu, tedy 18,3 %. Spektrum mutací bylo širší než v jiných studiích, možná vlivem použití přímé sekvenace [17].
O rok dříve byla publikována studie u 189 pacientů se sporadickým a u 44 jedinců s familiárním deficitem růstového hormonu (GHD), a to buď ve formě IGHD nebo CPHD. Záchyt PROP1 mutací v této skupině byl výrazně nižší: v homozygotní nebo složeně heterozygotní formě byly nalezeny u 1,1 % pacientů ze skupiny sporadického a 29,5 % ze skupiny familiárního GHD [18].
V naší populaci byla provedena studie v roce 2005 u 74 pacientů s CPHD v dětském i dospělém věku. Probandi byli testováni na přítomnost mutací v genech HESX1, PROP1 a POU1F1 (PIT1). Byl nalezen jeden pacient s mutací v genu POU1F1 a 18 jedinců s mutací v PROP1 genu, žádný pacient neměl mutaci v HESX1 genu. Prevalence nosičů mutací v analyzovaných transkripčních faktorech byla tedy 25,7 %, z toho 24,3 % v PROP1 genu. V tomto genu byly identifikovány pouze tři různé mutace: 301delGA, 150delA a 349T>A (model genu a proteinu PROP1 s označením nalezených mutací je zobrazen na obr. 3). První dvě zastupovaly 97 % všech mutací v PROP1 genu, ať už v homozygotní nebo složeně heterozygotní formě [13].
![PROP1 gen a PROP1 protein. Šipkami jsou vyznačenymutace popsané v předcházející české studii
[13].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/eeff3605653cecd1cfa013ae9e930f00.jpg)
Naše studie navazuje na tento projekt. Naším cílem byla identifikace pacientů s PROP1 defektem ve skupině pacientů léčených pro idiopatický deficit růstového hormonu (skupina byla rozdělena na pacienty s kombinovaným deficitem nebo izolovaným deficitem GH) a eventuální přínos jeho rutinní diagnostiky v této skupině. Oproti předpokladu a v rozporu s předcházející studií nebyl zachycen žádný pacient s homozygotní ani s heterozygotní mutací v PROP1 genu ze všech 50 českých probandů s kombinovaným deficitem. Při bližší zpětné analýze můžeme spekulovat o možných důvodech tohoto negativního výsledku: 1) především k němu pravděpodobně vedly omezené šance na záchyt nových pacientů s PROP1 deficitem po uplynutí krátkého času od předešlé studie; 2) kohorta vyšetřovaných probandů byla fenotypově hetorogenní.
Z našich výsledků a z výsledků studií výše uvedených vyplývá, že indikaci vyšetření mutací PROP1 genu je vhodné posuzovat u každého konkrétního pacienta na základě jasně definovaného fenotypu. V úvahu přicházejí pacienti s idiopatickým CPHD, tedy s jasně vyloučenou získanou příčinou – porodem koncem pánevním, kraniotraumatem, postradiačním postižením žlázy, tumorem. Z hlediska spektra hormonálních deficitů by měl být v dětství přítomen deficit GH a TSH, později se může přidružit deficit gonadotropinů a ACTH. Z hlediska morfologie hypofýzy je typická hypoplazie až „empty sella“, ale i přechodná hyperplazie. Do fenotypické charakteristiky však nepatří další anomálie (přerušená hypofyzární stopka nebo septo-optická dysplazie). Vyšší záchyt mutací lze předpokládat u familiárního výskytu CPHD. Těmto kritériím odpovídal fenotyp pěti litevských pacientů, kdy u čtyř byla potvrzena homozygotní mutace PROP1 genu.
Ze všech 94 českých pacientů s idiopatickým izolovaným deficitem GH byl jeden heterozygot pro PROP1 mutaci. Morfologie hypofýzy tohoto pacienta byla normální. Jelikož dědičnost PROP1 mutací je výhradně autozomálně recesivní, tento nález považujeme za náhodný, tedy nesouvisející s deficitem GH u tohoto pacienta.
Naše studie nepotvrdila výtěžnost rutinní diagnostiky PROP1 mutací u rozsáhlé skupiny pacientů s idiopatickým deficitem růstového hormonu, ale svůj význam bezesporu má v její specificky charakterizované podskupině.
V roce 2007 byl publikován zahraniční článek rozšiřující poznatky o regulaci genové exprese PROP1 genu. Metodou komparativní genomiky byly identifikovány tři nekódující konzervativní oblasti, u nichž se předpokládá význam v regulaci transkripce PROP1 genu. Byly lokalizovány v proximální části promotoru genu, uvnitř jeho prvního intronu a za III. exonem PROP1 genu [19]. Můžeme spekulovat, že mutace v této oblasti by mohly být zodpovědné za CPHD u těch fenotypicky definovaných jedinců, u kterých nebyly nalezeny homozygotní či složeně heterozygotní mutace v kódujících oblastech PROP1 genu.
Závěr
Klinický význam vyšetřování PROP1 mutací pro konkrétního pacienta s kombinovaným hypofyzárním hormonálním deficitem splňujícího uvedená fenotypická kritéria je jistě nesporný. U pacientů s homozygotní nebo složeně heterozygotní mutací tohoto genu je možno předvídat postupně se rozvíjející deficit dalších hypofyzárních hormonů (gonadotropinů a ACTH), což umožní včasné zahájení léčby. Dalším klinickým přínosem je, že případný nález hyperplazie hypofýzy nebude zaměněn za nádor a pacient nebude zbytečně operován. Nesporný význam má analýza PROP1 mutací v rodině. U sourozenců postižených jedinců pozitivní nález umožní včas stanovit diagnózu všech hormonálních deficitů a zahájit jejich substituci.
Problematika byla řešena za podpory GAUK č. 50707/2007-2008 a VZ MSM 0021620814.
Děkujeme MUDr. Ondřejovi Cinkovi, PhD., za metodické vedení laboratorní části studie a slečně Kláře Veselé za pomoc v její realizaci.
Došlo: 1. 1. 2009
Přijato: 12. 3. 2009
MUDr. Barbora Obermannová
Pediatrická klinika UK 2. LF
FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: obermannova@seznam.cz
Zdroje
1. Dattani MT, Robinson IC. The molecular basis for developmental disoders of the pituitary gland in man. Clin. Genetics 2000;57 : 337–346.
2. Dattani MT. Novel insights into the aetiology and pathogenesis of hypopituitarism. Hormone Res. 2004;62(3): 1–13.
3. Ingraham HA, Chen RP, Mangalam HJ, et al. A tissue specific transcription factor containing a homeodomain specifies a pituitary phenotype. Cell 1988;55 : 519–529.
4. Wu W, Cogan JD, Pfaffle RW, et al. Mutation in PROP1 cause familiar combined pituitary hormone deficiency. Nature Genet. 1998;18(2): 147–149.
5. Cohen LE, Radovick S. Molecular basis of CPHD. Endocrine Rev. 2002;23(4): 431–442.
6. Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clin. Endocrinol. 2005;63 : 121–130.
7. Mullis PE. Genetic control of growth. Eur. J. Endocrinol. 2005;152 : 11–31.
8. Lebl J, Zapletalová J, Koloušková S, et al. Dětská endokrinologie. 1. vyd. Praha: Galén, 2004.
9. Tajima T, Ohtake A, Hoshino M, et al. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J. Clin. Endocrinol. Metab. 2009;94(1): 314–319.
10. Dattani MT, Preece M. Growth hormone deficiency and releated disorders: insights into causation, diagnosis and treatment. Lancet 2004;363(6): 1997–1985.
11. Jacobson EM, Li P, Leon-del RA, et al. Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev. 1997;11 : 198–212.
12. Duquesnoy P, Roy A, Dastot F, et al. Human Prop-1: cloning, mapping, genomic structure: mutations in familial combined pituitary hormone deficiency. FEBS Lett. 1998;437 : 216–220.
13. Lebl J, Vosáhlo J, Pfaeffle RW, et al. Auxological and endocrine phenotype in a population-based cohort of patients with PROP1 gene defects. Eur. J. Endocrinol. 2005;153 : 389–396.
14. Deladoey J, Fluck C, Buyukgebiz A, et al. „Hot Spot“ in the PROP1 gene responsible for combined pituitary hormone deficiency. J. Clin. Endocrinol. Metab. 1999;84 : 1645–1650.
15. Agarwal G, Bhatia V, Cook S, et al. Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion. J. Clin. Endocrinol. Metab. 2000;85 : 4556–4561.
16. Mendonca BB, Osorio MGF, Latronico AC, et al. Longitudinal hormonal and pituitary imaging changes in two females with combined pituitary hormone deficiency due to deletion of A301,G302 in the PROP1 gene. J. Clin. Endocrinol. Metab. 1999;84 : 942–945.
17. Reynaud R, Magali G, Alexandru S, et al. Genetic screening of combined pituitary hormone deficiency: experience in 195 patients. J. Clin. Endocrinol. Metab. 2006;91(9): 3329–3336.
18. Turton JP, Mehta A, Raza J, et al. Mutations within the transcription factor PROP1 are rare in a cohort of patients with sporadic combined pituitary hormone deficiency (CPHD). Clin. Endocrinol. (Oxf.) 2005;63(1): 10–18.
19. Ward RD, Davis SW, Cho M, et al. Comparative genomics reveals functional transcriptional control sequences in the Prop1 gene. Mamm. Genome 2007;18(6–7): 521–537.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2009 Číslo 6
- Léčba bolesti a horečky u dětí
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
- Gastroezofageální reflux a gastroezofageální refluxní onemocnění u kojenců a batolat
- Očkování nejvíc potřebuje ten, kdo sám být očkován nemůže − kazuistika
- Pokrok v boji s malárií − první vakcína poskytující přijatelnou ochranu proti nemoci
Najčítanejšie v tomto čísle
- Kortikosteroidy a azathioprin v prvním roce léčby Crohnovy nemoci u dětí
- Inhalovaný oxid dusnatý v léčbě těžkého respiračního selhání dětí
- Klinický význam vyšetřování mutací v genu pro transkripční faktor PROP1 u dětí s vrozeným deficitem růstového hormonu
- Dny dětské endokrinologie 2009