
Identifikace rodiny s nosičstvím germinální delece genu SUFU na podkladě diagnózy desmoplastického meduloblastomu u batolete
Identification of a Family with SUFU Germline Deletion Based on a Case of Desmoplastic Medulloblastoma in an Infant
Background:
Medulloblastoma, an embryonal neuroectodermal tumor of the cerebellum, is the most common malignant brain tumor in children. There are approximately 15 cases diagnosed in the Czech Republic each year. The recent World Health Organization classification recognizes several histopathological subtypes of medulloblastoma: classical, desmoplastic/ nodular with its extensive-nodularity variant, and anaplastic/ large-cell variant. Further molecular analysis identified four basic subgroups of medulloblastoma: WNT, SHH, Group 3, and Group 4. The subgroup of SHH meduloblastoma is associated with somatic mutations of SHH, PTCH1, SUFU, SMO and TP53, while the most common mutations found in infants up to three years of age were PTCH1 and SUFU. The majority of medulloblastomas are sporadic diseases, whereas only about 5– 10% of all cases occur in connection with hereditary genetic syndromes.
Case:
We present a case of a 21-months old girl diagnosed with a localized posterior fossa tumor. The histopathological examination revealed a desmoplastic/ nodular medulloblastoma. The treatment comprised a radical exstirpation of the tumor followed by adjuvant chemotherapy. With the use of array-CGH, a partial biallelic deletion of the SUFU gene (locus 10q24.32) was detected in the tumor DNA, whereas a monoallelic deletion was found in the peripheral lymphocyte DNA of the patient. These findings were confirmed by an independent qPCR method. Monoallelic germline deletion of SUFU was also identified in the patient’s mother, who was a healthy carrier. Pedigree of the family suggested a transition of the germline deletion of SUFU, since another brain tumors (including one case diagnosed before the age of three years) were identified in previous generations.
Conclusion:
Germline mutations in SUFU gene are believed to predispose to infant desmoplastic/ nodular medulloblastomas, basal cell carcinomas and meningiomas. The susceptibility gene shows autosomal dominant inheritance with an incomplete penetrance. There is no evidence-based surveillance strategy suggested for the carriers of germline SUFU mutations/ deletions so far. Our recommendation is based both on a family history of our patient and similar cases described in the literature. Since the germinal mutations in SUFU are responsible for up to 50% of all desmoplastic medulloblastomas in children under three years of age, genetic testing of SUFU should be encouraged in this population of patients.
Key words:
medulloblastoma – hereditary cancer syndromes – genetic testing – gene deletion – SUFU gene
This publication was written at Masaryk university as part of the project MUNI/A/1552/2014 with the support of the Specific University Research Grant, as provided by the Ministry of Education, Youth and Sports of the Czech Republic in the year 2015, with the support by funds from the Faculty of Medicine MU to junior researcher K. Z., and by Ministry of Health of the Czech Republic, grants AZV NV15-30657A and RVO (FNBr, 6526705).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
19. 8. 2015
Accepted:
30. 9. 2015
Autoři:
J. Šoukalová 1; K. Vejmělková 2; T. Cermanová 3; K. Kašíková 1; A. Mikulášová 1,4; H. Janyšková 4; K. Melichárková 2; Z. Pavelka 2; M. Ježová 5; Š. Pospíšilová 3; P. Kuglík 1,4; I. Valášková 1; R. Gaillyová 1; J. Štěrba 2; K. Zitterbart 2
Působiště autorů:
Oddělení lékařské genetiky, FN Brno
1; Klinika dětské onkologie LF MU a FN Brno
2; Centrum molekulární biologie a genové terapie, Interní hematologická a onkologická klinika LF MU a FN Brno
3; Ústav experimentální biologie, PřF MU, Brno5 Ústav patologie, LF MU a FN Brno
4
Vyšlo v časopise:
Klin Onkol 2016; 29(Supplementum 1): 83-88
Kategorie:
Kazuistika
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amko2016S83
Souhrn
Východiska:
Meduloblastom, embryonální neuroektodermální tumor mozečku, je nejčastější zhoubný nádor mozku u dětí. Ročně je diagnostikováno v České republice přibližně 15 případů. Dle histopatologické klasifikace Světové zdravotnické organizace z roku 2007 jsou rozeznávány následující tři podtypy meduloblastomu: klasický, desmoplastický (a jeho krajní varianta, meduloblastom s převažující nodularitou) a velkobuněčný/ anaplastický meduloblastom. Molekulárně-genetické analýzy ukázaly, že existují čtyři hlavní podskupiny meduloblastomu označované WNT/ Wingless, SHH/ sonic Hedgehog, Group 3 a Group 4. V nádorové tkáni meduloblastomů podskupiny SHH se vyskytují somatické mutace genů SHH, PTCH1, SUFU, SMO a TP53, přičemž u dětí mladších tří let jsou nacházeny nejčastěji mutace genů PTCH1 a SUFU. Převážná většina meduloblastomů jsou sporadická onemocnění, jen v asi 5– 10 % případů se nádor vyskytuje jako součást dědičného genetického syndromu.
Případ:
Prezentujeme případ pacientky, u které byl ve věku 21 měsíců diagnostikován nádor zadní jámy lební. Histologické vyšetření prokázalo desmoplastický/ nodulární typ meduloblastomu. Léčba zahrnovala radikální neurochirurgickou operaci a adjuvantní chemoterapii, dítě zůstává po léčbě v kompletní remisi onemocnění. Metodou array-CGH byla v DNA izolované z tkáně tumoru detekována bialelická parciální delece genu SUFU (lokus 10q24.32), přičemž monoalelická delece části genu SUFU byla nalezena i v DNA izolované z lymfocytů periferní krve pacientky. Nálezy byly u pacientky ověřeny nezávisle metodou kvantitativní PCR (qPCR). Vyšetření genu SUFU bylo provedeno také u rodičů a starší sestry pacientky. Ztráta jedné alely v oblasti genu SUFU byla prokázána u matky pacientky. Výsledky DNA analýzy tak ukazují na germinální charakter mutace a její hereditární přenos. Předpokládáme souvislost tohoto nálezu s opakovaným výskytem nádorů mozku v rodě matky pacientky.
Závěr:
Zárodečné mutace SUFU genu jsou zmiňovány jako predispozice ke vzniku desmoplastických meduloblastomů u dětí mladších tří let, bazaliomů, bazocelulárních karcinomů a meningeomů. Dědičnost predispozice je autozomálně dominantní s neúplnou penetrancí. Dosud ale nebyla stanovena jednotná, na důkazech založená doporučení pro sledování rizik u nosičů této zárodečné mutace. Náš návrh vychází jak z rodinné situace, tak z podobných familiárních případů popsaných v odborné literatuře. Vzhledem k tomu, že germinální mutace genu SUFU jsou zodpovědné za přibližně 50 % všech desmoplastických meduloblastomů u dětí do tří let věku, je genetické testování mutací genu SUFU doporučeno u všech dětí mladších tří let s desmoplastickým meduloblastomem, resp. meduloblastomem molekulární podskupiny SHH.
Klíčová slova:
meduloblastom – hereditární nádorové syndromy – genetické testování – delece genu – gen SUFU
Úvod
V ČR je ročně diagnostikován nádor přibližně u 350 dětí. Nádory mozku představují až 30 % všech nádorů u dětí, spolu s akutními leukemiemi jsou v této věkové kategorii nejčastějšími nádorovými onemocněními [1]. Nádory mozku u dětí jsou velmi heterogenní skupinou, jednotlivé histologické typy se odlišují obvyklou lokalizací nádoru, typickým věkem dítěte při diagnóze, symptomatologií i biologickým chováním tumoru [2,3].
Meduloblastom je nejčastější zhoubný nádor mozku dětí. Ročně je diagnostikováno v ČR přibližně 15 případů. Z histopatologického hlediska jde o primitivní neuroektodermální nádor vyrůstající z mozečku (grade 4), se schopností vytvářet vzdálené metastázy v mozkomíšním moku, na mozkových obalech či v parenchymu centrální nervové soustavy (CNS). Dle histopatologické klasifikace Světové zdravotnické organizace z roku 2007 jsou rozeznávány následující podtypy: klasický meduloblastom, velkobuněčný/ anaplastický meduloblastom a desmoplastický meduloblastom s jeho krajní variantou, s převažující nodularitou [4,5].
Multimodální léčba meduloblastomu zahrnuje chirurgickou resekci, radioterapii a chemoterapii. Intenzita terapie závisí na věku dítěte, rozsahu resekce a přítomnosti metastáz. Moderní neurochirurgické postupy, nové techniky radioterapie a kombinovaná adjuvantní chemoterapie zlepšily historicky neuspokojivou prognózu nemoci. Dlouhodobě dnes přežívá přibližně 70– 81 % pacientů standardního rizika (dítě starší tří let, s nádorem lokalizovaným a radikálně odoperovaným) a 30– 70 % pacientů vysokého rizika [2,3,6]. Přesto však, zejména u dětí mladších tří let s nálezem klasického meduloblastomu a metastatického meduloblastomu, nelze považovat léčebné výsledky za uspokojivé. Závažná je pozdní toxicita terapie u většiny přeživších v oblasti tělesné, kognitivní i psychosociální [7].
Poznání molekulární podstaty nádorového onemocnění vedlo k definici čtyř molekulárních podskupin meduloblastomu: WNT/ Wingless, SHH/Sonic Hedgehog, Group 3 a Group 4 [2,6,8]. Dětští pacienti s meduloblastomem podtypu WNT mají nejlepší prognózu, zatímco pacienti s meduloblastomem podskupiny 3 mají v retrospektivních analýzách prognózu nejhorší. Z pohledu níže prezentované kazuistiky je významné, že desmoplastická varianta meduloblastomu se vyskytuje především u kojenců a batolat: dle metaanalýzy z roku 2012 ve 42 % případů ve srovnání s 28 % v souboru bez věkového rozlišení [6]. Molekulárně-geneticky jde u desmoplastické varianty meduloblastomu kojenců a batolat v cca 90 % o nádor s dysregulací signální dráhy Hedgehog (podskupina SHH) [2]. U meduloblastomů podskupiny SHH se setkáváme v nádorové tkáni s mutacemi genů SHH, PTCH1, SUFU, SMO a TP53, přičemž u kojenců a batolat jsou nejčastější mutace v genech PTCH1 a SUFU [8,9].
Převážná většina meduloblastomů jsou sporadická onemocnění. Z hlediska nádorových predispozičních stavů je však zcela zásadní skutečnost, že část meduloblastomů se vyskytuje u dětí s hereditárními genetickými syndromy (definovaná germinální mutace nádorových supresorů). Procento těchto případů není doposud zcela jasně určeno, dle střízlivých odhadů jde o 5– 10 % dětských pacientů s meduloblastomem [10]. Jedná se především o Gorlinův syndrom, nazývaný též syndrom bazocelulárního névu (zárodečné mutace PTCH1, PTCH2). Přítomny u tohoto syndromu bývají kraniofaciální a skeletální anomálie, bazaliomy kůže a desmoplastický meduloblastom. Zárodečné mutace genu SUFU jsou taktéž asociovány s některými fenotypovými rysy Gorlinova syndromu, případně též s familiárními meningeomy [11– 14]. Dědičnost je autozomálně dominantní s neúplnou penetrancí. Dalšími ze známých hereditárních nádorových syndromů, u nichž bývá meduloblastom přítomen, jsou Li-Fraumeniho syndrom (TP53), syndrom konstitučního deficitu mismatch repair (MMR) genů (CMMR-D, Turcotův syndrom typ 1), Fanconiho anémie (BRCA2, PALB2) a Turcotův syndrom typ 2 (familiární adenomatózní polypóza – APC) [10].
Popis případu
Ve věku 1 roku a 9 měsíců se u dívky rozvíjí mozečková symptomatologie (ztráta dosavadního pohybového stereotypu, nejistota sedu a chůze), přidávají se symptomy intrakraniální hypertenze (zvracení, bolesti hlavy). Zobrazovacím vyšetřením byl diagnostikován nádor zadní jámy lební, došetření prokázalo lokalizované onemocnění. Primární nádor byl neurochirurgem radikálně odstraněn. Histologicky byl popsán desmoplastický/ nodulární typ meduloblastomu. Adjuvantní léčba byla vedena s kurativním záměrem dle mezinárodního protokolu Children‘s Oncology Group (ACNS0334), zahrnovala tři indukční bloky konvenční kombinované chemoterapie (vinkristin, etoposid, cyklofosfamid, cisplatina) a tři bloky konsolidační chemoterapie (thiotepa, karboplatina) v myeloablativních dávkách s podpůrnou reinfuzí autologních progenitorů krvetvorby. Adjuvantní léčba radioterapií nebyla protokolárně v této věkové kategorii v případě dosažení kompletní remise onemocnění indikována. Akutní hematologická a infekční toxicita léčby stupně 3 a 4 byla očekávaná. Dívka zůstává v remisi, doba sledování od ukončení léčby je v tuto chvíli krátká, čtyři měsíce.
Z osobní anamnézy pacientky je třeba zdůraznit následující skutečnosti: dívka se rodí ze 4. rizikové gravidity, od 23. týdne těhotenství byla matka opakovaně hospitalizována pro inkompetenci děložního hrdla. Porod proběhl v 38. týdnu gravidity, byl indukován pro preeklampsii a ukončen akutně sekcí při alteraci ozev plodu. Porodní hmotnost byla 2 650 g a porodní délka 48 cm, skóre dle Apgarové 2-8-10, dívka byla krátce uložena do inkubátoru v kyslíkové atmosféře, následná poporodní adaptace již byla bez komplikací, novorozenecký ikterus byl léčen krátkodobě fototerapií. Do domácího ošetřování mohla být dívka propuštěna již šest dní po porodu. Psychomotorický vývoj probíhal do manifestace onemocnění bez pozoruhodností.
Údaje z rodinné anamnézy jsou následující: matka dívky má 31 let, je sledována „se štítnou žlázou“, bez nutnosti hormonální substituce, v dětství prodělala obrnu lícního nervu. V gynekologické anamnéze uvádí kromě dvou porodů další dvě neúspěšná těhotenství, první gravidita skončila spontánním abortem v 17. týdnu při septickém onemocnění, druhé těhotenství zamlklo v 10. týdnu. Otec pacientky má 36 let, je zdravý. Starší sestra pacientky má nyní 3,5 roku, byla narozena předčasně v 26. týdnu těhotenství, porodní délka 34 cm, porodní hmotnost 700 g. Za nejdůležitější skutečnost ve vztahu k prezentované kazuistice považujeme udávaný opakovaný výskyt nádorů CNS v rodě matky, viz genealogické schéma (obr. 1).
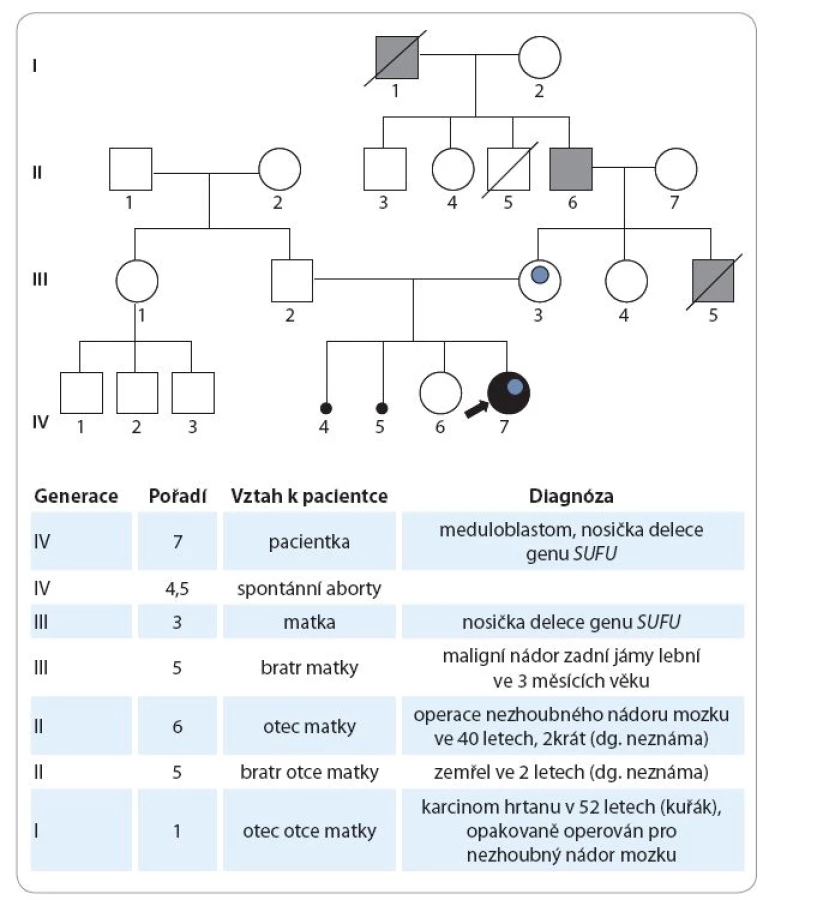
Rodina byla vyšetřena v genetické ambulanci. Eutrofická pacientka má relativní makrocefalii (98. percentil), s frontální prominencí. Přítomen je lehký hypertelorizmus, další somatické nápadnosti nejsou patrny. Faciálně je významná podoba s matkou, která má také makrokranii. Otec i starší sestra pacientky mají obvod hlavy normální. U pacientky bylo indikováno klasické cytogenetické vyšetření (karyotyp), celogenomové vyšetření metodou array-CGH a sekvenování genu TP53, zvažována byla i diagnostika genu PTCH při podezření na Gorlinův syndrom. Cytogenetickým vyšetřením byl detekován normální ženský karyotyp. Molekulárně-genetické vyšetření (DNA izolovaná z lymfocytů periferní krve) neprokázalo žádnou mutaci genu TP53, která by byla kauzální příčinou Li-Fraumeniho syndromu.
Současně byla zajištěna i vyšetření z tkáně meduloblastomu. Zásadní informaci přineslo vyšetření nádorumetodou celogenomové hybridizace.Metoda array-CGH odhalila v tkáni nádoru bialelickou deleci části genu SUFU na chromozomu 10, lokus 10q24.32 (104,315,002kb-104,402,706kb). Vzhledem ke známé skutečnosti, že germinální mutace či delece genu SUFU jsou zodpovědné za přibližně 50 % všech des-moplastických meduloblastomů u dětí mladších tří let věku [9], a vzhledem ke genealogickým údajům v rodině probandky jsme zvažovali hereditární nádorovou predispozici a byla vyšetřena i DNA izolovaná z lymfocytů periferní krve pacientky. Zde metoda array-CGH prokázala monoalelickou deleci části genu SUFU. Grafický výstup vyšetření array-CGH ukazuje obr. 2.

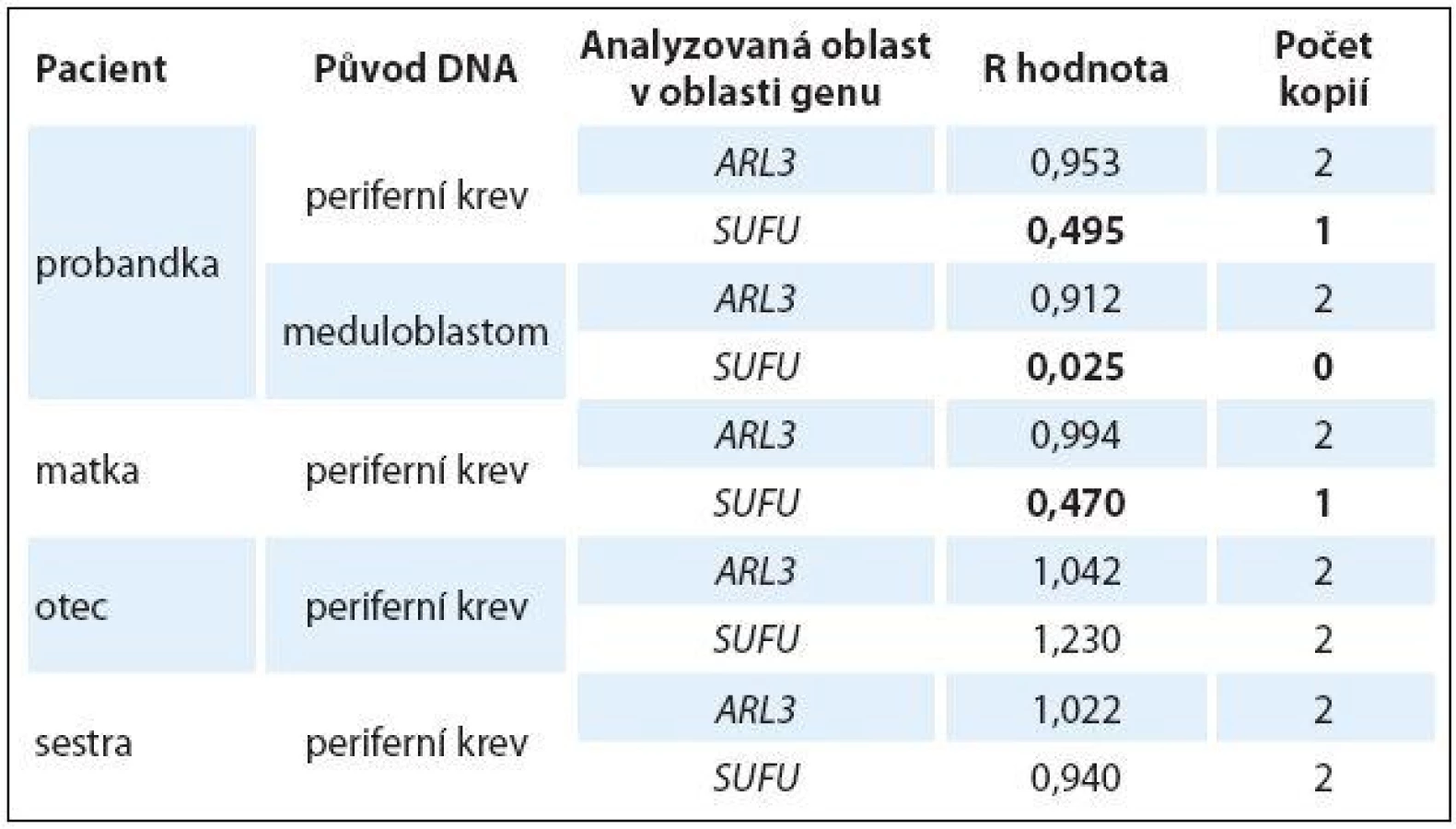
Ztráta detekovaná v oblasti genu SUFU byla následně testována a ověřena pomocí kvantitativní PCR (qPCR) u pacientky, rodičů a sestry pacientky. Zjištěné hodnoty R jsou shrnuty v tab. 1. Pomocí qPCR byla potvrzena ztráta jedné alely v oblasti genu SUFU ve vzorku DNA z periferní krve pacientky (R = 0,495) a ztráta obou alel této oblasti ve vzorku DNA z tumoru pacientky (R = 0,025). Ztráta jedné alely oblasti genu SUFU byla rovněž detekována ve vzorku DNA z periferní krve matky (R = 0,470). Ve vzorcích DNA z periferní krve otce a sestry pacientky byl potvrzen diploidní charakter testované oblasti genu SUFU. Ve všech vzorcích byl potvrzen diploidní charakter kontrolní oblasti v genu ARL3 (R ~ 1,000).
Detailně je metodika obou vyšetření popsána v následujícím textu:
Array-CGH: vyšetřována byla hluboce zamražená tkáň tumoru a periferní krev pacientky. Z těchto materiálů byla izolována DNA přes kolonky kitu Invisorb® Spin Tissue Mini Kit (Qiagen). Jednotlivé kroky vyšetření DNA pomocí celogenomové hybridizace byly provedeny podle manuálu výrobce použitého čipu (CytoSure™ Array Handbook; 4 × 180 k formats). Získaná DNA tumorové tkáně pacientky i referenční DNA (Human Genomic DNA, Female; Promega) byly značeny fluorochromy Cy5, resp. Cy3 (CytoSure Genomic DNA Labelling Kit; Oxford Gene Technology). Značené DNA byly společně hybridizovány při 65 °C po dobu 40 hod na oligonukleotidové fragmenty uchycené na čipu CytoSure Cancer + SNP Array 4 × 180 k (Oxford Gene Technology). Čip byl scanován na přístroji SureScan (Agilent) a vyhodnocen pomocí softwaru CytoSure Interpret Software (Oxford Gene Technology). Tento software automaticky identifikuje každý spot na čipu a stanoví poměr obou fluorochromů. Jestliže je log2 normalizovaného poměru fluorochromů větší než 0, hovoříme o ziscích, v opačném případě o ztrátách genetického materiálu.
qPCR: Počet kopií genu SUFU byl ověřován pomocí relativní kvantifikace DNA metodou qPCR. Testovány byly vzorky DNA izolované z periferní krve a tumoru pacientky, z periferní krve rodičů a sestry pacientky. Celkem 10 ng každé testované DNA a 10 ng komerčně dostupné referenční DNA (Agilent Technologies, Santa Clara, CA, USA) bylo smícháno s 12,5 µl Power SYBRgreen PCR Master Mix (Life Technologies, Paisley, UK), 2,5 µl přímého primeru (5 µM) a 2,5 µl zpětného primeru (5 µM) do celkového objemu 25 µl. V každém vzorku DNA byly amplifikovány odděleně tři genomové oblasti ohraničené třemi páry primerů (Sigma-Aldrich, St. Louis, MO, USA; referenční genom GRCh37/ hg19): 1. oblast v provozním genu GAPDH (lokus 12p13.31; přímý primer 5’-CTCCCACCTTTCTCATCC-3’, zpětný primer 5’-CCACATCACCCCTCTACC-3’), 2. kontrolní diploidní oblast v blízkosti chromozomové aberace v genu ARL3 (lokus 10q24.32, přímý primer 5’-CGGTCATCACCAACAATCAG-3’, zpětný primer 5’-ATTTTACGCCGTCTTGGTGT-3’) a 3. oblast v genu SUFU (lokus 10q24.32, přímý primer 5’-GGAAGAGCCTCCCCTTCTTA-3’, zpětný primer 5’-AGGACAAAGTGCTCCGGATA-3’). Kvantifikace byla provedena pomocí přístroje StepOne™ Real-Time PCR System (Life Technologies, Paisley, UK) při následujícím nastavení průběhu reakce: 10 min při 95 °C, 40 cyklů 15 sekund při 95 °C a 60 sekund při 60 °C. Vznik specifických amplifikačních produktů byl testován pomocí 1% agarové elektroforézy. Získaná data qPCR byla analyzována pomocí softwaru StepOne Software v2.3 (Life Technologies, Paisley, UK) generující pro každou amplifikační reakci hodnotu CT (počet cyklů, při kterém fluorescence překročí prahovou hodnotu). Počet kopií dané testované oblasti byl pak stanoven na základě výpočtu hodnoty R = 2–∆∆CT, kde ∆∆CT = ∆CT vzorku – ∆CT reference, kde ∆CT = CT analyzované oblasti – CT provozního genu. Jako mezní hodnoty R byly použity > 1,3 pro přítomnost třech kopií testované oblasti a < 0,7 pro přítomnost jedné kopie testované oblasti.
Diskuze
V našem případě desmoplastického meduloblastomu u batolete jsme metodou array-CGH prokázali v periferní krvi pacientky ztrátu části jedné alely genu SUFU a ztrátu obou alel v nádorové tkáni, tato zjištění jsme následně potvrdili pomocí techniky qPCR. Ztráta jedné alely v oblasti genu SUFU byla detekována rovněž v periferní krvi matky pacientky. Z těchto výsledků lze usuzovat, že ztráta jedné alely genu SUFU u probandky má germinální charakter oocytárního původu, zatímco ztráta druhé alely tohoto genu má charakter somatický. Delece části genu SUFU je v příčinné souvislosti se vznikem desmoplastického/ nodulárního meduloblastomu. Většina případů germinální nádorové predispozice je dána heterozygotními mutacemi v genu SUFU vedoucími ke ztrátě funkce proteinu. Stejně jako v našem případě však byly identifikovány i větší delece: je popsána 2,5 Mb delece zahrnující SUFU u pacienta s meduloblastomem a Gorlin-like fenotypem [11,12] i velká heterozygotní delece SUFU v rodině s familiárním meduloblastomem [14].
Z genealogických údajů předpokládáme, že zárodečná mutace genu SUFU byla příčinou výskytu maligního tumoru mozku v kojeneckém věku u zemřelého bratra matky pacientky a že matka pacientky získala tuto zárodečnou mutaci od svého otce. Tento předpoklad by bylo potřeba ověřit vyšetřením příbuzných matky pacientky. Bylo proto doporučeno vyšetření obou rodičů a sestry matky pacientky vzhledem k 50% riziku nosičství této zárodečné mutace. Tato vyšetření zatím neproběhla.
Zárodečné mutace SUFU jsou příčinou nejen predispozice ke vzniku desmoplastických meduloblastomů u dětí mladších tří let, ale také predispozicí k bazaliomům a basocelulárním karcinomům nastupujícím už v druhé dekádě života a meningeomů nastupujícím již v třetí dekádě života [14,15]. Dědičnost predispozice je autozomálně dominantní s neúplnou penetrancí. S ohledem na zvýšené riziko výskytu nádorů spjatých s germinální mutací či delecí genu SUFU je důležitá sekundární onkologická prevence u jejích nositelů. Dosud ale nebyla stanovena jednotná, na důkazech založená doporučení k časné detekci nádorů spjatých s nosičstvím této zárodečné mutace. U malých dětí se známým nosičstvím germinální mutace/ delece SUFU lze odůvodněně doporučit neurologické vyšetření každé tři měsíce a při atypickém nálezu, resp. při nově vzniklých symptomech pak neodkladné provedení magnetické rezonance (MRI) mozku. I asymptomatickým nosičům mutace je navrhováno screeningové vyšetření MRI mozku každé tři měsíce od narození do ukončeného druhého, resp. třetího roku života [14]. Dále lze nosičům odůvodněně doporučit zejména celoživotní důslednou radioprotekci a fotoprotekci a preventivní prohlídky kožním lékařem jedenkrát ročně jako primární a sekundární prevenci kožních nádorů. U fertilních žen se doporučuje roční preventivní gynekologické vyšetření a ultrazvukové vyšetření malé pánve a břicha (riziko ovariálních fibromů), v dospělosti pak lze navrhnout již od konce druhé, resp. začátku třetí dekády u všech nosičů mutace každoroční neurologické vyšetření a screeningové MRI vyšetření mozku k včasné identifikaci dalších možných nádorů CNS, meningeomů [15].
Závěr
Meduloblastom je nejčastější zhoubný nádor mozku u dětí. U části případů, dle střízlivých odhadů v 5– 10 %, se vyskytuje ve spojitosti s hereditárními genetickými syndromy. Vedle Li-Fraumeniho syndromu, syndromu konstitučního deficitu mismatch repair (MMR) genů, Fanconiho anémie a Turcotova syndromu typu 2 jde především o Gorlinův syndrom (syndrom bazocelulárního névu). Nejnovější práce uvádějí, že riziko meduloblastomu u Gorlinova syndromu je relativně nízké, pouhá 2 %. Naopak, germinální mutace genu SUFU jsou zodpovědné až za 50 % všech desmoplastických meduloblastomů u dětí mladších tří let věku, a je proto doporučeno genetické testování této hereditární nádorové predispozice u všech dětí mladších tří let s desmoplastickým meduloblastomem, resp. meduloblastomem molekulární podskupiny SHH.
Publikace vznikla na Masarykově univerzitě v rámci projektu MUNI/A/1552/2014 podpo-řeného z prostředků účelové podpory na specifický vysokoškolský výzkum, kterou poskytlo MŠMT v roce 2015, a dále s podporou prostředků IRP z LF MU (K. Z.) a MZ ČR: AZV NV15-30657A a RVO (FNBr, 6526705).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Karel Zitterbart, Ph.D.
Klinika dětské onkologie
LF MU a FN Brno
Černopolní 9
613 00 Brno
e-mail: kzitter@med.muni.cz
Obdrženo: 19. 8. 2015
Přijato: 30. 9. 2015
Zdroje
1. Bajčiová V, Šmelhaus V, Kodytková D et al. Dětská onkologie se musí opírat o spolehlivá data. Medical Tribune 2011; 7(3): C2– C3.
2. Gajjar A, Bowers DC, Karajannis MA et al. Pediatric brain tumors: innovative genomic information is transforming the diagnostic and clinical landscape. J Clin Oncol 2015; 33(27): 2986– 2998. doi: 10.1200/ JCO.2014.59.9217.
3. Pavelka Z, Zitterbart K. Nádory centrálního nervového systému u dětí. Neurol Prax 2011; 12(1): 52– 58.
4. Louis DN, Ohgaki H, Wiestler OD et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114(2): 97– 109.
5. Sumerauer D. Neuropathological diagnostics in pediatric oncology from the clinical point of view. Cesk Patol 2012; 48(2): 72– 74.
6. Kool M, Korshunov A, Remke M et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012; 123(4): 473– 484. doi: 10.1007/ s00401-012-0958-8.
7. Moxon-Emre I, Bouffet E, Taylor MD et al. Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J Clin Oncol 2014; 32(17): 1760– 1768. doi: 10.1200/ JCO.2013.52.3290.
8. Kool M, Jones D, Jager N et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014; 25(3): 393– 405. doi: 10.1016/ j.ccr.2014.02.004.
9. Brugières L, Remenieras A, Pierron G et al. High frequency of germline SUFU mutations in children with desmoplastic/ nodular medulloblastoma younger than 3 years of age. J Clin Oncol 2012; 30(17): 2087– 2093. doi: 10.1200/ JCO.2011.38.7258.
10. Scheinemann K, Bouffet E (eds). Pediatric neuro-oncology. New York: Springer-Verlag 2015.
11. Taylor MD, Liu L, Raffel C et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet 2002; 31(3): 306– 310.
12. Pastorino L, Ghiorzo P, Nasti S et al. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 2009; 149A(7): 1539– 1543. doi: 10.1002/ ajmg.a.32944.
13. Brugières L, Pierron G, Chompret A et al. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J Med Genet 2010; 47(2): 142– 144. doi: 10.1136/ jmg.2009.067751.
14. Kijima C, Miyashita T, Suzuki M et al. Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer 2012; 11(4): 565– 570. doi: 10.1007/ s10689-012-9548-0.
15. Smith MJ, Beetz C, Williams SG et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 2014; 32(36): 4155– 4161. doi: 10.1200/ JCO.2014.58.2569.
Štítky
Detská onkológia Chirurgia všeobecná OnkológiaČlánok vyšiel v časopise
Klinická onkologie

2016 Číslo Supplementum 1
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Nejasný stín na plicích – kazuistika
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Tramadol a paracetamol v tlumení poextrakční bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
Najčítanejšie v tomto čísle
- PALB2 jako další kandidátní gen pro genetické testování u pacientů s hereditárním karcinomem prsu v České republice
- Hepatoblastom, etiologie, kazuistiky
-
Seznam onkologických pracovišť a onkologů zajišťujících prevenci pro osoby (i zdravé) s dědičným rizikem nádorů
Seznam genetických ambulancí při KOC - Genetika tumorigenézy nádorov kolorekta (možnosti testovania a screeningovej predikcie dedičnej formy ochorenia – Lynchovho syndrómu)