
Mírné hyperhomocysteinémie z deficitu MTHFR (C677T a C1298A) u dospělých a adolescentů v metabolické ambulanci. Je třeba je diferencovat a léčit?
Mild Hyperhomocysteinemias From Deficiency of MTHFR (C677T and C1298A) in Adults and Adolescents Attending Metabolic Unit: Is There Any Necessity for Their Differentiation and Treatment?
Goal:
incidence and monitoring of mild hypeorhomocysteinemias in patients with cardiovascular risk attending the metabolic unit.
Type of Study:
retrospective analysis of a metabolic study.
Patients and Methods:
44 patients – from metabolic unit- selected homozygotes or heterozygotes confirmed by molecular-genetics methods for polymorphism of methylenetetrahydrofolate reductases (MTHFR C677T and A1298C) and accompanied with mild hyperhomocysteinemia (> 30 µmol/l of homocysteine in blood) were further metabolically analyzed and documented.
Results:
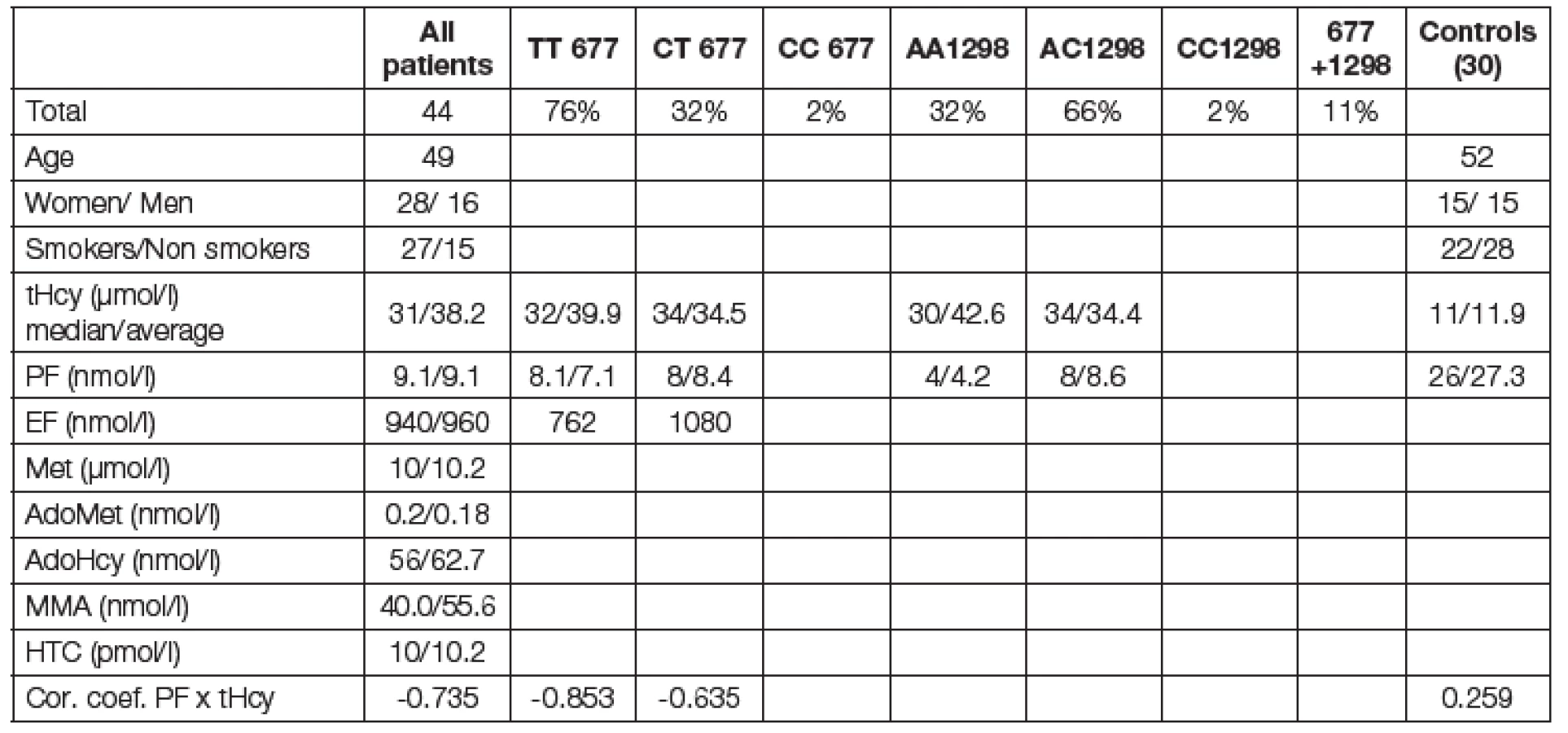
authors discuss and postulate till now not described form of mild hyperhomocysteinemia caused only by decreased enzymatic activity of MTHFR as a relatively often metabolic disturbance of folate metabolism. Due to defficient enzyme aktivity of MTHFR C677T in TT homozygotes (< 40%) the levels of plasmatic folates are decreased and the remethylation of homocysteine in the methionine cyclus is diminished.The negative correlation of lowered plasmatic folate with hyperhomocysteinemia in TT homozygotes (r= -0.853) and in CT heterozygotes (r = -0.635) was proved. In healthy control (r = +0.259). Supplementation of these patients with folic acid effectively increased folate plasmatic levels and decreased homocysteine levels. Typical clinical cases are documented with metabolic schemes or pedigrees. The therapeutical effect of homocysteine decrease on prognosis of cardiovascular risk has not been till now observed.
Conclusion:
mild hyperhomocysteinemias from folate defficiency due to homozygozity or heterozygozity of MTHFR in the Czech population represent the 3rd most cause of hyperhomocysteinemias: 1st kidney diseases, 2nd deficiency of holotranscobalamin. All mild hyperhomocysteinemias detected within routine laboratory practice must be differenciated and with adequate therapeutical effort established. Otherwise the routine estimation of homocysteine could be missused only for economical purposes.
Key words:
Hyperhomocysteinemia, Methylenetetrahydrofolate Reductase, MTHFR, Plasma Folate Cardiovascular Risk, Spina Bifida.
Autoři:
J. Hyánek; V. Maťoška; L. Dubská; B. Míková; H. Pejznochová; J. Dvořáková; L. Táborský; L. Košan; V. Martiníková; J. Privarová; J. Brtnová
Působiště autorů:
Metabolická ambulance, OKBHI a Lékárna Nemocnice na Homolce, Praha
Vyšlo v časopise:
Klin. Biochem. Metab., 25, 2017, No. 1, p. 18-26
Souhrn
Cíl studie:
určení výskytu a sledování mírné hyperhomocysteinémie (mHHC) u pacientů z metabolické ambulance s kardiovaskulární a perinatální zátěží.
Typ studie:
retrospektivní analýza dat z metabolické studie.
Materiál a metody:
44 pacientů vybraných homo- a heterozygotů polymorfismu metylentetrahydrofolát reduktázy (MTHFR C677T a A1289C) s mHHC (>30 µmol/l) bylo vyšetřeno geneticky na určení homo- a heterozygotie MTHFR, a tito byli dále klinicko-biochemicky a metabolicky diferencováni a dokumentováni.
Výsledky:
autoři dokumentují a diskutují málo známou nosologickou jednotku mHHC ze snížené enzymatické aktivity MTHFR jako samostatné a relativně časté metabolické onemocnění. Následkem je nedostatek aktivního folátu pro enzymový systém remetylace, který toxický homocystein odstraňuje. Snížení enzymové aktivity MTHFR nastává u nositelů homozygotní TT až o 40 % . Prokázali jsme snížení plazmatického folátu, které negativně koreluje s jeho metabolickými důsledky - HHC (r = -0,853 u homozygotů; r = -0,635 u heterozygotů; u zdravých r = +0,259). Suplementace kyselinou listovou efektivně zvýšila plazmatický folát a snížila hladinu homocysteinu. Pozorování je doloženo kazuistikami typických pacientů, jejich rizikových rodokmenů a efektivity léčby.
Závěr:
mHHC z deficitu folátu následkem homozygozity nebo heterozygozity MTHFR je v naší populaci třetí nejčastější příčinou HHC hned po ledvinné insuficienci a po deficitu vitaminu B12 (holotranskobalaminu). Každá mHHC musí být diferencována, vysvětlena a odstraněna, aby se snížilo riziko možných vitaminových deficitů a vývoje nežádoucích vaskulárních či perinatálních komplikací. O preventivním účinku snížení homocysteinu na rozvoj kardiovaskulárních komplikací se pochybuje.
Klíčová slova:
hyperhomocysteinemie, metylentetrahydrofolát reduktáza, MTHFR, genotyp 677 T<T a C<T; plazmatický folát, kardiovaskulární a perinatální komplikace, spina bifida.
Úvod
Podrobné studie jako Norwegian Vitamin Trial (NORVIT), Westnorwegian Vitamin Trial (WENBIT), Hordaland Homocysteine Studies, které následovaly 10 let po povzbudivých studiích preventivního podávání B-vitaminů (SEARCH, WACS, VISDPO,TITATOPS, VISP, HOPE-1 a 2, CHAOS), dokládají negativní význam snižování zvýšené hladiny celkového homocysteinu (tHcy) - hyperhomocysteinemie (HHC) podáváním vitaminů skupiny B jako možnou prevenci kardiovaskulárních komplikací [1, 2, 3, 5, 12, 18, 26, 27]. Důsledkem těchto závěrů je ztráta zájmu kardiologů a dalších kliniků o praktický význam stanovení tHcy v plazmě. Dosud však nebylo předloženo žádné racionální vysvětlení skutečného rizika tohoto toxického metabolitu v etiologii cévního onemocnění, ačkoliv poškozuje endotel, snižuje metylační kapacitu DNA a argininu. Experimentálně opakovaně doložená in vitro a in vivo homocysteinová etiopatogeneze endoteliálních změn byla po uplynulých 50 letech zamítnuta [6, 9, 10, 12, 22, 29, 30, 33]. Nadále se tolerují i pozitivní názory na patogenitu mírného zvýšení tHcy před lety publikované americkými i evropskými autory jako např. zjištění Walda a spol., že zvýšení hladiny tHcy o 5 µmol/l zvyšuje riziko příští cévní příhody o16 % a nebo závěry Homocysteine Collaborative Study dokládající snížení rizika ischemické choroby o 11 %, pokud se tHcy sníží o 3 µmol/l [1, 11, 12, 15, 30, 31]. Indikace vyšetření tHcy byla zpochybněna, interpretace klinicko-biochemického významu HHC zůstala nedořešená a využití v diferenciální diagnostice je nedoceňováno. Pomůckou k řešení neutěšené situace jsou vyčerpávající studie o klinicko-biochemickém významu tHcy, externích vlivech na metodiku stanovení i interpretaci nálezů od Refsumové a Uelanda [34].
Příčiny a druhy hyperhomocysteinemií
Referenční hodnoty pro tHcy jsou podle našich 50ti letých zkušeností doporučené u pacientů (< 60 let a normální kreatininémie): < 15 µmol/l pro ženy a < 20 µmol/l pro muže; u dětí jsou hodnoty nižší (< 10 µmol/l), nejnižší hodnoty pak nacházíme u těhotných (6 -10 µmol/l). U populace starší 60 let je třeba brát zřetel na kreatininémii, zvýšení tHcy průměrně o 8 µmol/l je způsobeno vzrůstem kreatininémie o 10 µmol/l. U mladších osob naopak s ohledem na možný externí příjem folátu očekáváme hodnoty průměrně o 6,7 µmol/l nižší [17, 24].
Podle Guttormsenové a Bostoma [6, 21] se HHC rozlišují spíše historicky na mírné HHC (< 30 µmol/l), intermediární (30 - 100 µmol/l) a výrazné HHC (> 100 µmol/l). Rozumnější je dělení podle etiopatogeneze, na té se podílí především geny, vitaminy a léky. Mírné zvýšení tHcy je způsobeno už i lehkou ledvinnou dysfunkcí jakéhokoliv původu, deficitem vitaminů skupiny B, toxickým poškozením jater nejrůznějším agens, účinkem některých vzácnějších léků (např. methotrexát); nejvyšší hodnoty tHcy pak nalézáme u vzácné nemoci z dědičného deficitu cystathionin-beta-syntázy (CBS) - klasické homocystinurie [9, 27]. Mírná hyperhomocysteinémie (mHHC) je považována stále za experimentálně prokázaný, ale stále „nedořešený“ faktor předčasného endoteliálního poškození, trombopatií, perinatálního a onkologického vlivu. Patogenní hladina tHcy však není definována a iniciace vzniku flebotrombózy, či tromboembolie se může pohybovat až kolem 100 µmol/l tHcy, u neurogenního, onkologického a perinatálního poškození není zatím definována [23]. Kontroverzní metabolické, molekulárně genetické i epidemiologické studie o škodlivém účinku homocysteinu a pozitivním účinku folátu však nadále pokračují [4, 5, 10, 12, 13, 15, 30, 33].
Všechny příčiny HHC spočívají v jeho nedostatečném metabolizování v remetylačním enzymatickém cyklu methioninsyntázy a její reduktázy buď jako následek genového defektu jednotlivých enzymových bílkovin nebo deficitního enzymatického mechanismu z nedostatku koenzymů tj. vitaminů B12,B6 ,B2 a aktivního folátu [9]. Obr. 1.

Nedostatečně zdůrazňovaná je zatím nejčastější příčina vzniku mHHC vznikající už při mírné ledvinné insuficienci. Hladina tHcy velmi dobře koreluje se zvýšenou kreatininémií, a to už následkem mikroskopických lézí endotelu ledvinných kapilár [7, 8, 17]. Dobře prozkoumaným je už skoro 50 let známý dědičný deficit cystathionin-beta-syntázy u klasické homocystinurie s více než padesáti známými genovými mutacemi [9, 26, 27]. Relativně častá v naší populaci je HHC z potravinového deficitu vitaminu B12 u veganů a vegetariánů nebo způsobená chronickým deficitním vstřebáváním s odlišnou manifestací v závislosti na věku [9, 23].
K HHC dochází také deficitem ve skupině vitaminu B12 následkem změn v jeho molekulárně genetické struktuře. Cobalaminy (Cbl) C, D, E, F, G vyvolávají deficit enzymového makrokomplexu methioninsyntázy a metylmalonyl-CoA-mutázy, zatímco CblE a G jen deficit methioninsyntázy. Deficit haptocorrinu se projevuje stejně, liší se normální hladinou HTC. Sem patří i málo známé HHC z deficitu jednouhlíkatých methenylových skupin vznikajících při oxidaci serinu, které jsou potřebné k navázání na tetrahydrofolát, aby se vytvořil 5,10-metylentetrahydrofolát. Ten je zredukován MTHFR na aktivní 5-metyltetrahydrofolát. Deficit aktivního folátu spolu s deficitem methioninadenosyltransferázy se v současné době považují za příčinu poruch demyelinizačních pochodů vedoucí již v útlém věku k psychomotorické retardaci [9].
Vzácnou příčinou HHC je dědičný nebo získaný deficit vstřebávání folátů (malabsorpce) ve střevě. Zde se folátové polyglutamáty z potravy enzymaticky konvertují folátreduktázou z jejunální mukózy na monoglutamáty, aby se mohly vstřebat [32]. Léčebně podávaná kyselina listová se proti polyglutamátům vstřebává dvakrát vydatněji.
Dosti prostudovaný, ale dosud nedostatečně klinicky doceněný, je funkční deficit enzymového systému termolabilní metylentetrahydrofolátreduktázy (MTHFR) následkem genových mutací C677T a A1298C (OMIM 236250) lokalizovaných na 1p36. 3. Protože primáti nedovedou syntetizovat folát, jsou odkázáni na jeho přívod potravou. Klinická manifestace představuje škálu od bezpříznakových dospělých pacientů (jen s metabolickým nálezem) až po těžkou neurologickou symptomatologii s retardací u malých dětí.
Podmínky enzymatické reakce MTHFR
V naší práci se věnujeme metabolickým variantám polymorfismu MTHFR C677T a A1298C (OMIM 236250) z deficitu N-5,10-metylentetrahydrofolátreduktázy, E.C.1.5.1.20. Zjednodušené schéma je uvedeno na Obr. 1. Jedná se o klíčový enzymatický komplex redukující 5,10-metylentetrahydrofolát na N-5-metyltetrahydrofolát, z jehož nedostatku rezultuje deficit tzv. aktivního folátu (folate trap) nezbytného k remetylaci cytotoxického homocysteinu na methionin v enyzymovém makrokomplexu methioninsyntázy a její reduktázy. Esenciální aminokyseliny methioninu jsou potřebné kromě syntézy bílkovinných struktur také k syntéze DNA a pro remetylační a imprinting procesy.
Nekovalentně vázaný flavinadenindinukleotid (FAD) jako kofaktor přijímá redukční ekvivalenty z NAD(P)H a přenáší je na CH2-H4 folát. Redukce CH2H4 folátu je jedinou možností, jak v lidském organismu syntetizovat k životu nezbytný „aktivní 5- metylfolát“. Enzymová bílkovina MTHFR existuje ve formě tetraméru složeného z dimérových podjednotek o molekulové hmotnosti 70000 - 77000 kDa [20]. Vazba FAD, NAD(P)H a CH2-H4 folátu je lokalizovaná na N-terminálním konci molekuly. Molekula enzymu může být deficitní následkem polymorfismu dvou genetických mutací: 677 C<T a 1298 A<C. V prvém případě jde v molekule o záměnu v nukleotidu C za T s následkem, že se kódovaná aminokyselina alanin222 vymění za valin (A222V); ve druhém případě se v nukleotidu zamění A za C a místo glutamátu 429 vzniká alanin (G429 V). Mutace posunuje reakční rovnováhu mezi hladinami holotetraméru na jeho apoenzymy diméry směrem k dimérům, které mají nižší enzymovou aktivitu. Disociace závisí na předchozím uvolnění FAD, který katalyzuje elektronový transfer z NAD(P)H na CH2-H4 folát a vzdaluje se tak od katalytické domény. Následkem je tepelná nestabilita enzymové molekuly při 37ºC se snížením enzymové aktivity až o 40 %, ta se výrazněji projevuje u TT homozygotů než CT heterozygotů. Přidání CH3-H4 folátu (např. podáním folátmonoglutamátu nebo kyseliny listové) zpevňuje vazbu FAD na N-terminálním konci molekuly a zlepšuje enzymovou aktivitu. Závislost na externím podání folátu není vždy lineární, zřejmě kvůli dalším možným enzymatickým zevním vlivům. I když syntéza Hcy při deficitu MTHFR stoupá, celková remetylační kapacita methioninového cyklu není postižena, což znamená, že se tato metabolická situace projevuje různým zvýšením Hcy i u téhož pacienta, zřejmě podle jeho aktuálního metabolického stavu a okamžitého nasycení vitaminy z potravy a toto sycení zase závisí na druhu příjmu potravy i na potravinářských módních vlivech [13].
Prevalence polymorfismu MTHFR a kauzální léčba HHC
Prevalence C677T mutace MTHFR v severoamerické, severoevropské, holandské a welšské populaci se různí v rozsahu 5 - 12 - 20 % a Kožich a spol. v pražské populaci našli podobné % výskytu (IGA MZCR, NM 26-3, 2000), [16, 21]. Zvýšené kardiovaskulární riziko u tohoto polymorfismu popsal první Frosst a Blom, riziko výskytu spina bifida první v. d. Put a Kluijtsmans [5, 19, 23, 25]; jejich výsledky jsou pozdějšími nálezy zpochybňovány, ne však zcela vyvráceny. Podání kyseliny listové zvyšuje významně hladinu plazmatického folátu a tím i enzymovou aktivitu a snižuje hyperhomocysteinémii u homo- i heterozygotů. Kvantitativní stanovení 5-metylfolátu v plazmě je zatím velice náročné, a rutinně se neprovádí. Zůstává orientace jen podle plazmatického folátu (PF) a jeho ještě méně informativního zásobního formylfolátu v erytrocytech (EF). Podávání aktivního 5-metylfolátu je rovněž účinné. Firemní studie jsou velmi povzbudivé, ale neuvádí bohužel hladinu aktivního folátu v krvi [29]. Naše vlastní zkušenosti u těhotných jsou zatím nedostatečné.
Pacienti a metody
Metabolickou ambulancí prochází ročně zhruba 500 dospělých a adolescentních pacientů s lipidovou symptomatologií a kardiovaskulárním rizikem. Pro studii bylo vybráno 44 pacientů se zvýšenou hladinou tHcy > 30 µmol/l a nálezem homozygotní nebo heterozygotní mutace MTHFR 677C>T a 1298A>C. Hladina tHcy byla zvolena podle používaného rozdělování HHC Guttormsenové a Bostoma [6, 21]. Ze studie byli vyloučeni pacienti se zvýšenou hladinou kreatininu, metylmalonátu (MMA), izolovaným deficitem holotranskobalaminu (HTC), léčení diabetici, vegetariáni, pacienti s m. Crohn, výrazné obezity a pacienti přijímající jakékoliv vitaminové tablety či doplňky. U všech vybraných pacientů byly přítomny kromě výrazné dyslipidemie nejméně dva rizikové faktory (IM, AP, TE, PE, stenóza či klaudikace DK, DVT, CMP, ACB, diagnostická nebo terapeutická PTCA, rozléčená hypertenze, tromboembolie, aj.) a Risk Score >10 % (podle ČSAT). Pět pacientů ze souboru bylo léčeno sodantonovými antiepileptiky, u tří pacientů byl současně léčen prostý deficit holotranskobalaminu (HTC) z nedostatku vnitřního faktoru. Homocystinurie s typickými nálezy Met, cystathioninu, adoHcy, adoMet byly odesílány do ÚDMP Praha. Referenční skupinou (n = 30) byli pacienti vyšetření při preventivních prohlídkách v ambulaci závodního lékaře NNH. K odhadu průměrné hladiny folátů v populaci sloužilo 1000 náhodně vybraných hodnot pacientů z LISu NNH.
Stanovení lipidového spektra a tHcy bylo provedeno rutinními biochemickými metodami na analyzátorech Unicel DxC800 firmy Beckman Coulter, krevní obraz na přístroji XE 5000 firmy Sysmex. Holotranscobalamin byl stanovován imunochemicky metodou MEIA na analyzátoru Architect 2000 firmy Abbott. Celkové foláty v plazmě a erytrocytech byly měřeny metodou ECLIA na analyzátoru Cobas e411 firmy Roche. Pyridoxalfosfát stanovován na HPLC systému firmy Agilent. Metylmalonát v krvi metodou GC/MS. Stanovení methioninu, cystathioninu, adoMet, adoHcy bylo prováděno v laboratořích ÚDMP 1. LFUK Praha. Molekulárně-genetická vyšetření MTHFR C677T a A1298C byla provedena PCR amplifikací a HRM (high resolution melting) analýzou. Ultrasonografické vyšetření karotid na VIGMED SOUND, sondou 7,0 - 7,5 -10 MHz za standardních podmínek. Ke statistickému zpracování byl použit STATA Data Analyzer (RNDr. J. Dostál, DrSc., ÚBČAV Praha).
Pacientům byla podávána léčebně kyselina listová (Acidum folicum 10 mg/tbl, Léčiva/Zentiva). Při zahájení léčby se vždy vycházelo z aktuálních plazmatických hladin folátu a hladin zásobního erytrocytárního folátu, podáváno bylo obvykle 2,5 - 5 - 10 mg/týden. Denní potřeba kyseliny listové (RDA) činí jen 400 µg/d u dospělých, u gravidních 450 µg/d. Léčebné podání vitaminu B6 (Pyridoxin, 20 mg/tbl, Léčiva) bylo dávkováno podle aktuální hladiny v krvi 1 - 2 tbl týdně pouze u epileptiků na kontinuální léčbě sodantonovými antiepileptiky. U vybraných pacientů byl podáván vitamin B2 v dávce 25 mg /týden (Riboflavin, Léčiva).
Perorální zátěže L-Methioninem 100 mg/kg hmotnosti prováděné úspěšně v 90tých letech, které dobře diferencovaly homo-i heterozygoty MTHFR, byly pro mírné vedlejší reakce po podání L-Met u některých pacientů zastaveny.
Grantová studie MZČR NE-54908 byla schválena etickou komisí NNH.
Výsledky
Metabolicko-demografická charakteristika souboru vyšetřovaných pacientů je uvedena v Tabulce 1. Podle molekulárně-genetického schématu uvedeného na obr. 2. lze vyšetření mutací MTHFR genotypu TT 677 považovat za kauzální pro mHHC v 69 % pacientů a modulační dokonce v 88 % případů. Kohorta homo- nebo heterozygotních pacientů má signifikantně vysoký tHcy a nižší hladinu PF. Vyhodnotili jsme korelační koeficienty negativní závislosti hladiny tHcy a PF pro TT mutaci r = -0,853 a CTr = -0,652; zatímco u kontrolní skupiny zdravých byla hodnota r = +0,259; (p < 0,01), obr. 3. Hodnoty zásobního EF nebyly diagnosticky signifikantní. Ostatní sledované metabolity (Met, adoMet, adoHcy, cystathionin, HTC, MMA, vitB6) nebyly v této kohortě pacientů významné. Léčebné podání 2,5 až 10 mg acidum folicum týdně bylo u nových pacientů velmi efektivní a je uvedeno na typickém grafu obr. 4. Rodokmen s vysokým rizikem je uveden na obr. 5.





Diskuse
Naše studie potvrzuje zvýšený výskyt mHHC ve slovanské populaci, který je u pacientů z metabolické ambulance často provázený zvýšeným kardiovaskulárním rizikem podobně, jako bylo prokázáno i v jiných evropských populacích [19, 25, 29]. Zvýšení bylo provázeno signifikantně sníženou hladinou PF nikoliv EF.
Předpokládáme, že deficit MTHFR představuje samostatnou nosologickou jednotku snadno léčitelnou podáním kyseliny listové. Pokud se k deficitu folátu připojují ještě další vitaminové deficity např. B12 či B6, je mHHC ještě výraznější a vyžaduje kombinovanou léčbu.
Skutečný výskyt mHHC by byl v naší populaci jistě mnohem vyšší vzhledem k zjišťované přesycenosti populace folátem; svůj podíl sehrává nepochybně vliv nerozumné a klamavé reklamy (posilovací a ozdravné nápoje, „spalovače“, vitaminové směsi a omlazovací lázeňské kůry, zajištovací „léčba fertilních žen tabletami pro optimální vývoj miminka“ aj.). V lékárně je dostupný metylfolát v několikerém provedení např.: Metafolin (Calcium L-metylfolát), Quatrefolic (glukosaminová sůl L-metylfolátu), Smartfolin 1 (Metafolin + acidum folicum + vitamin E), Superfolin 1 (acidum folicum+vitaminy+ jod).
Folátový deficit jsme pozorovali současně při deficitu vitaminu B6 u pacientů na intenzivní léčbě antiepileptiky jak popsala Dvořáková [16]. Neměli jsme možnost náročného stanovení vit B2 v krvi, ale bylo zajímavé, že ani dlouhodobé podávání vitaminu B2-FAD Riboflavinu v dávce 25 mg/d (ačkoliv je riboflavin koenzymem MTHFR) hladinu tHcy ani PF u žádného z našich pacientů signifikantně neovlivnilo. Ve dvou rodinách byly nalezeny spina bifida s homo- nebo heterozygotními mutacemi obou MTHFR a mHHC u vnuček obou probandek. Současný výskyt obou homozygotních forem MTHFR mutací 677 a 1298 jsme u žádného z pacientů nenalezli, asi není slučitelný se životem. Kombinace heterozygotních forem obou MTHFR však nebyly vzácností a nacházeli jsme je u 22 % vyšetřovaných.
Bylo zarážející, že pacienti s deficitem MTHFR konzumují málo zeleniny a odmítají hlavně čerstvou listovou zeleninu, kde je přítomno nejvíce folátu ve formě polyglutamátu. Poruchy vstřebávání folátu, popisované u chronických dysfunkcí tenkého střeva nebo následkem hereditálního deficitu pteroylhydrolázy tvořící ve střevě z polyglutamátu vstřebatelný monoglutamát, jsme u našich pacientů nepozorovali. Rovněž patologické změny krevního obrazu typu makrocytární anémie či maskování perniciózní anémie jsme u žádného z našich pacientů neprokázali [9].
Průměrné hodnoty folátu v naší dospělé i adolescentní populaci pacientů z NNH odpovídají evropským normám a neliší se ani od průměrných vzorků dospělých pacientů z ambulance závodního lékaře NNH (PF = 23 ± 14 nmol/l a EF 1050 ± 422 nmol/l). Často jsme však v naší ambulanci pozorovali hodnoty folátů z předávkování PF > 1000 nmo/l a EF > 10000 nmol/l, tyto hodnoty byly naměřeny hlavně u pacientů z hematologických či gynekologických ambulancí. Léčebné podání při každém podezření na HHC ambulantními lékaři spočívající v nadměrném dávkování až 3 tbl. kyseliny listové denně nebylo vzácností. Žádné toxické příznaky z předávkování jsme dosud nepozorovali, avšak laboratorní stanovení 1000x vyšších hladin PF a EF je metodicky i finančně náročné. Trvá i několik měsíců než se hladiny folátů takto předávkovaných pacientů sníží k terapeutické hladině. Zneklidňuje nás obava z podpory možného souběžně probíhajícího nádorového růstu hlavně u starších pacientů, jak upozorňují rozporuplné onkologické studie [35]. Prof. Farber a Wolff z Harwardu, kteří v r.1943 pozorovali u dětí léčených folátem intenzivnější růst lymfoblastomu než u neléčených. Prof. Jordi Folch-Pi později připojil aminoskupinu a metylovou skupinu k molekule kyseliny listové a byl objeven dodnes velmi účinný antifolát - methotrexát.
Přímá asociace mezi deficitní aktivitou MTHFR z polymorfismu a tíží aterosklerózy nebyla nikdy potvrzena, až na výjimečnou japonskou studii [33]. Zvýšené riziko KVO u TT homozygotů MTHFR dokládají práce z různých etnik -Japonska,Turecka, Israele aj. [14, 33]. Vyšší výskyt mHHC jsme prokázali i v naší grantové studii IGA MZČR Nr5489-3 (2001) u dětských a adolescentních pacientů narozených z rizikových rodičů, kteří měli častější TT polymorfismus. Existují však i práce, kde byl sice nalezen častější polymorfismus TT s vyšším rizikem KVO, ale bez zvýšení tHcy [28]. Žádná ze studií uvedených v úvodu nevysvětlila negativní účinky vitaminů na snížení tHcy, zda mHHC je příčinou nebo následkem nastupujících změn cévního endotelu. Byli to až Brattstrom a Wilcken, kteří se nebáli prohlásit, že zvýšený tHcy není příčinou aterosklerotických změn, nýbrž už následkem mikroskopických cévních aterosklerotických změn především ledvinných kapilár [8]. Homocysteinový „veterán a bojovník“ za etiologický význam tHcy u KVO Bostom po 20 letech považuje zvýšený tHcy jen za „drahé“ vyšetření mikroskopického poškození ledvin nahrazující nedostatečně citlivou clearanci kreatininu nebo cystatinu C [7]. Redukční účinek folátu na hladinu tHcy nezávisí na věku ani na kreatininu [17]. Už mnohem dříve prokázala Pullinová, že podáním folátu se sice sníží tHcy, ale funkce poškozeného endotelu se už nezlepší [28]. Po 3leté neúspěšné sekundární prevenci KVO komplikací Amer. College of Cardiology podávaní B-vitaminů pacientům nedoporučuje [29].
Přesto však zvýšený tHcy nadále zůstává užitečným pomocníkem při detekci vitaminových deficitů skupiny B a zůstává vážným metabolickým markerem stupně cévního poškození jako např. zvýšený CRP, glykémie nebo krevní tlak. Při hodnocení rizika, např. budoucí komplikace, se s ní musí počítat a co nejdříve příčinu mHHC identifikovat a odstranit. V patogenezi kardiovaskulárního poškození však významu např. hypercholesterolémie nedosahuje.
Při nejasném zvýšení hladiny tHcy i přes podávání folátu je třeba pomyslet na některou z vzácných dědičných metabolických poruch molekuly kobalaminu a nebo jednouhlíkatých metylmetabolitů, zvláště při nálezu časného psychomotorického opoždění a neurologické symptomatologie dítěte. Tyto pacienty s mHHC je třeba neprodlenně odeslat k diferenciaci do ÚMPD Praha, kde mají k dispozici u nás nejdostupnější metodiku. Výsledky výzkumu vzácných remetylačních poruch provázených HHC avizují další dědičná onemocnění, která zatím na žádnou léčbu nereagují [36].
Závěr
Výsledky rutinního stanovení tHcy uplynulých 20 let potvrzují zvýšený výskyt mHHC z deficitu aktivity MTHFR vlivem polymorfismu 677 a 1298 také ve slovanské populaci a shodují se s nálezy severoevropských i severoamerických studií. mHHC z deficitu MTHFR je tak po ledvinné insuficienci a deficitu holotranskobalaminu třetí nejčastější metabolickou příčinou hyperhomocysteinémie spojenou s vysokou incidencí KVO, neurogenních a perinatálních komplikací. Patogeneze pro rozvoj kardiovaskulárních, trombogenních či perinatálních komplikací zůstává nedostatečně potvrzena a terapeutické snižování tHcy je tudíž neefektivní. Přesto každá hyperhomocysteinémie > 30µmol/l musí být jako metabolický rizikový faktor diferencována, vysvětlena a odstraněna! Vyžaduje proto větší pozornosti nejenom laboratorních lékařů a metabologů, ale i kardiologů, perinatologů, neurologů, gerontologů a neuropsychiatrů, aby se stanovení homocysteinu nestalo záležitostí obchodníků s laboratorní analýzou.
Použité zkratky
domet adenosylmethionin
adoHcy adenosylhomocystein
CBS cystathionin-beta-synthasa
CMP centrální mozková příhoda
EF erytrocytární folát
FAD flavinadenindinukleotid (vitamin B2 riboflavin)
HHC hyperhomocysteinémie
HTC holotranskobalamin (aktivní vitamin B12)
IM infarkt myokardu
KVO kardiovaskulární onemocnění
mHHC mírná hyperhomocysteinémie
MMA metylmalonová kyselina
MTHFR metylentetrahydrofolátreduktáza
NNH Nemocnice Na Homolce, Praha
OMIM Online Mendelian Inheritance in Man
PE plicní embolie
PF plazmatický folát
PTCTA perkutánní transluminární angioplastika
RDA Recommended Daily Allowance (doporučené denní množství)
TE tromboembolie
ÚPMD Ústav dědičných metabolických poruch 1. lékářské fakulty UK Praha
tHcy celkový homocystein
Další literatura u autora.
Do redakce došlo 16. 2. 2016
Adresa pro korespondenci:
Prof. MUDr. Josef Hyánek, DrSc.
Nemocnice na Homolce, OKBHI
Roentgenova 2
150 30 Praha 5
e-mail: josef.hyanek@homolka.cz
Zdroje
1. The homocysteine studies collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA, 2002, 288, 2015-22. (Editorial).
2. The Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular diseases. NEJM, 2006, 354, 1567-77.
3. Homocysteine and coronary vascular risk: considering the evidence in the context of study design, folate fortification and statistical power. Clin. Chem., 2007, 53, 807-9 (Editorial).
4. Bonaa, K. H., Njolstad, I., Ueland, P. M. et al.: Homocysteine lowering and cardiovascular events after acute myocardial infarcts. NEJM., 2006, 354, 1578-88.
5. Blom, H, J.: Overview of homocysteine and folate metabolism with special reference to cardiovascular disea-ses and neural tube defects. J. Inher. Metab. Dis., 2011, 34, 75-81.
6. Bostom, A. G., Selhub, J. Homocysteine and atherosclerosis: subclinical and clinical disease associations. Circulation, 1999, 89, 2361-3.
7. Bostom, A. G. Homocysteine:expensive creatinine or important modifiable risk factor for arteriosclererotic outcomes in renal transplant recipients? Editorial. J. Am. Soc. Nephrol. 2000, 11, 149-151.
8. Brattstrom, L. B., Wilcken, D. E.: Homocysteine and cardiovascular disease:cause of effect? Amer. J. Clin. Nutr., 2000, 72, 315-323.
9. Carmel, R., Jacobsen, D. W.: Homocysteine in health and disease. Cambridge Univ. Press, 2001.
10. Castro, R., Rivera, L. Blom, H. J. et al.: Homocysteine metabolism, hyperhomocysteinemia and vascular disease: an review . J. Inher. Metab. Dis., 2006, 28, 3, 20.
11. Clarke, R., Armitage, J., Lewington S. et al.: On behalf of B-Vitamin treatment trialist collaboration. Homocysteine-lowering trials for prevention of cardiovascular events: a review of the design and power of the large randomized trials . Am. Heart J., 2006, 151. 282-7
12. Cronin, S., Furie, K. L., Kelly, P. J. et al.: Dose-related association of MTHFR 677T allele with risk of ischemic stroke:evidence from a cummulative meta-analysis. Stroke, 2005, 36, 1581-87.
13. Davis, S. R., Quinlivan, E-P-. Shelnutt, K. P. et al.: Homocysteine synthesis is elevated but total remethylation is unchanged by the methylentetrahydrofolate reductase 677C>T polymorphism and by dietary folate restriction in young women. J. Nutr., 2005, 135, 1045-50.
14. Delaughery, G. m Evans, A., Sageshi, A. et al.: Common station in methyleneterahydrofolate reductase. Circulation. 1996, 94, 3076-78.
15. Doshi, S. N., McDowell, I. F., Moat, S. J. et al.: Folic acid improves endothelial function in coronary artery diseases via mechanisms largely independent of homocysteine lowering. Circulation, 2002, 105;22-26.
16. Dvořáková, J., Kolínová, M., Hyánek, J. et al.: Hyperhomocysteinemie u nemocných léčených antiepileptiky- první zkušenosti. Klin. Biochem. Metab., 2000, 8, 190-194.
17. Elshorbagy, A. K., Oulhaj, A., Konstatinova, S. et al.: Plasma creatinine as a determinant of plasma total homocysteine concentrations in the Hordaland Homocysteine Study;Use of statistical modeling to determine reference limits. Clin. Biochemistry, 2007, 40, 1209-1218.
18. Fredericksen, J., Jul, K. Grande, P. et al.: Methy-lenetetrahydrofolate reductase polymorphism (C677T), hyperhomocysteinemia, and risk of ischemic cardiovascular disease and venous thromboembolism: prospective and case-control studies from the Copenhagen City Heart Study. Blood, 2004, 104, 3046-3051.
19. Frosst P., Blom, H. J., Milos, R. et al.: A candidate genetic risk factor for vascular disease: a common mutation in MTHFR. Nature Genetics, 1995, 10, 111-113.
20. Guenther, B. D., Shepphard, C. A., Tran, P. et al.: The structure and properties of methyleneterahydrofolate reductase from Escherichia coli suggest how folate ame-liorates human hyperhomocysteinemia. Nature Structural Biology, 1999, 6, 359-366.
21. Guttormsen, A. B., Ueland, P. N., Nesthus, I. et al.: Determinants and vitamin responsiveness of intermediate hyperhomocysteinemia (> 40 µmol/l). J. Clin. Invest., 1996, 98, 2174-83.
22. Hladovec, J.: Experimental homocysteinemia: endothelial lesions and thrombosis. Blood Vessels, 1979, 16, 202-205.
23. Hubacek, J. A.: Association of MTHFR genetic variants C677T and A1298C on predisposition to spontaneous abortion in Slavonic population. Clin. Chim. Acta, 2015, 44O, 104-107.
24. Hyánek, J., Pejznochová, H., Dvořáková, J.: Naše zkušenosti s diferencováním mírných hyperhomocysteinemií. Klin. Biochem. Metab., 2000, 8, 58-63.
25. Kluijtmans L. A. J., Kastelein, J. J. P, . Lindemans, J. et al.: Molecular genetic analysis in mild hyperhomocysteinemia:a common mutation in the MTHFR gene is genetic risk factor for cardiovascular disease. Am. J. Hum. Genet., 1996, 58, 35-41.
26. Kožich, V., Kraus, J, De Franchi, R. et al.: Hyperhomocysteinemia in premature arterial disease:examination of CBS alleles at the molecular level. Human. Mol. Genet. 1995, 623-629.
27. Kožich, V., Kraus, J., Hyánek, J.: Homocystein, geny a vitaminy: souvislost s kardiovaskulárním onemocněními a komplikacemi v těhotenství. DMEV, 1999, 3, 113-120.
28. Pullin, Ch., Ashfield, J. et al.: Optimalization of dietary folate or low dose folic acid supplements lower homocysteine but do not enhance edothel function in healthy adults, irrespective of the MTHFR genotype. J. Amer. Coll. Cardiol., 2001, 38, 1799-1805.
29. Liem, A., Reynierse-Buitenwwert, G. . H., Aeilko, H., et al.: Secondary Prevention with folic acid: effects on clinical outcomes., J. Amer. CollCard. 2003, 41, 2150-13.
30. Loscalzo, J.: Homocysteine trends-clear outcomes for complex reasons. NEJM, 2006, 354, 1629-32.
31. Malinow, M. R. Homocysteine and arterial occlusive diseases. J. Int. Med., 1994, 236, 603-609.
32. Milman, N.: Intestinal absorption of folic acid – new physiologic and molecular aspects. Indian. J. Med. Res., 2012:136, 725-728.
33. Ou, T., Yamakaw-Kobayashi, K., Arinami, T. et al.: Methylenetetrahydrofolate reductase and apolipoprotein E polymorphism are independent risk factors for coronary heart disease in Japanese: a case control study. Atherosclerosis, 1998, 18, 1465-9.
34. Refsum, H., Smith, A. D., Ueland, P. M. et al.: Facts and reccommendations about total homocysteine determinations:An expert opinion. Clin. Chem., 2004, 50, 3-32
35. Sanjoaquin, M., A., Allen, N., Conto, E. et. al.: Folate intake and colorectal carcinom risk: a meta-analytical approach. Int. J. Canc., 2005, 113, 825-28.
36. Schiff, M., Benoist, J. F., Tilea, B. et al.: Isolated remethylation disorders:do our treatments benefit patiens? J. Inher. Metabol. Dis., 2011, 34, 137-145.
Štítky
Biochémia Nukleárna medicína Nutričný terapeutČlánok vyšiel v časopise
Klinická biochemie a metabolismus

2017 Číslo 1
Najčítanejšie v tomto čísle
- Mírné hyperhomocysteinémie z deficitu MTHFR (C677T a C1298A) u dospělých a adolescentů v metabolické ambulanci. Je třeba je diferencovat a léčit?
- Doporučení k vyšetřování mozkomíšního moku
- Otrava etylenglykolem a falešně vysoká hladina laktátu
- Výživa a kostní metabolismus