
Periodické horečky a další syndromy s poruchou regulace zánětlivé odpovědi
Autori:
A. Šedivá
Pôsobisko autorov:
Ústav imunologie UK 2. LF a FN Motol, Praha
Vyšlo v časopise:
Čes-slov Pediat 2011; 66 (3): 187-191.
Kategória:
Vybrané kapitoly z nové učebnice Klinická pediatrie
1. Periodické horečky a „autoinflammatory syndromes“ – nomenklatura
Oblast periodických horeček a asociovaných zánětlivých stavů představuje nejnovější jasně vymezenou kapitolu v rámci imunodeficiencí, do kterých patří svým jasně definovaným defektem v imunitních mechanismech u některých ze zařazených jednotek. Jedná se však o oblast překrývající se s dalšími, hlavně zánětlivými a autoimunitními onemocněními, a v některých pramenech je možné najít její zařazení do jiné oblasti.
Periodické horečky tvoří podskupinu jednotek, nazývaných v anglicky psané literatuře „autoinflammatory syndromes“. Anglický originál klade velké překážky správné nomenklatuře v české literatuře. Vzhledem k tomu, že názvosloví není jasně upraveno, budeme se v tomto textu držet anglického originálu, který zaručuje jasné zařazení a srozumitelnost.
2. Definice
„Autoinflammatory syndromes“ mají již ve svém názvu obsaženu podstatnou informaci, která říká, že k vzplanutí zánětlivého stavu dochází bez jasné vnější příčiny.
Podstata onemocnění tkví ve složitých mechanismech, které regulují větev nespecifické imunity, vedoucí ke spuštění zánětlivé odpovědi. Tyto velmi účinné mechanismy jsou za normálních okolností pečlivě koordinovány, nicméně u diskutovaných patologických stavů dochází na genetickém podkladě k mutacím genů a následným poruchám molekul účastnících se v procesech aktivace zánětu.
Kritéria zařazení jednotlivých jednotek do této skupiny nejsou jednoznačná s výjimkou Familiární středozemní horečky, u ostatních jednotek se jedná o kombinaci typických klinických příznaků, potvrzených v případech, kdy je genová mutace známa, genetickým vyšetřením.
3. Epidemiologie
Vzhledem k recentním objevům řady onemocnění v této kapitole není epidemiologie až na vzácné výjimky známa a je u některých jednotek výrazně podmíněna etnickou skupinou. U Familiární středozemní horečky je frekvence udávána 0,1 % v turecké populaci, kde byla vzhledem k podstatě onemocnění nejvíce zkoumána.
4. Etiopatogeneze
Patonegeze celé skupiny onemocnění je spojena s mechanismy vrozené imunity. Poruchy vznikají většinou v regulaci zánětlivé odpovědi, která je za normálních okolností po rozpoznání patogenu spuštěna imunitním systémem. U diskutovaných stavů dochází k samovolné aktivaci zánětu, vedoucí posléze ke klinickým příznakům.
Detailní patogeneze je u různých jednotek této skupiny různá a závisí na konkrétní mutaci v daném genu a následné poruše funkce dané molekuly. Základní fakta budou zmíněna u jednotlivých onemocnění.
5. Klinický obraz
Klinický obraz je důsledkem patogenetických pochodů a je tudíž také odlišný u jednotlivých onemocnění. Soubor klinických příznaků vede k diagnóze. Ve většině situací se jedná o známky extrémně vystupňované zánětlivé odpovědi, což se odráží i v názvu celé kapitoly „Periodické horečky“.
Příznaky typické pro dané jednotky jsou uvedeny u každého onemocnění.
6. Diagnostika
Diagnostika těchto stavů je většinou složitá a stojí na pečlivém zhodnocení klinických příznaků typických pro danou jednotku. Pomocnými ukazateli jsou velmi významně zvýšené markery zánětu, hlavně CRP, které dosahuje v době ataky stovkových hodnot. Nespecifickou pomocí je laboratorní průkaz excesivní sekrece interleukinu 1 stimulovanými buňkami pacienta. Klinický obraz vede k podezření na nozologickou jednotku, kdy je poté jedinou možností potvrzení diagnózy genetické vyšetření a nalezení příčinné mutace. Tato vyšetření nejsou však široce dostupná a i v případech, kdy jsou provedena, není mutace nalezena u většiny pacientů. Tato oblast je v dynamickém vývoji a dá se očekávat velmi rychlé zlepšení situace.
7. Diferenciální diagnóza
Diferenciální diagnostika těchto stavů je mimořádně obtížná. Na prvním místě stojí horečky způsobené infekčními činiteli, kdy se uplatňují všechna vyšetření indikovaná u infekčních onemocnění. Ve složitých rozvahách může pomoci vyšetření hladiny prokalcitoninu, markeru bakteriální infekce, který se však do určité míry může zvyšovat i u periodických horeček.
Z dalších diferenciálně diagnostických stavů připadají v úvahu zánětlivá onemocnění ze skupiny systémových autoimunitních onemocnění, zvláště systémové formy revmatoidní artritidy nebo počáteční stadia dalších systémových autoimunit. V těchto případech může pomoci vyšetření autoprotilátek, které však zvláště v dětském věku nemusí být přítomny ani u autoimunitních onemocnění.
8. Terapie
Terapie uvedených stavů je složitá a vedla dosud pouze k omezenému efektu. V léčbě byla zkoušena celá řada preparátů s cílem zabránit atakám aktivace zánětu. Zprávy o těchto léčebných strategiích ukazují na omezený a individuální efekt terapie. Uvedená nová fakta o patogenetických podkladech onemocnění otevřela cestu biologickým terapiím, a to v prvé řadě blokádě IL-1. Anakinra, rekombinantní antagonista lidského IL-1beta receptoru, je v současné době nejúspěšnější strategií v prevenci atak a nese příslib do budoucna v možném omezení rizika vzniku amyloidózy. Léčebné použití bylo dokumentováno u skupiny MWS, FCU a NOMID/CINCA, dále u TRAPS a PAPA u jednotlivých pacientů (specifikace jednotlivých onemocnění jsou uvedeny v tabulce 1). Vzhledem k dominantní úloze IL-1beta v patogenezi onemocnění lze očekávat další zprávy o léčebném potenciálu této terapie. Podobný mechanismus má látka nazývaná IL-1 Trap, inhibitor IL-1.
Pozitivní efekt byl zaznamenán i u inhibice TNF, hlavně u použití etanerceptu, p75TNFR:Fc fúzního proteinu, u TRAPS. Nadějný je hlavně preventivní účinek v rozvoji amyloidózy, který se ukazuje u některých léčených pacientů. Stejná léčba se podle literárních údajů používala i u HIDS, kde jsou možnosti terapie omezené, s částečným individuálním efektem. U HIDS byl též zkoušen Simvastatin, zasahující do stejné metabolické cesty. Efekty léčby HIDS jsou velmi rozporuplné.
Klasicky uznávanou léčbou je použití kolchicinu u FMF, kde se s úspěchem využívá k potlačení akutních atak i k prevenci amyloidózy. Kolchicin se váže na beta-tubulin a inhibuje tvorbu mikrotubulů v buňce. Kromě toho má nezávislé protizánětlivé účinky, přesný mechanismus ovlivnění patogeneze FMF však znám není.
Přechodné nespecifické potlačení zánětu lze dosáhnout krátkou kúrou kortikoidů. Strategie této aplikace se liší, hlavně u PFAPA je možné i jednorázové podání, u dalších jednotek po individuálním zvážení 3–4denní kúra s velmi rychlým poklesem dávek, počáteční dávky nepřesahují 1 mg Prednisonu na kg váhy a dávku.
9. Prognóza krátkodobá/dlouhodobá
Prognóza onemocnění je odlišná podle jednotlivých postižení a tíže klinických příznaků. Obecně se tíže a frekvence příznaků lepší s věkem. Hlavním faktorem, ovlivňujícím prognózu, je možný vznik amyloidózy, která je rizikem hlavně u Familiární středozemní horečky. Vznik této komplikace je třeba pečlivě sledovat a správnou léčbou nemoci jí předcházet.
10. Primární a sekundární prevence
Prevence je u geneticky podmíněných onemocnění složitá. U jasně prokázaných případů Familiární středozemní horečky či dalších jednotek se uplatňují zásady genetického poradenství. V případě již vzniklé choroby je třeba řádné léčby a potlačení zánětu k omezení rizika vzniku amyloidózy.
11. Přehled onemocnění spadajících do kategorie Periodické horečky a „Autoinflammatory syndromes“
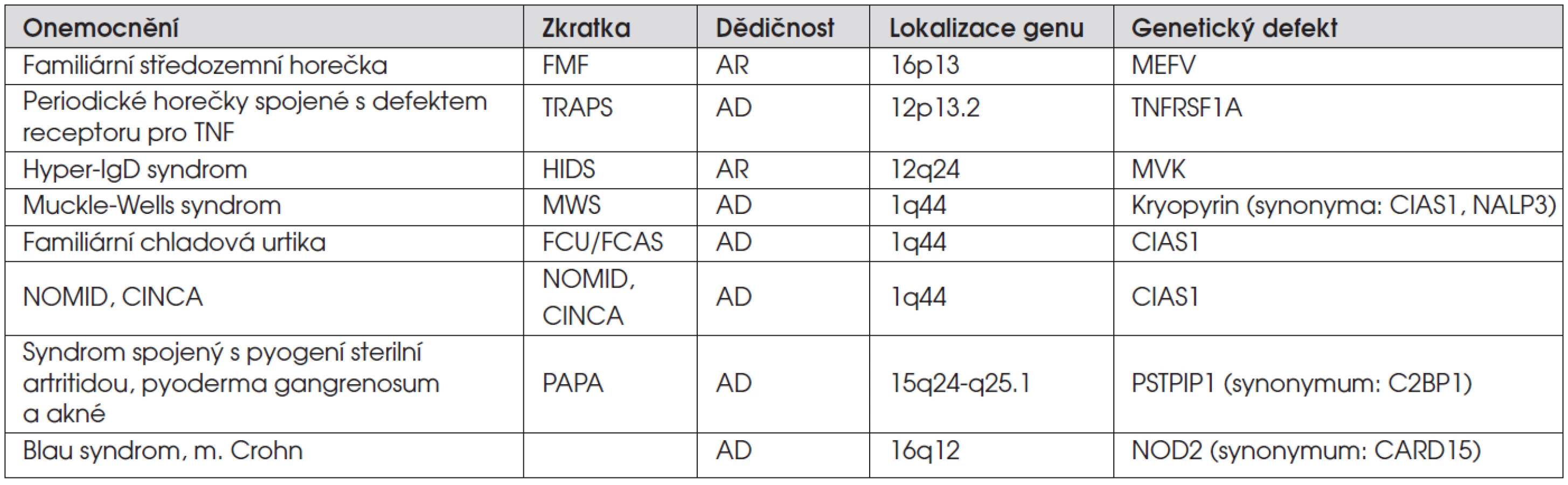
Vlastní skupina periodických horeček je představována Familiární středozemní horečkou (Familiar Mediteraneen Fever, FMF), hyper-IgD syndromem (HIDS), periodickými horečkami spojenými s defektem receptoru pro TNF (TNF-receptor associated periodic syndrome, TRAPS). K těmto klasickým stavům se dále řadí tak zvané kryopyrinopatie, spojené dohromady jednotnou genetickou etiologií s různou tíží klinických příznaků: syndromy Muckle-Wells (MWS), familiární chladová urtika (FCU) a systémové zánětlivé nemocnění se začátkem v novorozeneckém věku, charakterizované neurologickým, kožním a kloubním postižením (neonatal onset multisystem inflammatory disease NOMID, se synonymem chronic infantile neurologic cutaneous and articular syndrom, CINCA).
Později se do celé skupiny zařadil syndrom spojený s pyogenní sterilní artritidou, pyoderma gangrenosum a akné (PAPA). Posledním příspěvkem do klasického rámce periodických horeček je Periodická granulomatózní artritida. Tento název v sobě spojuje onemocnění nazývaná v literatuře Blau syndrom nebo „early onset sarcoidosis“. Podle posledních poznatků se však jedná o totožné stavy pouze s historicky různým popisem a v literatuře byl navržen výše uvedený název zahrnující tyto granulomatózní záněty. Onemocnění jasně spadají do diskutované kapitoly a jsou správně uváděna mezi periodickými horečkami, i když paradoxně horečka nepatří k jejich základním klinickým příznakům.
Familiární středozemní horečka (Familiar Mediteraneen Fever, FMF)
FMF je nejdéle známým onemocněním z celé skupiny, zmínky o této jednotce byly publikovány již před 70 lety. FMF je autozomálně recesivní onemocnění, charakterizované opakovanými atakami horeček a serositidami. Epizody horečky bývají relativně kratší, od několika hodin do několika dní. U naprosté většiny pacientů bývá udávána bolest břicha, postiženy bývají klouby, změny mohou být i na kůži v podobě exantému. Nejzávažnějším příznakem, který může u části pacientů komplikovat průběh onemocnění a který určuje prognózu onemocnění, je amyloidóza. Kromě genetického průkazu mutací nejsou pro diagnostiku FMF k dispozici žádná specifická laboratorní vyšetření.
Gen, jehož mutace způsobují FMF, byl objeven v roce 1997 (tab. 1). Gen pro MEFV je lokalizován na krátkém raménku 16. chromozomu. Frekvence mutací v populaci lze jen těžko stanovit, neboť se výrazně liší v jednotlivých populacích. Alespoň některé z dosud identifikovaných mutací daného genu mají základ v původních mutacích, vzniklých přibližně před 2500 lety na Středním východě. Z této lokality docházelo ke stěhování národů, včetně větve směřující na severozápad, a tudíž není vyloučen výskyt onemocnění i v našich krajích. Gen kóduje protein nazývaný pyrin. Pyrin je v buňkách asociován se strukturami cytoskeletu a zasahuje do tří velmi důležitých funkčních celků buňky – moduluje apoptózu, produkci cytokinů, hlavně IL-1, a účastní se na funkci cytoskeletu.
Hyper-IgD syndrom (HIDS)
Hyper-IgD syndrom byl poprvé popsán v roce 1984. Jedná se o autozomálně recesivní onemocnění, způsobené mutacemi genu pro mevalonát kinázu. Onemocnění poněkud vybočuje z řady ostatních jednotek. Jde vlastně o vrozenou poruchu metabolismu, která sdílí genový defekt s mevalonovou aminoacidurií, OMIM 251170. Podstatou jsou mutace v genu pro mevalonát kinázu, MVK, která řídí jeden z kroků biosyntézy cholesterolu a isoprenoidů. Mevalonová acidurie a HIDS jsou vlastně totožná onemocnění, s rozdílně vyjádřeným fenotypem, způsobené mutacemi v totožném genu. Zatímco mutace způsobující HIDS jsou rozptýleny po celém genomu, ty, které způsobují mevalonovou acidurii, jsou soustředěny do oblasti, která kóduje oblast mezi aminokyselinami 243-334 mevalonát kinázy. Horečky jsou přítomny u obou jednotek, jsou však dominantním znakem u HIDS.
Horečky a další klinické příznaky HIDS vznikají většinou v útlém dětství. Bývají velmi vysoké, trvají většinou několik dní. Ataka bývá provázena krční lymfadenitidou, někdy exantémem, bolestmi břicha, bolestmi svalů a kloubů, někdy i zvracením. U části pacientů se mohou objevit i slizniční ulcerace, hlavně v dutině ústní. Vysoká hladina IgD je sice typická pro onemocnění, nicméně hlavně u malých dětí může být hladina IgD v mezích normy, na druhou stranu vyšší IgD se může vyskytnout i u jiných typů periodických horeček. Zvýšení IgD bývá provázeno i zvýšením IgA. Ataky se mohou vyskytovat celý život, ale většinou jsou nejčastější v dětství a dospívání. Amyloidóza provází HIDS spíše výjimečně.
Periodické horečky spojené s defektem receptoru pro TNF (TNF-receptor associated periodic syndrome, TRAPS)
Autozomálně dominantně je děděna skupina nemocí nazývaných nyní TRAPS. TRAPS shrnuje již dříve popisovaná onemocnění, jako je například Familial Hibernian Fever či Familiar Periodic Fever. První popis této jednotky byl zveřejněn právě pod názvem Familiar Hibernian Fever. Popis rozsáhlé rodiny umožnil také zjištění autozomálně dominantní dědičnosti.
Onemocnění je způsobeno mutacemi v genu kódujícím podjednotku receptoru pro TNF, nyní nazývanou TNFRSF1A. Mutace jsou soustředěny v extracelulární části 55kD podjednotky TNFRSF1A. Klinický obraz TRAPS zahrnuje opakované ataky horeček provázené u většiny pacientů stěhovavými myalgiemi. Bývá přítomna bolest břicha, dále konjunktivitida a periorbitální edém. Kožní projevy ve formě makul a erytému jsou taktéž velmi častou manifestací. Nicméně klinické projevy mohou být velmi variabilní a u některých pacientů jen velmi mírně či částečně vyjádřeny. Na rozdíl od hyper-IgD syndromu může být TRAPS provázen amyloidózou. V laboratorním obraze bývají nespecifické známky zánětu, vyšší CRP, vyšší FW. Může být i mírná aktivace komplementu. I při tomto onemocnění můžeme zaregistrovat zvýšení IgD či IgA.
Kryopyrinopatie – MWS, FCU a NOMID/CINCA
Muckle-Wells syndrom (MWS), familiární chladová urtika (FCU, někdy také FCAS, familiar cold autoinflammatory syndrome) a systémové zánětlivé onemocnění se začátkem v novorozeneckém věku, charakterizované neurologickým, kožním a kloubním postižením (neonatal onset multisystem inflammatory disease, NOMID, se synonymem chronic infantile neurologic cutaneous and articular syndrome, CINCA)
Tato tři autozomálně dominantní, zdánlivě odlišná onemocnění jsou spojena svou genovou podstatou. Jsou způsobena mutacemi v jednom genu nazývaném CIAS1 (zkratka z cold induced autoinflammatory syndrome), v současné době spíše citovaný pod synonymem NALP3.
Patogeneze MWS, FCU a NOMID/CINCA je úzce spojena s funkcí proteinu kódovaného uvedeným genem, jímž je kryopyrin, klíčová molekula v iniciálních fázích zánětlivé odpovědi. Gen CIAS1/NALP3 byl původně spojen s MWS, dále s FCU a posléze s NOMID/CINCA.
MWS
MWS je syndrom opakovaných horeček spojený často s artralgiemi, bolestmi břicha, urtikou a v pozdějším věku se senzorineurální hluchotou. Nemoc může také být komplikována amyloidózou hlavně s renálním postižením. Začátek onemocnění je variabilní, může být kdykoli v průběhu dětství a dospívání, vzácněji i v dospělosti. Někdy mohou být přítomny i další klinické příznaky, jako jsou konjunktivitidy a uveitidy, únava a afty. Hluchota je pro MVS velmi typická a vzniká u více než 70 % pacientů.
FCU (FCAS)
FCU je velmi podobné onemocnění, včetně možné přítomné hluchoty, navíc však s chladem jako jasným precipitujícím faktorem urtiky a dalších popsaných klinických příznaků. Ataky vyvolává celková expozice chladu, ale na rozdíl od získané chladové urtiky pouze lokální expozice chladu nevyvolá typické klinické symptomy. Typickým nálezem jsou výrazné známky zánětu v době ataky, hlavně velmi výrazná leukocytóza.
NOMID/CINCA
Syndrom NOMID/CINCA je spojen s chronickým postižením kůže ve formě urtikariálních až vaskulitických lézí, s meningitidou, posléze stejně jako u předchozích jednotek se senzorineurální hluchotou a dále s deformující artropatií. Typickým projevem je začátek v neonatálním či velmi časném věku. Často se jedná o nedonošené děti, u kterých se velmi časně objevuje kožní postižení, s pozdějším rozvojem dalších typických neurologických, kloubních a kostních příznaků. Typické známky časné osifikace pately a epifýz dlouhých kostí může pomoci ve složitém diagnostickém procesu.
Onemocnění je nejzávažnější formou z celé skupiny periodických horeček a má významnou úmrtnost již v dětském věku.
Pyogenní sterilní artritida, pyoderma gangrenosum a akné (PAPA)
Poslední z autozomálně dědičných poruch regulace zánětu je PAPA. Jak je již obsaženo v názvu, jedná se o pyogenní sterilní artritidu, pyoderma gangrenosum a akné.
Zařazení do této skupiny onemocnění umožnilo objevení genové podstaty v roce 2002. Mutace byly objeveny v genu CD2BP1, zkratka z CD2 binding protein 1. Myší analog tohoto genu je PSTPIP1 (proline-serine-threonin phosphatase interacting protein), gen je tedy někdy nazýván CD2BP1/PSTPIP1 a odpovídající protein CD2BP1/PSTPIP1. Tento protein se přímo váže na pyrin, protein spojený s FMF, a stejně jako pyrin je spojený s cytoskeletálním aparátem buňky.
Crohnova nemoc
Blau syndrom, EAS (early onset sarcoidosis)
Výzkumné směry vedoucí k odhalení patogeneze těchto onemocnění mapují složité pochody, ve kterých složky nespecifické imunity zahajují zánětlivou odpověď. Složité uspořádání těchto procesů a jejich kontrolní kroky jsou jemně regulovány. V genezi i v regulaci zánětu se zde podílí celá řada molekul. Při postupném odhalování těchto pochodů dochází i k dalším objevům s návazností na klinické jednotky. Ve spojitosti s objevem NOD2, který je jedním z rodiny NOD proteinů (nucleotide-binding oligomerization domain proteins), se do celé skupiny zařadila dvě onemocnění spojená právě s NOD2, a to Blau syndrom, onemocnění, které nyní prakticky splynulo s jednotkou nazývanou EOS, early onset sarcoidosis, a Crohnova nemoc. NOD2 je zapojen podobně jako kryopyrin do iniciace zánětlivé odpovědi, jejíž porucha je již delší dobu dávána do souvislosti s patogenezí Crohnovy nemoci.
Blau syndrom, či EOS, je familiární granulomatózní artritida, neuropatie, urtika. Právě trias artritida, uveitida a dermatitida je typickým znakem těchto onemocnění. Většinou se m. Blau považuje za familiární formu a EOS za případy na podkladě de novo mutací, ale toto rozdělení lze někdy jen těžko sledovat.
PFAPA
Nejasnosti panují kolem syndromu nazývaného „periodic fever, aphtous stomatitis, pharyngitis, cervical lymphadenopathy“ (PFAPA), který dosud není asociován se žádným genem z dané skupiny a jehož patogeneze není známa, ale dá se očekávat, že doplní diskutované spektrum onemocnění.
PFAPA je definován pouze na klinickém podkladě a zahrnuje opakované horečky s lymfadenitidou, faryngitidou a přítomností aftů. Při takovém popisu je však spektrum stavů spadajících pod danou definici široké a syndrom stále čeká na své upřesnění.
Schnitzler syndrom
Ještě větší záhadou je onemocnění nazývané Schnitzler syndrom, také zařazené do periodických horeček. Jedná se o onemocnění spíše dospělého věku, charakterizované typickým spojením horeček, kožního postižení ve formě urtikariální dermatitidy a monoklonální gamapatie IgM. Molekulární podstata není vůbec známa, což komplikuje terapeutické zásahy. Jedná se o vzácné onemocnění, na celém světe bylo pospáno pouze několik desítek pacientů.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2011 Číslo 3
- Gastroezofageální reflux a gastroezofageální refluxní onemocnění u kojenců a batolat
- I „pouhé“ doporučení znamená velkou pomoc. Nasměrujte své pacienty pod křídla Dobrých andělů
Najčítanejšie v tomto čísle
- Skríning kritických vrodených chýb srdca u novorodencov pulznou oxymetriou v regióne severného Slovenska
- Poruchy príjmu potravy z pohľadu pedopsychiatra
- Vybrané kapitoly z dětské onkologie a transplantace kostní dřeně
- Periodické horečky a další syndromy s poruchou regulace zánětlivé odpovědi