
Funkce plic souboru českých kojenců a batolat s cystickou fibrózou
Lung function in a group of Czech infants with cystic fibrosis
Objective: To assess lung function in a group of Czech infants with cystic fibrosis (CF) and to evaluate safety of the infant pulmonary function testing (iPFT) in these patients.
Methods: Fifty-five infants (age range 5–118 weeks) with classic form of CF clinically indicated for iPFT in our lab were sedated with chloralhydrate and underwent multiple breath inert gas washout test, bodypletysmography, tidal breath analysis, single occlusion compliance-resistance measurement and tidal volume rapid thoracoabdominal compression. During iPFT and until full recovery from sedation, haemoglobin saturation, pulse rate, minute ventilation and clinical correlates of lung aspiration were monitored. Lung function was compared to the available international normative values and between clinically defined subgroups (based on patient’s age, microbial colonisation, occurrence of severe pulmonary exacerbations (PEx), history of meconium ileus and exocrine pancreatic function).
Results: Lung function impairment was detected in 63.9% of infants with the most frequent impairment being ventilation inhomogeneity (51.2% of all), followed by hyperinflation (48.6% of all) and peripheral airway obstruction (50.0% of all). Lung function was significantly diminished in older patients (mean age 16.6 vs 2.0 months), patients with at least one PEx, intermittent Pseudomonas aeruginosa and chronic Staphylococcus aureus colonization. Only 1 adverse event occurred – brief desaturation down to 85%.
Conclusion: Lung function impairment may be detected by iPFT as early as in 2 months-aged CF infants, its frequency increases with age. Lung function differs between clinically defined subgroups of patients. iPFT is safe.
Keywords:
Cystic fibrosis – Infants – toddler – multiple breath inert gas washout test – bodypletysmography – tidal breath analysis – single occlusion compliance-resistance measurement – tidal volume rapid thoracoabdominal compression – Safety
Autoři:
V. Koucky; V. Skalická; J. Bartošová; T. Doušová; V. Vávrová; P. Pohunek
Působiště autorů:
Pediatrická klinika 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Čes-slov Pediat 2019; 74 (7): 392-400.
Kategorie:
SYMPOZIUM: cystická fibróza
Souhrn
Cíl studie: Zhodnotit funkci plic českých kojenců a batolat s cystickou fibrózou (CF), posoudit bezpečnost metod pro vyšetření funkce plic nespolupracujících dětí (infant pulmonary function testing – iPFT) u CF pacientů.
Metody: Padesát pět dětí s klasickou formou CF (věkové rozmezí 5–118 týdne) klinicky indikovaných k iPFT v naší laboratoři bylo po navození sedace chloralhydrátem vyšetřeno pomocí testu vícedechového vyplavování inertního plynu z plic, bodypletysmografie, analýzy klidového dechového vzoru, měření pružných vlastností dýchacího traktu a thorakoabdominální komprese. Během vyšetření a do úplného probuzení byla u pacientů monitorována saturace hemoglobinu kyslíkem, pulz, minutová ventilace a klinické příznaky aspiračních epizod. Výsledky funkce plic byly porovnány s dostupnými mezinárodními normami a dále mezi klinicky definovaným podskupinami pacientů (věk, mikrobiální kolonizace, výskyt těžkých exacerbací, anamnestický údaj mekoniového ileu a pankreatická in/suficience).
Výsledky: Porucha funkce plic byla detekována u 63,9 % dětí. Nejčastěji se jednalo o zvýšenou nehomogenitu ventilace plic (51,2 % vyšetřených), dále hyperinflaci plic (48,6 % vyšetřených) a obstrukci periferních dýchacích cest (50,0 % vyšetřených). Funkce plic byla signifikantně horší u dětí starších (průměrný věk 16,6 měsíce) než mladších (věk 2,0 měsíce), dále u dětí s alespoň jednou těžkou exacerbací v anamnéze, intermitentním záchytem Pseudomonas aeruginosa nebo chronickou kolonizací Staphylococcus aureus. Zaznamenána byla pouze 1 nežádoucí epizoda – krátká desaturace.
Závěry: Porucha funkce plic může být detekována pomocí iPFT u významné části kojenců s CF již ve věku 2 měsíců. Ve vyšších věkových kategoriích její výskyt narůstá. Funkce plic se liší mezi klinicky definovanými podskupinami pacientů. Metody iPFT jsou bezpečné.
Klíčová slova:
cystická fibróza – kojenci – batolata – test vícedechového vyplavování inertního plynu z plic – bodypletysmografie – analýza klidového dechového vzoru – měření pružných vlastností dýchacího traktu – thorakoabdominální komprese – bezpečnost
ÚVOD
Progresivní postižení dýchacího traktu je hlavní příčinou morbidity i mortality pacientů s cystickou fibrózou (CF). Na základě recentních výzkumů u pacientů s CF diagnostikovaných pomocí novorozeneckého screeningu bylo zjištěno, že porucha funkce plic může být přítomna již u nejmladších kojenců ve věku kolem 3 měsíců věku [1]. Stejně tak strukturální změny na CT plic byly zjištěny již u relativně mladých dětí s CF [2, 3]. Jakkoliv je toto zjištění zajímavé, klinicky významnější se jeví otázka reverzibility těchto změn a možnosti jejich terapeutického ovlivnění. Odpověď na ni mohou poskytnout data z longitudinálních kohortových studií sledujících vývoj zdravotního stavu včetně funkce plic kojenců s CF diagnostikovaných na podkladě novorozeneckého screeningu. Aktuálně jsou taková data k dispozici především z Velké Británie (LCFC – London Cystic Fibrosis Collaboration) a z Austrálie (AREST CF – Australian Respiratory Early Surveillance Team for Cystic Fibrosis), jejichž závěry jsou poněkud rozporuplné a ne zcela aplikovatelné do českého prostředí (např. kvůli absenci dlouhodobé antibiotické profylaxe infekce Staphylococcus aureus).
Vyšetření funkce plic u nespolupracujících dětí ve věku do cca 2 let je velmi náročné, vyžaduje sofistikované přístrojové vybavení a zázemí specializované laboratoře. Centrum pro cystickou fibrózu při Pediatrické klinice 2. LF UK a FN Motol disponuje od roku 2013 plným spektrem diagnostických nástrojů umožňujících hodnocení funkčního stavu respiračního traktu v této věkové kategorii. V návaznosti na to byl ve FN Motol zaveden program sledování stavu funkce plic u kojenců a batolat s CF, v jehož rámci byli pacienti vyšetřováni ve věku jednoho roku. Následně byl protokol modifikován a v současné době podstupují pacienti první vyšetření krátce po stanovení diagnózy a poté s cca ročním odstupem.
V tomto článku předkládáme první výsledky průřezové studie hodnotící funkci plic souboru českých kojenců a batolat s CF vyšetřených v období mezi únorem 2014 a únorem 2019. Prezentována budou i pilotní data z longitudinálního sledování vybraných pacientů s CF. Vzhledem k limitovanému rozsahu článku pro podrobnější informace odkazujeme čtenáře na disertační práci hlavního autora s názvem „Detekce časných patofy-ziologických změn dýchání u dětí s chronickým plicním onemocněním“.
METODIKA
Pacienti
Do studie bylo zařazeno 55 českých kojenců a batolat s klasickou formou CF, kteří podstoupili vyšetření v laboratoři plicní funkční diagnostiky Pediatrické kliniky 2. LF UK a FN Motol v období mezi únorem 2014 a únorem 2019. Indikaci k vyšetření stanovovaly ošetřující lékařky výhradně na základě komplexního hodnocení klinického průběhu onemocnění (posouzení „rizikovosti pacienta“). Diagnóza CF byla stanovena podle mezinárodně platných kritérií [4]. Věk v době vyšetření se pohyboval od 5 týdnů do 29,5 měsíců (medián 53,4 týdne). Kromě základních antropometrických parametrů (věk, pohlaví, výška, váha a BMI včetně jejich Z-skóre vztaženého k české normě) byly hodnoceny další klinické charakteristiky – genotyp, mikrobiální kolonizace, počet těžkých exacerbací vztažený k věku dítěte při vyšetření, počet dní léčby perorálními antibiotiky vztažený k věku dítěte, zevně sekretorická funkce pankreatu (pankreatická insuficience či suficience) a mekoniový ileus v anamnéze (tab. 1 a 2).

Mutace vyskytující se ve vyšetřovaném souboru (legacy označení): F508d, Y1424X, 526_527delAT, 1717-2A>G; G542X; CFTRdele2.3(21kb);
1774delCT; N1301K; 2789+5G>A; 2143delT; 621+1G>T, 1204_1205delGT, 2789+5G>A; I507del; 1341+1G>A; G551D; G542X; 574delA, N1303K;
3659delC; 2184insA.

Mikrobiální kolonizace byla hodnocena v sekretech z horních cest dýchacích (hluboký nasofaryngeální aspirát) s ohledem na výskyt tří bakterií: Staphylococcus aureus, Pseudomonas aeruginosa a Burkholderia cepacia komplex. Chronická kolonizace byla definována jako pozitivita více než poloviny vzorků v průběhu jednoho roku (při vyšetření alespoň 4 vzorků za rok), intermitentní jako méně než polovina pozitivních vzorků. V případě komplexu Burkholderia cepacia byl hodnocen pouze její výskyt nehledě na frekvenci. Těžká exacerbace byla definována jako nový výskyt respiračních symptomů, resp. zhoršení stávajících příznaků vyžadující léčbu intravenózními antibiotiky (ATB) či hospitalizaci z jiných důvodů (těžká dušnost s nutností oxygenoterapie, neschopnost adekvátního perorálního příjmu, selhání dosavadní ambulantní léčby atp.). Perorální ATB léčba byla započítávána pouze v případě léčby respiračních příznaků (infekce jak horních, tak i dolních cest dýchacích podle klinických příznaků). Pankreatická suficience byla posuzována na základě hodnoty elastázy-1 ve stolici (hodnoty větší nebo rovné 200 μg/g značí pankreatickou suficienci).
Pro účely statistické analýzy byly v rámci celého souboru pacientů definovány klinicky relevantní podskupiny podle následujících kritérií:
- věk: skupina mladších dětí (CFmalí) – kojenci ve věku do 6 měsíců a skupina starších dětí (CFvelcí) – kojenci a batolata mezi 6 a 30 měsíci;
- mikrobiální kolonizace: chronická a intermitentní kolonizace Staphylococcus aureus, chronická a intermitentní kolonizace Pseudomonas aeruginosa, záchyt komplexu Burkholderia cepacia;
- výskyt těžkých exacerbací: skupina bez těžké exacerbace a skupina s alespoň jednou těžkou exacerbací;
- přítomnost mekoniového ileu (ano × ne);
- přítomnost pankreatické suficience (ano × ne).
Vyšetření funkce plic
Vyšetření funkce plic bylo provedeno v arteficiálně navozeném spánku (chloralhydrát 80–100 mg/kg p. r.), který zajišťuje dobrou toleranci obličejové masky a dostatečně pravidelný dechový vzor nezbytný pro spolehlivou interpretaci naměřených dat. Vyšetřováni byli kojenci a batolata v klinicky stabilním stavu (tzn. alespoň 2 týdny bez známek respirační exacerbace). Pacienti byli umístěni do supinní polohy s hlavou v neutrální poloze vzhledem k trupu. Obličejová maska Rendel–Barker č. 0, 1 nebo 2 byla utěsněna kolem úst a nosu speciální pastou.
Vyšetřovací protokol zahrnoval pět a od března 2016 šest metod prováděných ve fixním pořadí:
- test vícedechového vyplavování dusíku z plic (N2-MBW);
- test vícedechového vyplavování hexafluoridu síry z plic (SF6-MBW) – dostupný od března 2016;
- bodypletysmografie (BP);
- analýza klidového dechového vzoru (Tidal);
- měření pružných vlastností dýchacího traktu (RC) – poddajnost a odpor dýchacího traktu;
- klidová thorakoabdominální komprese (TV-RTC) – parciální usilovná křivka průtok-objem.
Výstupní parametry jednotlivých metod vyšetření včetně jejich definic jsou uvedeny v tabulce 3. Technika jednotlivých vyšetřovacích metod, hodnocení kvality měření a postup při interpretaci výsledků odpovídaly mezinárodním doporučením [5–11]. Vzhledem k časové náročnosti celého protokolu a limitované době arteficiálně navozeného spánku však nebylo možno u všech dětí dokončit celý protokol. Různá technická náročnost vyšetřovacích metod vedla též k jejich odlišné úspěšnosti (ne všechna naměřená data dosáhla akceptovatelné kvality pro spolehlivou interpretaci).

Získané výsledky funkčního vyšetření u našich pacientů s CF byly porovnány s dříve publikovanými referenčními hodnotami [12–15]. Ačkoliv je tento způsob porovnání v případě iPFT považován obecně za problematický, do předkládaného článku jsme tato hodnocení začlenili ze dvou důvodů. Prvním jsou etické okolnosti, pro které nejsou v naší funkční laboratoři sedovány zdravé děti (normální hodnoty parametrů funkce plic českých dětí ve věkové kategorii do 2 let tedy nejsou k dispozici). Porovnání s normálními hodnotami ze zahraničních pracovišť je tak jedinou cestou, jak získat přehled o poruše funkce plic kojenců s CF ve vztahu ke zdravým kontrolám. Druhým důvodem je fakt, že metodika vyšetření funkce plic kojenců je v současné době standardizována natolik, že byl minimalizován vliv technických aspektů na absolutní hodnoty měřených parametrů. K dispozici jsou aktuálně moderní referenční hodnoty, jejichž metodika plně odpovídá té, již používáme v naší laboratoři. Výjimku v této oblasti představuje N2-MBW, jehož použití u kojenců s CF je v našem centru unikátní.
Bezpečnost vyšetření byla hodnocena podle výskytu nežádoucích událostí, jakými jsou významné desaturace (pokles SpO2 pod 90 % alespoň na 15 sekund), epizody hypoventilace (pokles ventilace pod 150 ml/min/kg po dobu alespoň 30 sekund), apnoe (zástava dechu trvající déle než 15 sekund) a bradykardie (pokles tepové frekvence pod 80/min po dobu 30 sekund) a klinické koreláty odpovídající aspiračním příhodám. V průběhu celého vyšetření a dále do plného probuzení byli pacienti na kontinuálním monitoru pulzu a saturací.
Pro porovnání vyšetřeného souboru pacientů s obecnou českou populací kojenců a batolat s CF a zhodnocení reprezentativnosti výběru byla použita data z Českého registru cystické fibrózy. Vybrány byly všechny žijící děti s klasickou formou CF narozené v letech 2013 až 2017. Z výročních zpráv registru byly získány relevantní údaje o klinickém průběhu jejich onemocnění a tyto byly porovnány s vyšetřeným souborem pacientů.
Statistická analýza
V celém vyšetřovaném souboru pacientů bylo u spojitých proměnných ověřeno normální rozdělení dat pomocí Kolmogorovova-Smirnovova testu. V případě dat s normálním rozdělením byl pro porovnání jednotlivých skupin pacientů využit t-test, resp. párový t-test pro závislé vzorky. Data bez normálního rozdělení byla porovnávána pomocí Mannova–Whitneyova U testu. V případě kategorických proměnných bylo porovnání mezi dvěma klinicky definovanými skupinami provedeno pomocí testu rozdílu dvou podílů. Porovnání naměřených hodnot parametrů funkce plic vzhledem k referenčním hodnotám bylo provedeno pomocí Z-skóre vypočítaného jako:
Z-skóre = (x-μ)/σ,
kde x značí naměřenou hodnotu parametru, μ jeho průměr v referenční populaci a σ jeho směrodatnou odchylku v referenční populaci. V případě vyjádření parametrů jako Z-skóre byl za normální rozmezí považován interval ±2 směrodatné odchylky (SD). Výjimkou byly parametry tPTEF/tE a V’maxFRC, u nichž je klinicky významné pouze jejich snížení. V případě parametru Raw bylo použito relativní vyjádření v % normy, jelikož nejsou k dispozici data umožňující spočítat Z-skóre. U parametru Raw bylo za klinicky významné považováno pouze jeho zvýšení nad 145 % (~ +2 SD). Porucha výživy byla pro účely této práce definována jako Z-skóre BMI pod -1,3 a porucha růstu jako Z-skóre délky (výšky) pod -2.
VÝSLEDKY
Základní charakteristiky souboru
Celkem byly k dispozici výsledky funkčního vyšetření od 48 (87,3 %) kojenců a batolat s CF, z toho 21 (43,8 %) bylo chlapců. Průměrný věk ± směrodatná odchylka vyšetřených pacientů byl 50,7 ± 33,1 týdne s širokým rozmezím 5–118 týdne. Medián věku byl 53,4 týdne. Ve skupině pacientů do 6 měsíců věku bylo 13 dětí. Celkem 16 pacientů bylo homozygotních pro mutaci F508del, 26 pacientů bylo složenými heterozygoty (F508del v kombinaci s jinou CF způsobující mutací) a 6 pacientů bylo složenými heterozygoty pro CF způsobující mutaci jinou než F508del. V naprosté většině případů se jednalo o mutace I.–III. funkční třídy podle De Boeck a Amaral, predikující závažnější průběh onemocnění. Průměrná hmotnost, BMI a výška včetně jejich Z-skóre jsou uvedeny v tabulce 1. Tabulka 2 udává další klinické charakteristiky studovaného souboru (mekoniový ileus, zevně-sekretorická funkce pankreatu, mikrobiální kolonizace ve skupině nad 6 měsíců věku, výskyt poruchy růstu a výživy, počty těžkých exacerbací za rok a počet dní užívání perorálních ATB).
Porovnání vyšetřeného vzorku pacientů s obecnou populací pacientů s CF
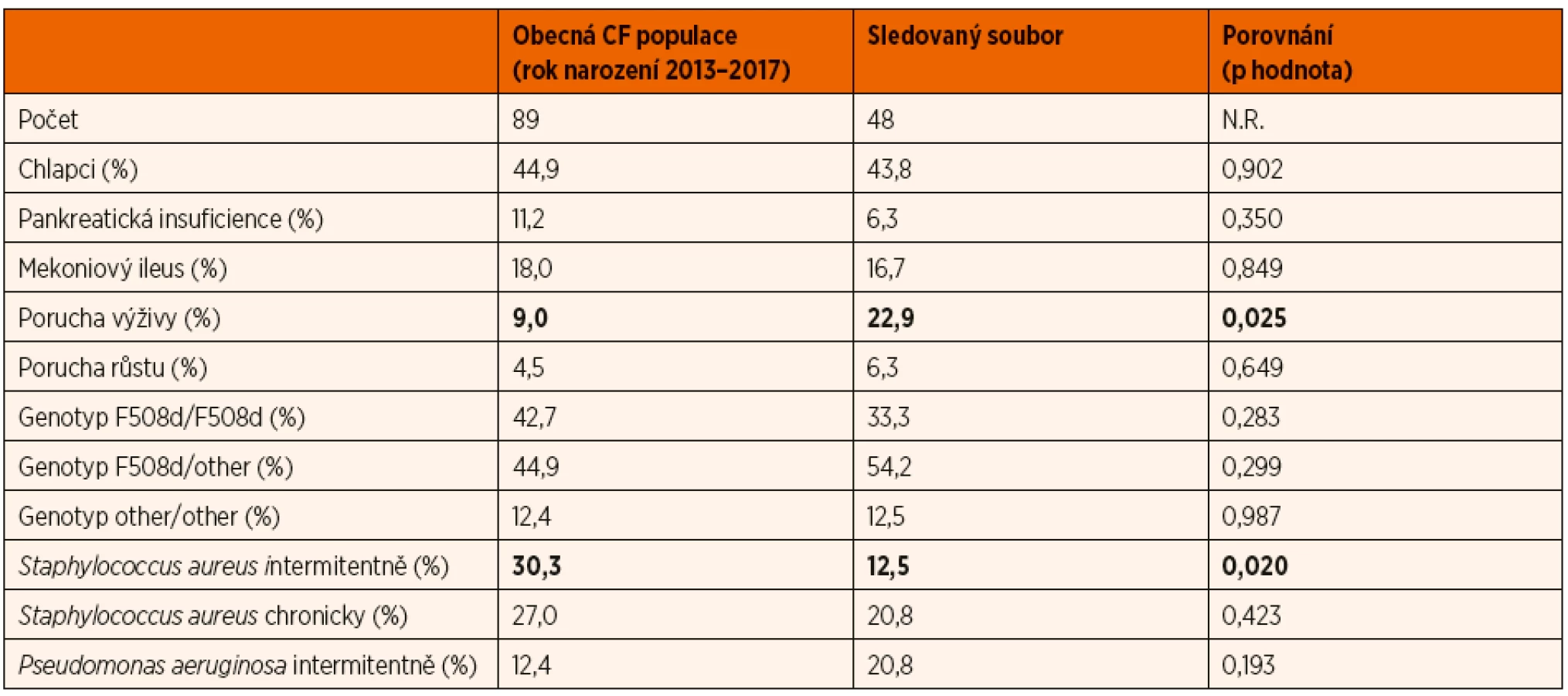
Reprezentativnost vyšetřeného vzorku pacientů pro obecnou českou populaci kojenců a batolat s CF byla hodnocena na základě jeho porovnání s daty z Českého registru CF. K dubnu 2019 bylo registru evidováno 89 pacientů s klasickou formou CF a rokem narození 2013 až 2017. V tomto vzorku bylo 44,9 % (40) chlapců, pankreaticky suficientních bylo pouze 11,2 % (10) pacientů, mekoniový ileus prodělalo 18,0 % (16). Nejčastější genotyp byl smíšený heterozygot (F508d/jiná CF způsobující mutace) – 44,9 %, homozygot pro mutaci F508d byl zjištěn ve 42,7 % a zbylých 12,4 % mělo genotyp jiná CF způsobující mutace/jiná CF způsobující mutace. Ve věku 12 měsíců bylo 27,0 % dětí chronicky kolonizováno Staphylococcus aureus, u 30,3 % byl tento záchyt intermitentní. Chronická kolonizace Pseudomonas aeruginosa nebyla v průběhu prvního roku zjištěna u žádného pacienta, zatímco intermitentní záchyt byl přítomen u 12,4 %. Komplex Burkholderia cepacia nebyl detekován u žádného sledovaného dítěte. Porucha výživy (podle Z-skóre BMI) byla detekována u 9,0 % pacientů a porucha růstu (podle Z-skóre výšky) u 4,5 %.
Vybrané klinické charakteristiky celého vyšetřeného souboru pacientů s průměrným věkem cca 1 rok a obecné populace českých pacientů s CF narozených v letech 2013 až 2017 se lišily jen minimálně. V obecné populaci byl signifikantně nižší výskyt poruchy výživy a signifikantně vyšší zastoupení pacientů s intermitentní kolonizací Staphylococcus aureus. Další rozdíly v mikrobiální kolonizaci nedosáhly statistické významnosti. Obdobně tak nebyly nalezeny signifikantní rozdíly mezi soubory v zastoupení chlapců, jednotlivých genotypů (homozygot F508d, smíšený heterozygot F508d/jiná CF způsobující mutace, nebo jiná CF způsobující mutace/jiná CF způsobující mutace), výskytu pankreatické suficience ani mekoniového ileu. Kompletní porovnání obou skupin pacientů včetně p-hodnot je uvedeno v tabulce. 4. Vyšetřený soubor pacientů nelze tedy považovat za plně reprezentativní pro obecnou českou populaci CF kojenců.
Funkce plic českých kojenců s CF
V období mezi únorem 2014 a únorem 2019 bylo v motolské laboratoři plicní funkční diagnostiky pro nespolupracující děti vyšetřeno 55 pacientů s CF. To odpovídá cca jedné třetině všech pacientů vyšetřených v této laboratoři za zmíněné období a více než polovině pacientů s CF diagnostikovaných v tomto období. Jedenáct dětí mělo dvě vyšetření s průměrným rozestupem 6 měsíců a jeden pacient měl vyšetření tři ve věku 11,6, 17,0 a 20,5 měsíce. Úspěšnost vyšetření se pohybovala v rozmezí 58,2–78,2 % podle metody a věku vyšetřovaného dítěte. Nejnižší úspěšnost byla v případě metody thorakoabdominální komprese, nejvyšší v případě analýzy klidového dechového vzoru. Významně nižší byla úspěšnost ve věkové kategorii do 6 měsíců oproti dětem starším. Sedace se nezdařila u 12,7 % pacientů (spánek nebyl navozen ani po podání maximální dávky chloralhydrátu v kombinaci s midazolamem v dávce 0,3 mg/kg během 1 hodiny). Výsledky z alespoň dvou metod funkčního vyšetření plic byly k dispozici u 48 dětí, tito pacienti tvořili základní soubor analyzovaný dále. Kromě neúspěšné sedace jsme se v celém souboru 48 vyšetřených pacientů setkali pouze s jedinou nežádoucí reakcí na vyšetření, a to desaturací k 85 %. Po napolohování hlavičky a úpravě pozice obličejové masky došlo k rychlé úpravě stavu. Zavedení oxygenoterapie nebylo nutné. Epizodu proto hodnotíme jako přechodnou obstrukci horních cest dýchacích. V celkovém počtu vyšetření byl tedy výskyt nežádoucích epizod hluboce pod 5 %.
V celém studovaném souboru byla jako nejčastější funkční porucha detekována nehomogenita ventilace plic (definovaná arbitrárně jako LCI2,5N2 nad 10) – postiženo bylo více než 50 % všech pacientů. Následovaly hyperinflace plic (definovaná jako Z-skóre FRCbox >2) a obstrukce periferních dýchacích cest (definována jako Z-skóre V´maxFRC <2), obě se vyskytly téměř u poloviny všech pacientů. V případě parametru V’maxFRC není k dispozici dostatek dat pro statistické hodnocení dětí mladších 6 měsíců (nízká úspěšnost měření). Méně častou funkční poruchu představovala obstrukce centrálních (velkých) dýchacích cest hodnocená podle klidového odporu dýchacích cest (Raw >145 % normy), která byla zjištěna u necelých 40 % pacientů. Zhruba u jedné čtvrtiny všech dětí byl zjištěn aktivní výdech (Z-skóre tPTEF/tE <-2) nebo porucha pružných vlastností dýchacího traktu (Z-skóre Crs >2 nebo <-2). Při komplexním hodnocení funkčního vyšetření plic bylo jako zcela bez funkční poruchy označeno 36,1 % dětí. Data jsou prezentována v grafu 1.
tPTEF/tE – čas dosažení maximálního průtoku během výdechu ku celkovému času
výdechu (podíl pacientů se Z-skóre <-2), značí aktivní výdech. Crs – poddajnost
dýchacího traktu (podíl pacientů se Z-skóre <-2 nebo >2), značí abnormální pružné
vlastnosti dýchacího traktu. Raw – odpor dýchacích cest (podíl pacientů s hodnotami
>145 % normy), značí obstrukci centrálních dýchacích cest. V´maxFRC – maximální
průtok na hladině funkční reziduální kapacity (podíl pacientů se Z-skóre <2),
značí obstrukci centrálních dýchacích cest. FRC – funkční reziduální kapacita (podíl
pacientů se Z-skóre >2), značí hyperinflaci plic. LCI – očišťovací index plic z dusíkového
washout (podíl pacientů s absolutními hodnotami LCI >10,0), značí postižení
nejperifernějších dýchacích cest.

Mezi skupinami dětí mladších (<6 měsíců) a starších byly nalezeny významné rozdíly ve funkci plic. Zatímco u mladších pacientů bylo bez poruchy funkce plic více než 50 % dětí, u starších to již bylo pouze 25 % (rozdíl statisticky signifikantní, p = 0,031). Obdobně signifikantní rozdíly v procentuálním zastoupení jednotlivých funkčních poruch byly nalezeny v případě nehomogenity ventilace (p = 0,022) a plicní hyperinflace (p = 0,039). Rozdíl ve výskytu obstrukce periferních dýchacích cest mezi skupinami nelze statisticky hodnotit. U 12 pacientů bylo provedeno funkční vyšetření plic opakovaně. Tito pacienti byli klinicky považováni za vysoce rizikové a byla u nich zavedena nadstandardně intenzivní léčba (diagnosticko-terapeutická bronchoskopie, i.v. ATB léčba, inhalace rekombinantní DNAázy, u jednoho pacienta léčba ivacaftorem atp.). V této podskupině bylo konstatováno celkové zlepšení funkce plic. Došlo k významnému poklesu nehomogenity ventilace (signifikantní pokles LCI2,5N2 o 2,6 jednotky, p = 0,003; obdobně tak i pokles LCI2,5SF6 o 2,0 jednotky, p <0,001 a Z-skóre LCI2,5SF6 o 2,1 SD, p = 0,022) a k hraničnímu zlepšení dalších funkčních parametrů (pokles Z-skóre RR o 0,9, p = 0,088; vzestup Z-skóre Vt o 1,1, p = 0,181, vzestup V´maxFRC% o 9,7 %, p = 0,203 atd.).
Výsledky funkčního vyšetření plic významně závisely na klinickém průběhu onemocnění. Pacienti, kteří prodělali alespoň jednu těžkou exacerbaci s i.v. ATB léčbou (n = 12), měli signifikantně horší funkci plic – konkrétně vyšší nehomogenitu ventilace (Δ LCI2,5N2 = 2,37; p = 0,003), vyšší Z-skóre FRC – hyperinflace (Δ FRCBOX = 1,75; p = 0,012), vyšší odpor velkých dýchacích cest (Δ Raw% = 65,6 %; p = 0,014) atp. Antropometrické parametry se však u nich nelišily. Počet dní na p.o. ATB významně koreloval s několika funkčními parametry: FRCBOX Z-skóre (r = 0,597, p <0,001), LCI2,5N2 (r = 0,509, p <0,001, graf 2), Raw% (r = 0,492, p = 0,003) a Z-skóre V´maxFRC (r = -0,452, p = 0,021). Rozdíly ve funkci plic byly nalezeny i mezi podskupinami pacientů s různou mikrobiální kolonizací. Např. u pacientů, kteří měli alespoň jeden kultivační záchyt Pseudomonas aeruginosa, byl zjištěn vyšší LCI2,5 než u pacientů bez tohoto záchytu. Naopak pacienti s intermitentním záchytem Staphylococcus aureus se od pacientů zcela bez záchytu Staphylococcus aureus nelišili ve funkci plic ani v antropometrických údajích. Děti s chronickou kolonizací Staphylococcus aureus však měly vyšší LCI2,5N2 než ty zcela bez této kolonizace.

DISKUSE
Naše výsledky funkčního vyšetření plic 48 kojenců a batolat s CF demonstrují bezpečnost, proveditelnost a klinický význam tohoto vyšetření. Pomocí kombinace 5 (resp. 6) metod funkčního vyšetření plic byly detekovány odchylky od normy již u dětí ve věku kolem 2 měsíců. U skupiny mladších dětí (věk <6 měsíců) se porucha funkce plic vyskytovala u 42 %, přičemž nejčastěji šlo o nehomogenitu ventilace (jako indikátor postižení nejperifernějších dýchacích cest – od 12. generace bronchiálního větvení dále), hyperinflaci plic, obstrukci periferních dýchacích cest a jejich různé kombinace (klasická obstrukční ventilační porucha). U pacientů starších (věkový průměr 16,6 měsíců) byl výskyt funkčních poruch ještě častější (75 %), přičemž jejich spektrum se od mladších dětí nelišilo, narůstala však jejich závažnost (např. vyšší výskyt hyperinflace plicní). Ačkoliv předkládaná studie nemá longitudinální charakter, lze na základě tohoto porovnání dvou skupin pacientů s různým věkem při vyšetření konstatovat, že již v kojeneckém a batolecím věku dochází k zřetelné progresi poruchy funkce plic. Naopak opakovaná vyšetření podskupiny rizikových pacientů podstupujících intenzivní léčbu dokládají příznivý efekt takové intervence a potenciál pacientů s CF ke zlepšení funkce plic. Dále naše data ukazují úzkou vazbu výsledků funkčního vyšetření na klinický průběh onemocnění – např. jejich závislost na počtu exacerbací, mikrobiální kolonizaci atd.
Předkládané průřezové výsledky jsou ve shodě s dříve publikovanými studiemi. Londýnští autoři [1] hodnotili výskyt poruchy funkce plic dětí záhy po stanovení diagnózy CF (tzn. ve věku cca 3 měsíců). Dvacet jedna procent tříměsíčních kojenců mělo v této studii zvýšenou nehomogenitu ventilace (LCI2,5SF6 vyšší než horní hranice normy zdravých kontrol). Funkční reziduální kapacita byla oproti zdravým kontrolám též signifikantně zvýšená, přičemž hyperinflace plic (zde definovaná jako FRC >1,96 SD) byla zjištěna u 18 % pacientů. Usilovné průtoky stanovené modifikací metody thorakoabdominální komprese byly oproti kontrolám významně snížené, přičemž obstrukce periferních dýchacích cest (Z-skóre FEV0,5 <-1,96 SD) byla detekována u 25 % pacientů. Tyto výsledky jsou plně srovnatelné s výsledky naší skupiny CFmalí (věk cca 2 měsíce). Úspěšnost vyšetření byla v případě londýnské studie využívající menšího počtu metod o něco vyšší než v našich podmínkách (81 % vs. cca 70 %). Poruchu funkce plic u nejmenších dětí dokládají i další obdobné studie [17, 18].
Naše výsledky též potvrzují dříve publikovaný význam testu vícedechového vyplavování inertního plynu z plic (MBW) u kojenců s CF [18, 19]. Prioritní jsou však data v případě dusíkové varianty MBW, kdy naše skupina jako první upozornila na možnost úspěšné implementace N2-MBW do vyšetřovacího protokolu funkce plic kojenců s CF [20]. Výsledky předkládané v tomto článku dále dokreslují klinický přínos N2-MBW a nepřímo i jeho srovnatelnost s SF6-MBW.
Vývoj funkce plic v průběhu prvních 2 let života zůstává stále předmětem diskusí, kdy různé studie poskytují protichůdné výsledky. Londýnská skupina (LCFC) udává při jejich standardní terapii (včetně dlouhodobé profylaxe Staphylococcus aureus flucloxacillinem) zlepšení obstrukce periferních dýchacích cest mezi 3. a 12. měsícem života (vzestup Z-skóre FEV0,5), zatímco parametry plicní hyper-inflace a nehomogenity ventilace (Z-skóre FRC a LCI2,5) zůstávají stacionární. Funkční nález ve 3 měsících věku podle jejich výsledků predikuje nález ve 12 měsících [21]. V roce 2017 byla tato data rozšířena o výsledky další sady měření (stejní pacienti ve věku 24 měsíců), která potvrdila zlepšující se trend parametru FEV0,5. Došlo k jeho zlepšení až na úroveň zdravých kontrol, přičemž Z-skóre LCISF6 zůstalo nadále snížené [22]. K opačným závěrům došli australští autoři (skupina AREST-CF), kteří detekovali významný pokles funkce plic (Z-skóre FVC a FEV0,5) v průběhu prvních 2 let života. Rychlost poklesu byla signifikantně vyšší u dětí s infekcí Staphylococcus aureus a Pseudomonas aeruginosa [23]. Australští autoři demonstrovali i progresi strukturálního postižení plic podle HRCT u dětí v rozmezí 0,2–6,5 let [3]. Naše longitudinální data podporují spíše australská zjištění, avšak je třeba brát v potaz skutečnost, že standardní léčba v ČR se zásadně liší jak od léčby australské, tak britské (nejvýznamnější rozdíl je v absenci antibiotické profylaxe infekce Staphylococcus aureus) a výsledky nejsou tedy plně srovnatelné.
Předkládaná studie má několik limitací. Naše práce nebyla koncipována jako kohortová populační studie, ale zahrnuje pacienty vybrané na základě jejich klinické prezentace. Vyšetřený vzorek tedy nelze považovat za zcela reprezentativní pro obecnou populaci českých kojenců a batolat s CF – viz např. vyšší výskyt poruchy výživy ve vyšetřeném souboru. Lze konstatovat, že byli vyšetřováni především pacienti rizikoví, což odráží tendenci ošetřujících lékařek odesílat k vyšetření především děti s těžším klinickým průběhem onemocnění (např. po prodělané těžké exacerbaci) či jinak problematické (non-compliance atd.). Jedná se tedy o selekční nevyváženost. Další limitací této práce je absence kontrolní skupiny, která by zajistila optimální porovnání se zdravou (českou) populací. Ačkoliv byly použity nejnovější dostupné normy, jejichž metodika vyšetření se plně shoduje s naší, nelze vyloučit určité odchylky absolutních hodnot jednotlivých parametrů jak směrem nahoru, tak i dolů. V důsledku toho se procentuální zastoupení jednotlivých poruch může mírně odlišovat, nicméně dobrá shoda procentuálního zastoupení jednotlivých funkčních poruch v naší skupině CFmalí a londýnské studii [1] svědčí o adekvátnosti použitých norem. Vzájemné porovnání daných parametrů mezi jednotlivými skupinami našich pacientů tímto dotčeno však není.
Dalším problémem naší práce je absence norem pro N2-MBW v populaci kojenců a batolat. Z tohoto důvodu byla empiricky stanovena horní hranice parametru LCI2,5N2 na 10,0 jednotky. Zvolená hodnota koresponduje i se zkušenostmi švédské skupiny, která se využitím N2-MBW u kojenců s CF též zabývá a má k dispozici vzorek zdravých kojenců vyšetřených simultánně pomocí N2-MBW i SF6-MBW. Ve shodě s našimi daty vychází LCI2,5N2 zhruba o 2,0 jednotky vyšší než LCI2,5SF6 i v jejich skupině zdravých dětí [24]. Již dříve zmíněná shoda ve výskytu zvýšené nehomogenity ventilace mezi naší a londýnskou skupinou malých kojenců s CF též dokládá adekvátnost této horní hranice. Předkládané výsledky funkčního vyšetření N2-MBW ve vazbě na klinické údaje našich pacientů s CF lze tedy považovat za jeden z dokladů potenciálního významu N2-MBW v běžné praxi. Navzdory určitým technickým limitacím [25, 26] se N2-MBW osvědčil jako vhodný nástroj pro monitorování stavu a vývoje funkce plic u kojenců s CF.
ZÁVĚR
Porucha funkce plic se začíná rozvíjet již u nejmladších dětí s cystickou fibrózou. Metody vyšetření funkce plic u nespolupracujících dětí jsou v tomto případě cenným a přitom bezpečným nástrojem pro detekci a monitorování plicního postižení u pacientů nejnižších věkových skupin. Oproti HRCT plic, které má srovnatelnou senzitivitu k plicnímu postižení a obdobně jako funkční vyšetření vyžaduje sedaci (či anestezii), není toto vyšetření spojeno s radiační zátěží a lze ho proto provádět frekventněji.
Hlavní funkční poruchou, kterou lze zjistit u nezanedbatelné části českých kojenců s CF již v průměrném věku 2 měsíců, je postižení nejperifernějších dýchacích cest (zvýšená nehomogenita ventilace). V dalším průběhu onemocnění se výskyt této poruchy i přes zavedenou standardní léčbu prakticky zdvojnásobuje. Zjištěná progrese poruchy funkce plic je sice vzhledem k designu naší práce pravděpodobně nahodnocena, nicméně si zaslouží pozornost lékařů pečujících o malé pacienty s CF a dokresluje význam časných terapeutických intervencí.
Vzhledem k demonstrovanému efektu rozšířených terapeutických intervencí u podskupiny vysoce rizikových pacientů vyšetřovaných opakovaně jsou ke zvážení úpravy standardního léčebného protokolu. Tato intenzivnější léčba by nebyla indikována plošně, ale cíleně pouze u pacientů, u nichž byla časným vyšetřením zjištěna porucha funkce plic. Kontrolní vyšetření s odstupem cca 12 měsíců by pak zhodnotilo efekt provedené intervence a přispělo k rozhodnutí o dalším směřování léčby. Tento cílený přístup by mohl přispět k dalšímu zlepšení zdravotního stavu českých dětí s CF ve vyšším věku, a předkládáme ho proto k diskusi v rámci české Pracovní skupiny pro CF. V této diskusi by bylo též třeba zhodnotit jeho proveditelnost v běžné praxi a kriticky zvážit jeho skutečné benefity pro pacienty.
Poznámka: Většina dat v článku byla převzata z disertační práce hlavního autora: „Detekce časných patofyziologických změn dýchání u dětí s chronickým plicním onemocněním“.
Poděkování:
Autoři děkují zdravotní sestře Petře Vančurové z Pediatrické kliniky 2. LF UK a FN Motol za asistenci při provádění funkčního vyšetření plic, dále doc. RNDr. Miroslavu Kouckému, CSc., z Technické univerzity v Liberci za pomoc při statistickém zpracování dat, MUDr. Marku Turnovcovi z Ústavu biologie a lékařské genetiky 2. LF UK a FN Motol a Mgr. Aleně Bílkové z Pediatrické kliniky 2. LF UK a FN Motol za export dat z CF registru a doc. RNDr. Karlu Olivovi, Dr., za jazykovou korekturu.
Podpořeno projektem koncepčního rozvoje výzkumné organizace 00064203 FN Motol Ministerstva zdravotnictví ČR.
MUDr. Václav Koucký
Pediatrická klinika 2. LF UK
a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: vaclav.koucky@fnmotol.cz
Zdroje
1. Hoo AF, Thia LP, Nguyen TT, et al.; London Cystic Fibrosis Collaboration. Lung function is abnormal in 3-month-old infants with cystic fibrosis diagnosed by newborn screening. Thorax 2012; 67 : 874–881.
2. Sly PD, Brennan S, Gangell C, et al.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF). Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med 2009; 180 (2): 146–152.
3. Mott LS, Park J, Murray CP, et al.; AREST CF. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 2012; 67 (6): 509–516.
4. Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros 2018; 17 (2): 153–178.
5. Hulskamp G, Pillow JJ, Dinger J, et al. Lung function tests in neonates and infants with chronic lung disease of infancy: functional residual capacity. Pediatr Pulmonol 2006; 41 : 1–22.
6. Gappa M, Colin AA, Goetz I, et al. Passive respiratory mechanics: the occlusion techniques. Eur Respir J 2001; 17 : 141–148.
7. Gappa M, Pillow JJ, Allen J, et al. Lung function tests in neonates and infants with chronic lung disease: lung and chest-wall mechanics. Pediatr Pulmonol 2006; 41 (4): 291–317.
8. Bates JH, Schmalisch G, Filbrun D, et al. Tidal breath analysis for infant pulmonary function testing. ERS/ATS Task Force on Standards for Infant Respiratory Function Testing. European Respiratory Society/American Thoracic Society. Eur Respir J 2000; 16 (6): 1180–1192.
9. Lum S, Hülskamp G, Merkus P, et al. Lung function tests in neonates and infants with chronic lung disease: forced expiratory maneuvers. Pediatr Pulmonol 2006; 41 (3): 199–214.
10. Sly PD, Tepper R, Henschen M, et al. Tidal forced expirations. ERS//ATS Task Force on Standards for Infant Respiratory Function Testing. European Respiratory Society/American Thoracic Society. Eur Respir J 2000; 16 (4): 741–748.
11. Robinson PD, Latzin P, Verbanck S, et al. Consensus statement for inert gas washout measurement using multiple - and single-breath tests. Eur Respir J 2013; 41 (3): 507–522.
12. Nguyen TT, Hoo AF, Lum S, et al. New reference equations to improve interpretation of infant lung function. Pediatr Pulmonol 2013; 48 (4): 370–380.
13. Jones M, Castile R, Davis S, et al. Forced expiratory flows and volumes in infants. Normative data and lung growth. Am J Respir Crit Care Med 2000; 161 : 353–359.
14. Ranganathan SC, Goetz I, Hoo AF, et al.; London Collaborative Cystic Fibrosis Group. Assessment of tidal breathing parameters in infants with cystic fibrosis. Eur Respir J 2003; 22 (5): 761–766.
15. Lum S, Stocks J, Stanojevic S, et al. Age and height dependence of lung clearance index and functional residual capacity. Eur Respir J 2013; 41 (6): 1371–1377.
16. De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med 2016; 4 (8): 662–674.
17. Kieninger E, Yammine S, Korten I, et al.; SCILD Study Group; BILD Study Group. Elevated lung clearance index in infants with cystic fibrosis shortly after birth. Eur Respir J 2017; 50 (5). Pii: 1700580.
18. Lum S, Gustafsson P, Ljungberg H, et al.; London Cystic Fibrosis Collaboration. Early detection of cystic fibrosis lung disease: multiple-breath washout versus raised volume tests. Thorax 2007; 62 (4): 341–347.
19. Davies G, Aurora P. The use of multiple breath washout for assessing cystic fibrosis in infants. Expert Rev Respir Med 2017; 11 (1): 21–28.
20. Koucký V, Skalická V, Pohunek P. Nitrogen multiple breath washout test for infants with cystic fibrosis. Eur Respir J 2018; 52 (2). Pii: 1800015.
21. Nguyen TT, Thia LP, Hoo AF, et al.; London Cystic Fibrosis Collaboration (LCFC). Evolution of lung function during the first year of life in newborn screened cystic fibrosis infants. Thorax 2014; 69 (10): 910–917.
22. Davies G, Stocks J, Thia LP, et al.; London Cystic Fibrosis Collaboration (LCFC). Pulmonary function deficits in newborn screened infants with cystic fibrosis managed with standard UK care are mild and transient. Eur Respir J 2017; 50 (5). Pii:1800015.
23. Pillarisetti N, Williamson E, Linnane B, et al.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF). Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am J Respir Crit Care Med 2011; 184 (1): 75–81.
24. Zatím nepublikovaná data z osobní komunikace Gustafsson P, Lindblad A.
25. Sullivan L, Forno E, Pedersen K, et al. Nitrogen back-diffusion during multiple-breath washout with 100% oxygen. Eur Respir J 2017; 50 (3). Pii: 1700679.
26. Gustafsson PM, Bengtsson L, Lindblad A, et al. The effect of inert gas choice on multiple breath washout in healthy infants: differences in lung function outcomes and breathing pattern. J Appl Physiol (1985) 2017; 123 : 1545–1554.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2019 Číslo 7
- Léčba bolesti a horečky u dětí
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
- Gastroezofageální reflux a gastroezofageální refluxní onemocnění u kojenců a batolat
- Očkování nejvíc potřebuje ten, kdo sám být očkován nemůže − kazuistika
- Pokrok v boji s malárií − první vakcína poskytující přijatelnou ochranu proti nemoci
Najčítanejšie v tomto čísle
- Novorozenecký screening cystické fibrózy a diagnostika CFSPID
- Vyšetrenie rotačnej tromboelastometrie (ROTEM) v manažmente krvácania a koagulopatie v pediatrii
- Historie cystické fibrózy u nás – editorial
- Alergická bronchopulmonálna aspergilóza u detských pacientov s cystickou fibrózou