
Teleangiectasia hereditaria haemorrhagica – syndrom Osler-Weber-Rendu. Popis případu a zkušeností s léčbou
Teleangiectasia hereditaria haemorrhagica –Osler-Weber-Rendu syndrome. Case study and treatment experience
Hereditary haemorrhagic telangiectasy is an inborn disease with autosomal dominant transmission. Nose bleeding usually occurs during the 2nd decade of life as the first sign of the disease. Later, during the 3rd or 4th decade of life, typical subtle, pinhead-sized (1–2 mm in diameter) vascular arteriovenous malformations occur. These are usually found on the oral mucosa and in the stomach and small intestine. During later stages of the disease, nose as well as gastrointestinal bleeding causes severe anaemia requiring transfusions. Advanced stages of hereditary hemorrhagic telangiectasy are associated with a development of ateriovenous vascular malformations in the liver, lungs and possibly the brain. Vascular ateriovenous malformations in the liver cause hyperkinetic circulation that may lead to heart failure. Blood within the pulmonary ateriovenous malformations bypasses filtration in the pulmonary capillary circulation and thus infected microtrombi may pass from the inferior vena cava to, for example, the brain. At first, local treatment – stopping epistaxis – is used. Symptomatic embolisation treatment and, sometimes, liver transplantation are used in advanced forms of the disease with anaemisation, despite iron substitution, and clinically significant ateriovenous malformations. Angiogenesis-inhibiting substances have been shown effective in patients with an advanced disease. Older clinical studies confirmed benefits of combined oestrogen-progesterone treatment, later also treatment with raloxifene or antioestrogens. Many post-2000 publications showed thalidomide and bevacizumab to be effective in this indication. Treatment with bevacizumab has led not only to increased haemoglobin concentrations but, through regression of ateriovenous malformations, provided control of hyperkinetic circulation. Discussion section provides an overview of treatment modalities. The main text describes a case of a 56 years old female patient with hypochromic anaemia despite maximum oral iron substitution. The patient lost blood through repeated epistaxes as well as continuous mild bleeding into gastrointestinal tract. The patient also had confirmed large ateriovenous malformations in the liver. Interferon alpha was used as the first line of treatment. The patient unexpectedly developed fast and pronounced myelosuppression. The number of neutrophils fell down from 1.15 x 109/l to 0.6 × 109/l as soon as after 3 injections of interferon alpha at a starting dose of 1.5 million units 3 times a week. Therefore, interferon alpha was discontinued. Blood count returned to normal following interferon discontinuation. The patient was started on thalidomide in December 2011. The patient reported lower incidence of epistaxes and smaller blood loss than before treatment as soon as during the first month of therapy. Regular administration of thalidomide reduced intensity and frequency of epistaxes in this patient.
Key words:
hereditary haemorrhagic telangiectasia – syndrom Osler-Weber-Rendu – haemangiomas – arteriovenous malformations – epistaxis – anaemia – estrogens – antiestrogens – raloxifen – thalidomide – bevacizumab – interferon α
Autori:
Z. Adam 1; G. Chlupová 2; A. Neumann 3; B. Jakubcová 3; J. Simonides 4; Z. Adamová 5; J. König 6; P. Krupa 7; P. Szturz 1; L. Pour 1; M. Krejčí 1; R. Hájek 1; Z. Král 1; J. Mayer 1
Pôsobisko autorov:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
2; Radiologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Vlastimil A. Válek, CSc., MBA
3; Hematologicko-transfuzní oddělení Nemocnice Znojmo, p. o., Znojmo, přednosta prim. MUDr. Jan Simonides
4; Ambulance pro děti a dorost Brno, Obilní trh
5; Oddělení ORL FN Brno, pracoviště Bohunice, přednosta prim. MUDr. Aleš Trnka
6; Oddělení zobrazovacích metod SurGal Clinic, s. r. o., Brno, přednosta prim. MUDr. Jan Křístek, Ph. D.
7
Vyšlo v časopise:
Vnitř Lék 2012; 58(6): 477-489
Kategória:
Kazuistiky
Súhrn
Hereditární hemoragická teleangiektazie je vrozené onemocnění s autozomálně dominantním přenosem. Prvním příznakem je krvácení z nosu, obvykle v 2. dekádě života. Teprve později, ve 3. či 4. dekádě, se vyvinou typické drobné cévní arteriovenózní malformace velikosti špendlíkové hlavičky (o průměru 1–2 mm). Bývají na kůži a na sliznicích dutiny ústní a samozřejmě také v žaludku a v tenkém střevě. Krvácivé projevy nejen z oblasti nosu, ale také z oblasti trávicí trubice způsobují v pokročilých případech těžkou anémii se závislostí na transfuzích. Pokročilejší formy hereditární hemoragické teleangiektazie jsou provázeny rozvojem arteriovenózních cévních malformací v oblasti jater, plic a případně v mozku. Cévní arteriovenózní malformace v oblasti jater způsobují hyperkinetický oběh, který může vyústit do srdečního selhání. V arteriovenózních plicních malformacích obchází krev filtraci plicním kapilárním řečištěm, a tak se mohou infikované mikrotromby dostal z oblasti dolní duté žíly třeba do mozku. V počátečních fázích je léčba lokální – zastavování epistaxe. U pokročilých forem s anemizací i při substituci železem a s rozvojem klinicky závažných arteriovenózních malformací se provádí symptomatická embolizační léčba a v některých případech transplantace jater. U pacientů s pokročilými formami nemoci se osvědčily léky, které inhibují angiogenezi. Starší klinické studie potvrdily přínos kombinované estrogen-progesteronové léčby, později i léčby pomocí raloxifenu či antiestrogenů. Po roce 2000 potvrdily četné publikace účinnost thalidomidu a bevacizumabu v této indikaci. Léčba bevacizumabem vedla nejen k vzestupu koncentrace hemoglobinu, ale díky regresi jaterních arteriovenózních malformací se upravil hyperkinetický oběh. V diskuzi přinášíme přehled léčebných možností. V textu popisujeme pacientku ve věku 56 let ve stadiu ztrátové hypochromní anémie i při maximální perorální substituci železem. Pacientka ztrácí krev jak opakovanými epistaxemi, tak i trvalým drobným krvácením do trávicí trubice. Tato pacientka měla prokázány velké arteriovenózní malformace v játrech. V rámci první linie léčby byl použit interferon α. Překvapením byla neobvykle rychlá a hluboká myelosuprese. Již po 3 injekcích interferonu α ve startovací dávce 1,5 milionu jednotek 3krát týdně poklesl počet neutrofilů z 1,15 × 109/l na 0,6 × 109/l. Proto musela být léčba interferonem α ukončena. Po přerušení dávek interferonu se krevní obraz vrátil do původních hodnot. Od konce prosince roku 2011 začala pacientka užívat thalidomid. Ihned v 1. měsíci udávala pacientka snížení četnosti epistaxí a menší ztráty krve v průběhu epistaxe než před léčbou thalidomidem. Pravidelné podávání thalidomidu snížilo intenzitu a frekvenci epistaxí u popisované pacientky.
Klíčová slova:
teleangiectasia hereditaria haemorrhagica – syndrom Osler-Weber-Rendu – hemangiomy – arteriovenózní cévní malformace – epistaxe – anémie – estrogeny – antiestrogeny – raloxifen – thalidomid – bevacizumab – interferon α
Úvod
Do ordinace přijde pacient krvácející z nosu. To není neobvyklý případ. První pomocí je lokální ošetření. Ne-specialista provede obvykle přední nosní tamponádu. Specialista ORL lékař má širší léčebné spektrum, může lokálně vyšetřit nosní dutinu a případně cíleně zakročil na krvácející cévce. A tím je krvácení, alespoň pro tuto chvíli, zastaveno.
Tím však práce lékaře nekončí. Je třeba položit si otázku, proč došlo ke krvácení z nosu. Co bylo příčinou krvácení. Nejběžnější příčiny jsou shrnuty v tab. 1.

Následující text je věnován vzácné vrozené chorobě, hereditární teleangiektazii, jejíž první příznak je krvácení z nosu. Teleangiektazie postihují kůži i sliznice. Na kůži představují jenom kosmetický problém, zatímco na sliznicích dýchacího a zažívacího traktu představují zdroj krvácení. S rozvojem teleangiektazií však souvisí i rozvoj arteriovenózních malformací, které také ohrožují nemocného člověka. Podezření na tuto chorobu může vzniknout jednak na základě otázky, zda krvácením z nosu netrpí rodiče či další příbuzní nemocného, a jednak z lokálního nálezu.
V poslední době jsou však pro tuto nemoc dostupné nové léky, které mohou pozitivně ovlivnit průběh choroby. Jedná se o thalidomid a bevacizumab. Proto čtenářům předkládáme popis případu dokumentovaný CT a sonografickým zobrazením arteriovenózních malformací a podrobným přehledem publikovaných informací o léčbě této nemoci.
Popis případu
Pacientka s typickými projevy hereditární hemoragické teleangiektazie (HHT) se narodila v roce 1955. Krvácivé teleangiektazie se vyskytovaly a vyskytují i u dalších členů rodiny, konkrétně u dědečka a u otce naší pacientky, u jedné ze 3 sester naší pacientky a dále u jednoho ze 2 dětí, u syna naší pacientky. Otec naší pacientky se dožil s touto nemocí překvapivě 89 let. Výskyt v rodině se pokusíme znázornit na schématu 1.

První příznaky – opakované krvácení z nosu, datuje naše pacientka do svých 35 let. Od té doby byla opakovaně v péči ORL lékařů. V roce 2003, tedy ve 48 letech, byla předána do péče hematologů nemocnice Znojmo. Důvodem byla zvyšující se frekvence krvácení z nosu a prohlubující se anemizace.
U syna naší pacientky se však první krvácení (epistaxe) objevilo již ve 20 letech.
Kromě hereditární hemoragické teleangiektazie se pacientka posledních 5 let léčí pro arteriální hypertenzi. Na Interní hematoonkologické klinice v Brně byla na první konzultaci v roce 2011.
Z předchozích vyšetření bychom chtěli zdůraznit výsledek posledního kardiologického vyšetření v nemocnici ve Znojmě, kde je mimo jiné uveden echokardiografický nález, popisující silně dilatované síně. Tloušťka stěn komor byla v normě, kontraktilita levé komory na dolní hranici normy, ejekční frakce byla asi 55 %. Byla přítomna mitrální regurgitace a stopa perikardiálního výpotku.
V září roku 2011 byla přijata na Interní hematoonkologickou kliniku v Brně ke krátkému přešetření a zahájení léčby. V době přijetí byly zřetelné mnohočetné angiektazie na konečcích prstů, na rtech, v obličeji, na jazyku, na břiše, na vnitřní straně stehen (obr. 1–4).
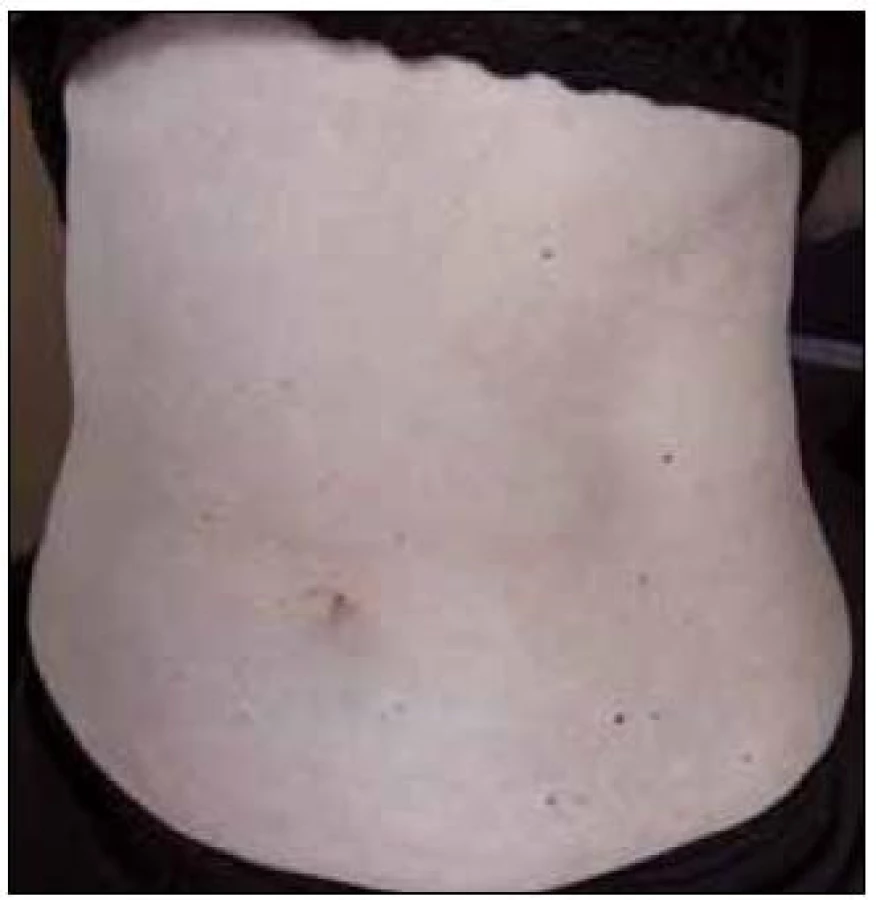
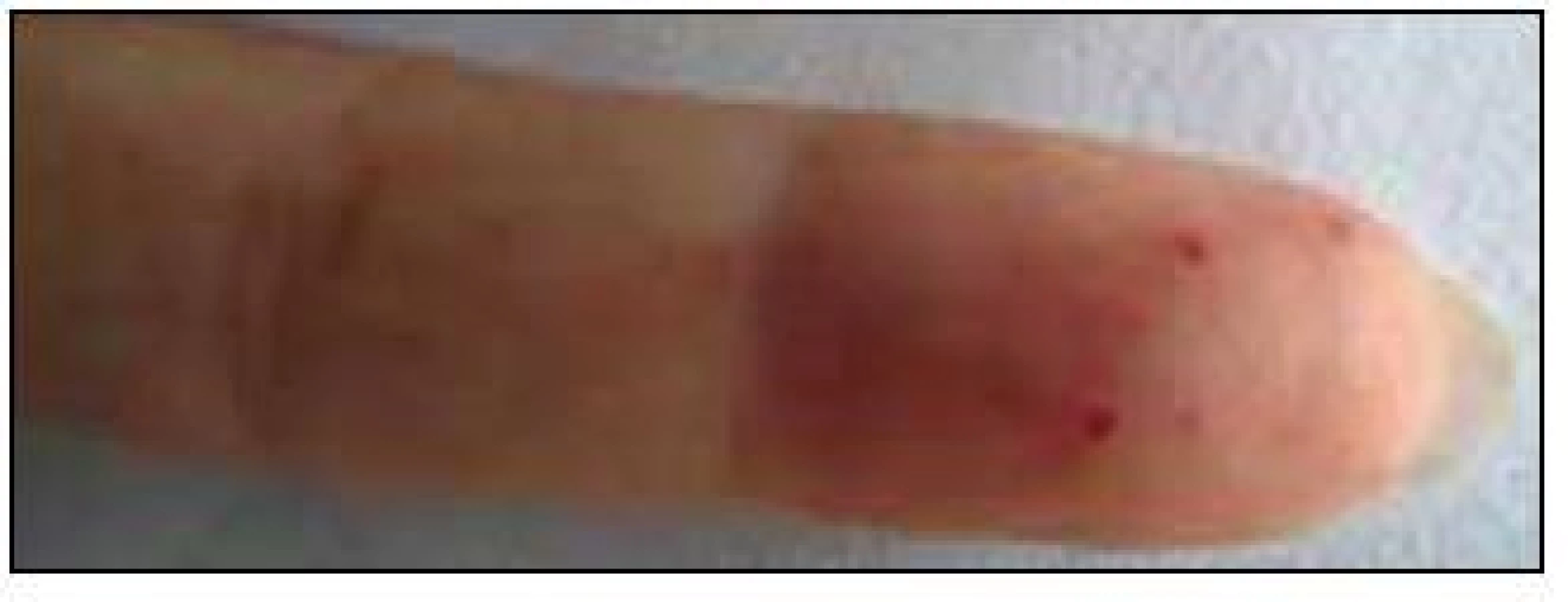
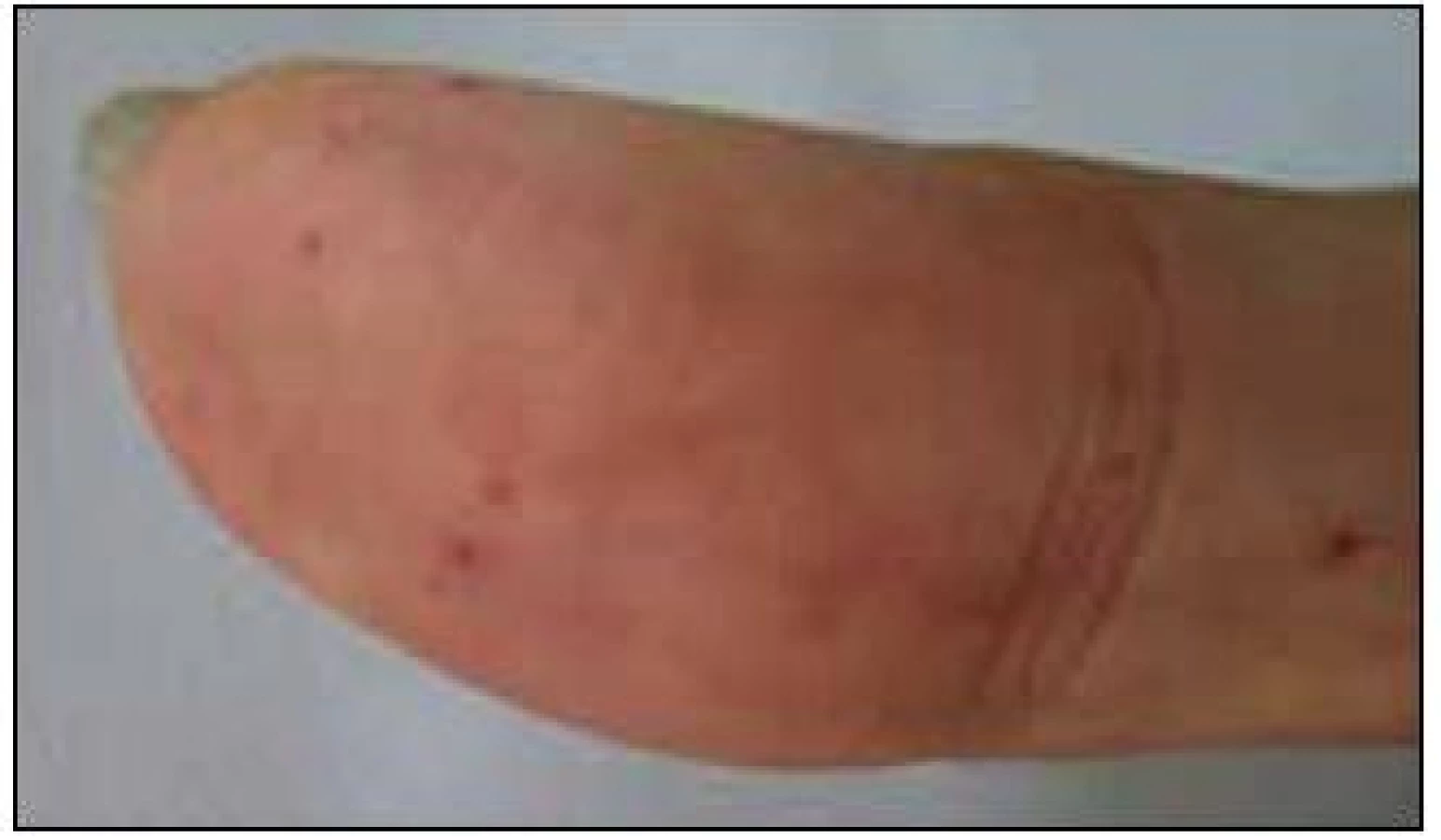
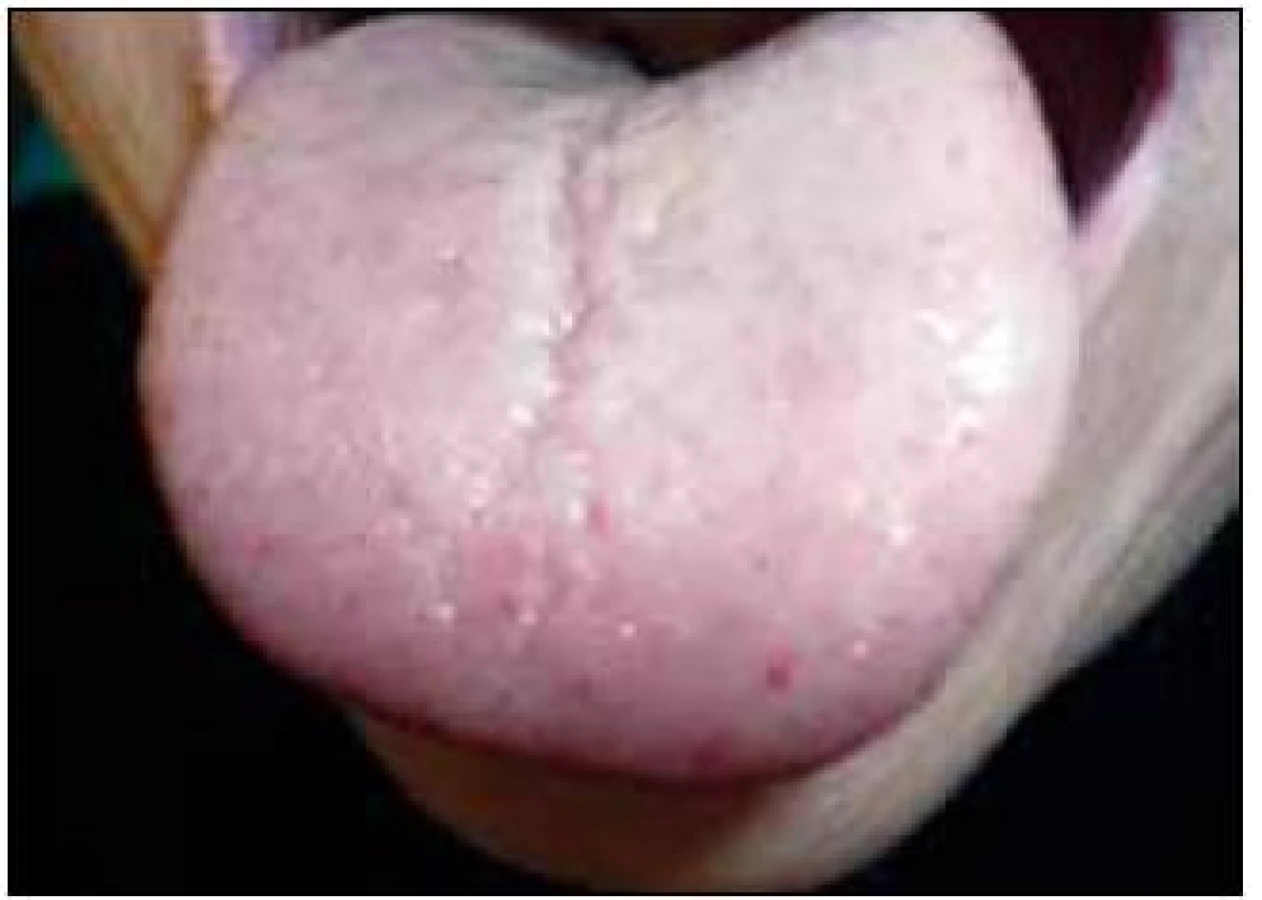
V době přijetí tato pacientka dlouhodobě užívala následující léky: Tenormin 50 mg 1 tabletu denně, Dicynone 2krát 1 tabletu denně, Ascorutin 3krát 2 tablety denně a Ferratab comp. 2 tablety denně, Paralen tablety při bolesti ramene v dávce do 3 g denně.
V době přijetí byla pacientka anemická, koncentrace hemoglobinu byla 76 g/l, koncentrace železa při dlouhodobé perorální substituce železem byla snížena na 2,7 µg/l (norma 6,6–26 µg/l) a koncentrace ferritinu byla snížena na 9,8 µg/l (norma 13–15 µg/l), koncentrace transferinu byla 3,17 g/l (norma 2,0–3,7 g/l) a saturace transferinu byla 0,03 (norma 0, 20–0,36). Ostatní biochemické hodnoty byly v normě. Koagulační parametry byly také v normě.
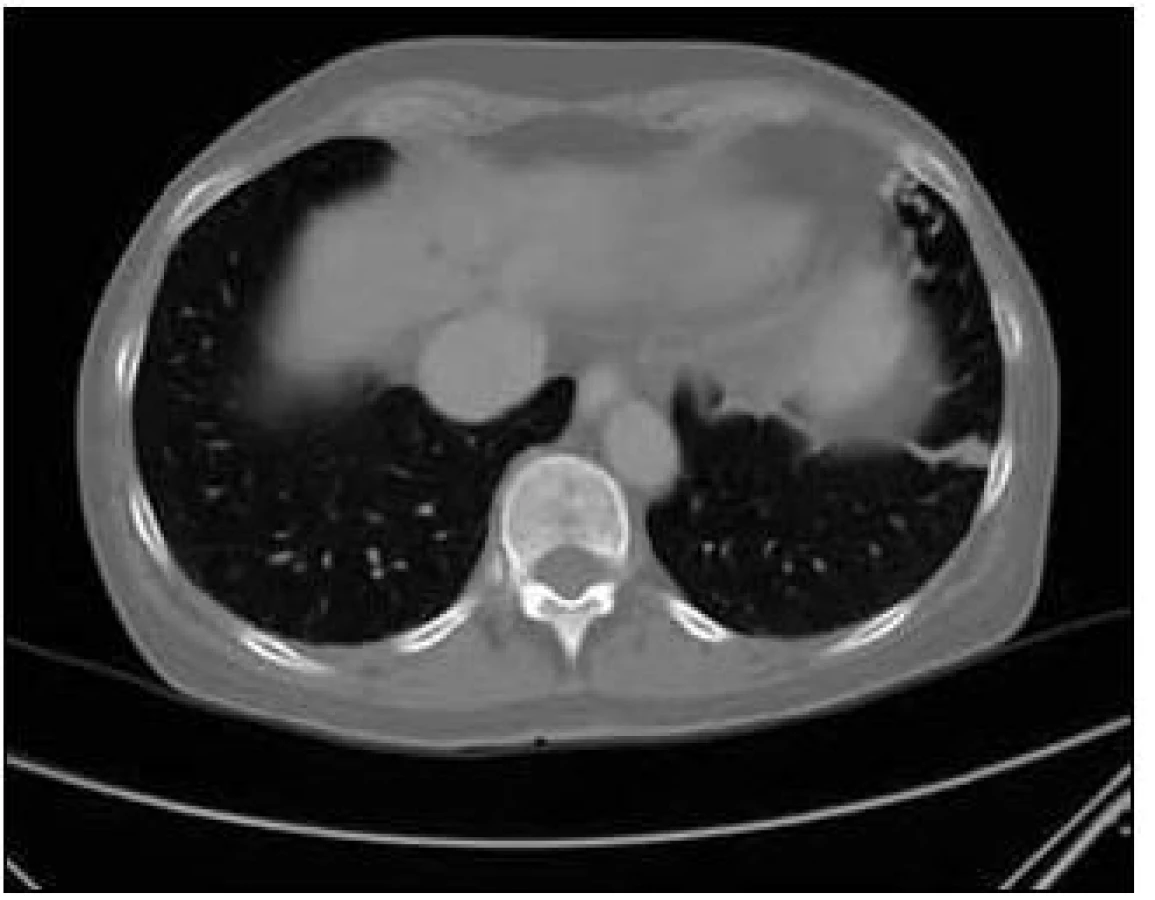
U pacientky jsme v rámci pátrání po rozsahu nemoci provedli CT plic. Na CT plic byla ve shodě s předchozími vyšetřeními ve Znojmě popsána arteriovenózní (A-V) malformace v lingule levé plíce. Dále bylo provedeno CT břicha s nálezem A-V malformací jater s popsanou dilatací cévních struktur a s časným sycením jaterních žil. Žlučové cesty nebyly dilatovány. Jako vedlejší nález při CT vyšetření břicha byly popsány jaterní cysty, myomy dělohy, angiomyolipom pravé ledviny, jemně zesílené intersticium na bazích plic a rarefakce osového skeletu (obr. 5–7).
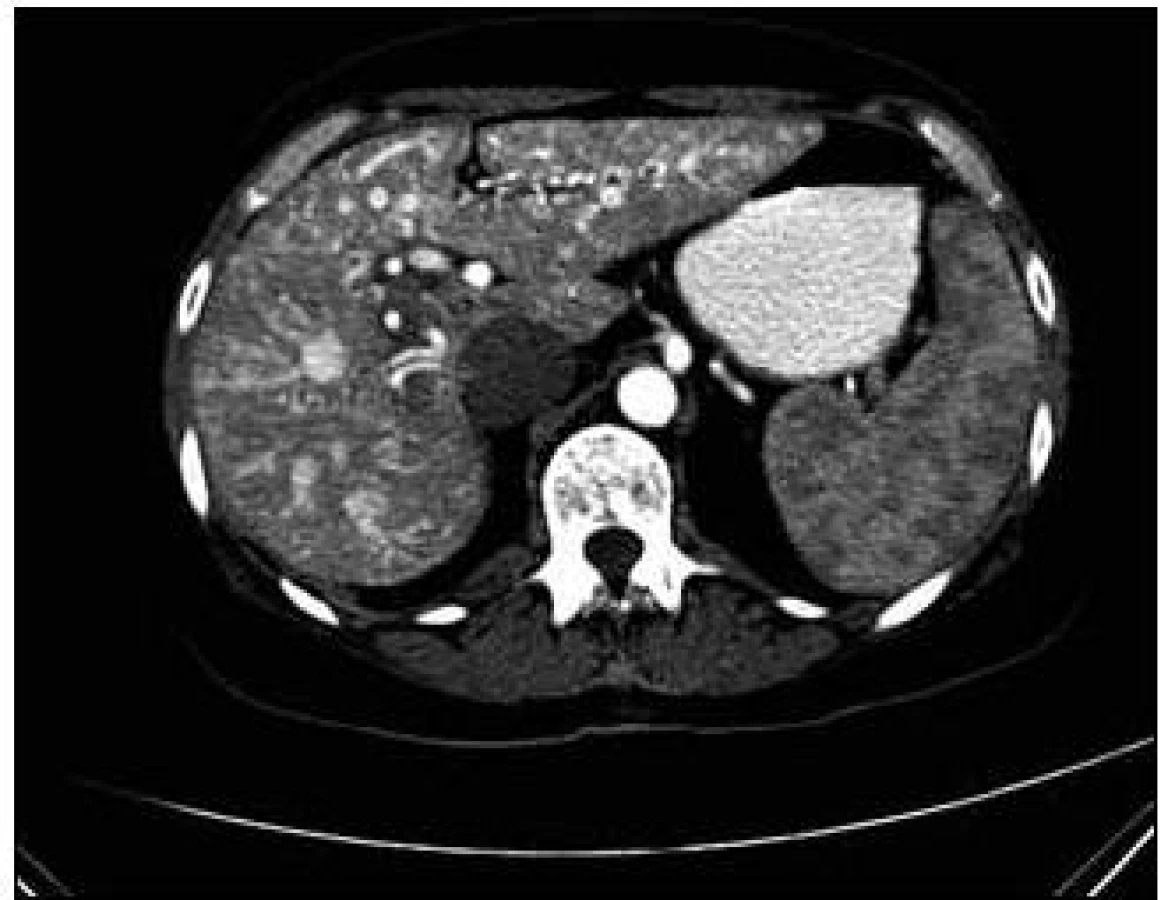
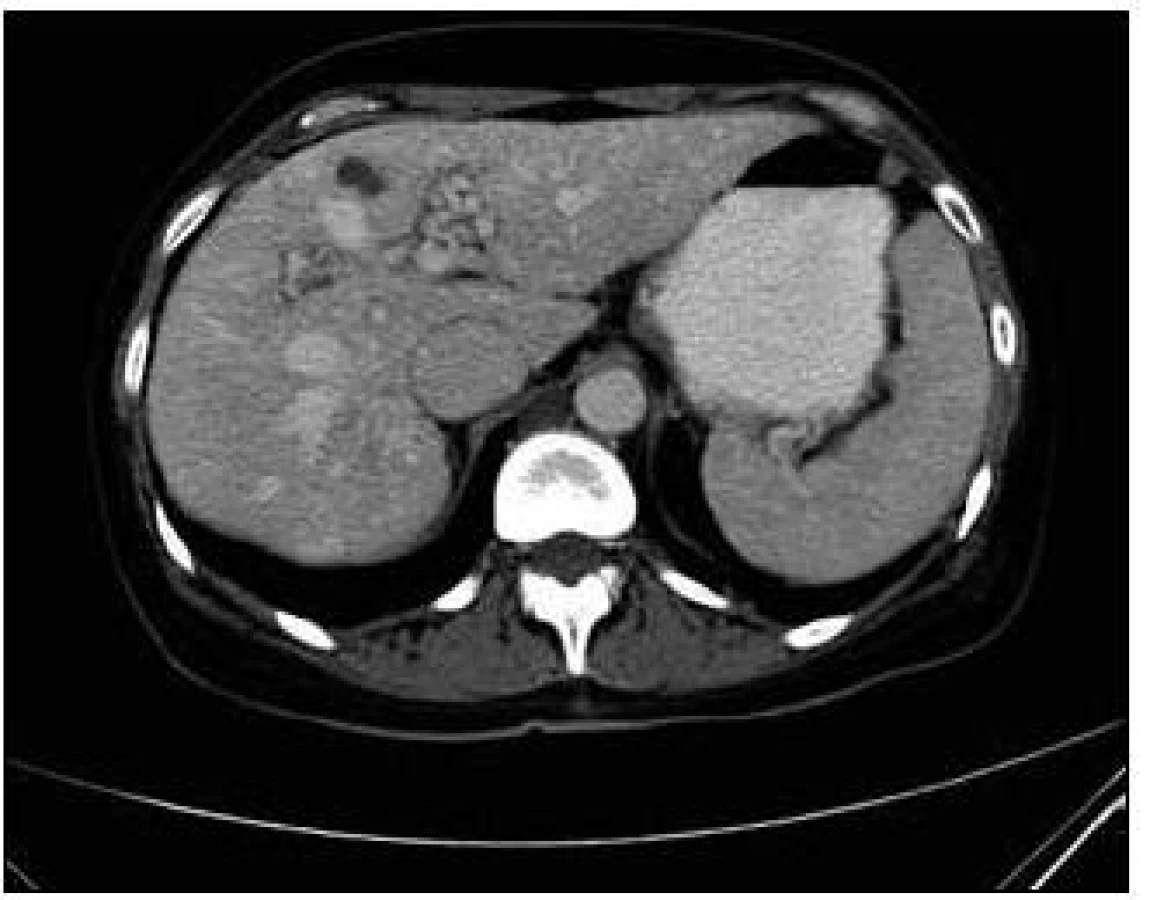
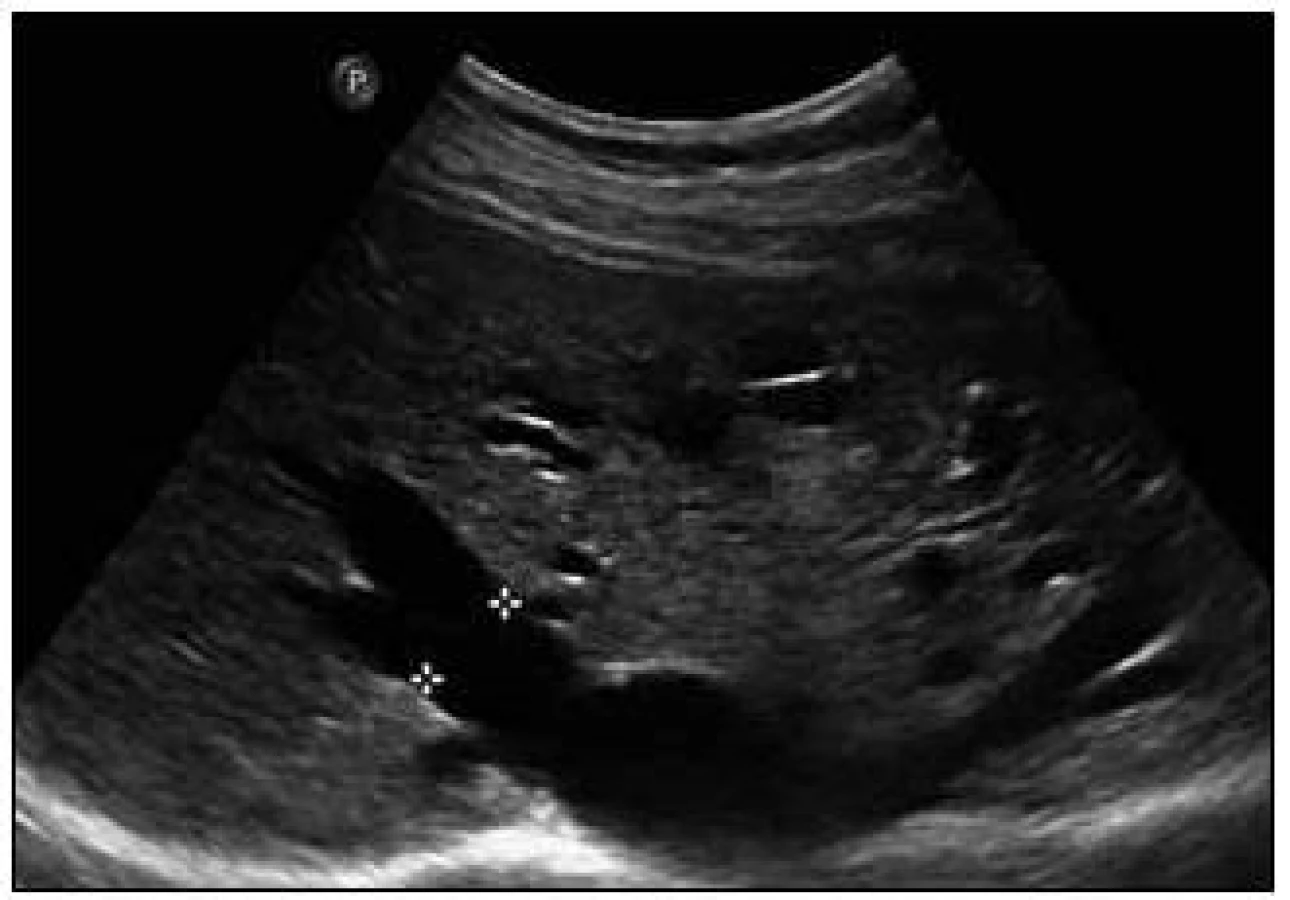
Sonografie jater popisuje podobný obraz ze svého úhlu pohledu. Játra se sonograficky jevila jako nezvětšená, v jaterním parenchymu byly zřetelné četné intrahepatické A-V spojky s maximem v oblasti portální žíly a při soutoku jaterních žil. V této oblasti byly rozšířené až na 16 mm. V. portae měla šíři 12 mm. Sonografický obraz odpovídal rozsáhlé arteriovenózní malformaci jaterního parenchymu, jak dokumentují obr. 8 a 9.
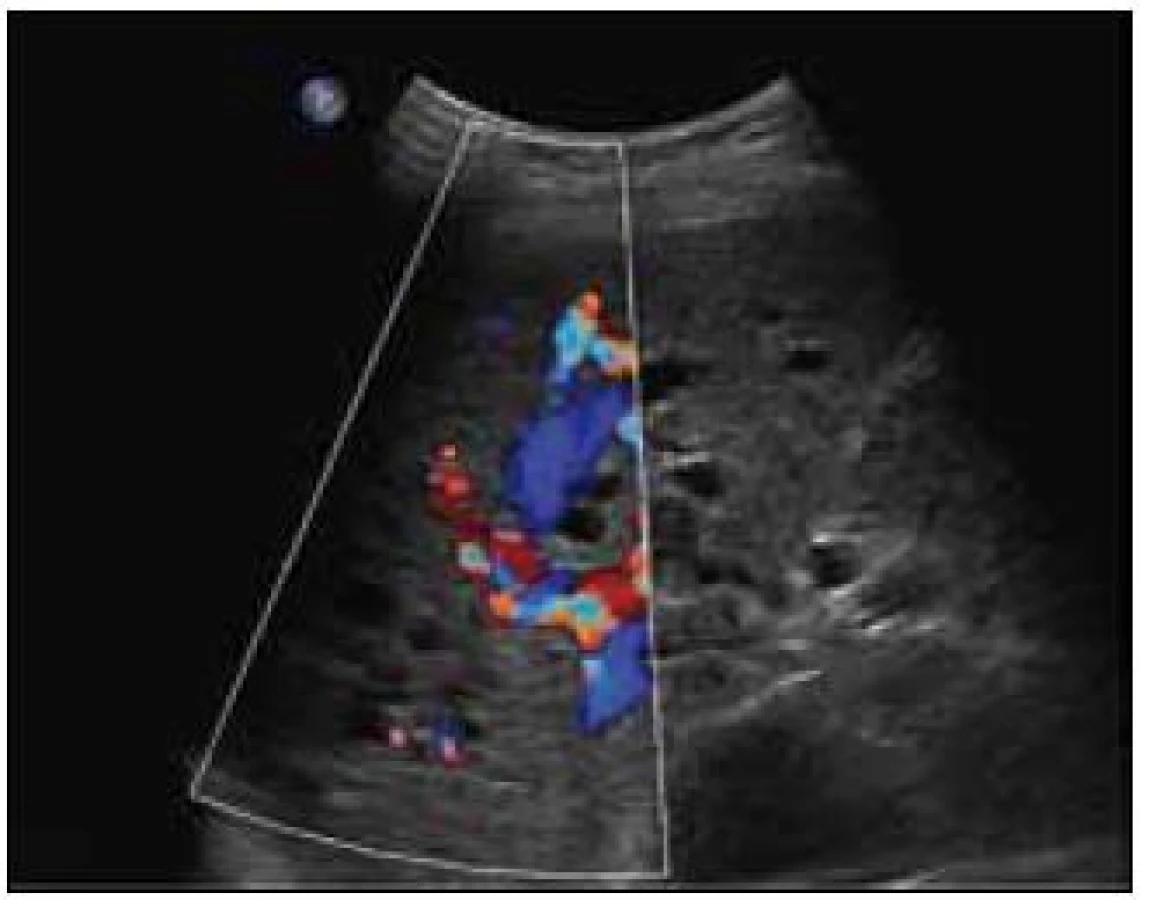
U pacientky bylo provedeno samozřejmě také MR mozku. Cituji nález: „Na vnitřní straně levého frontálního laloku je ovální útvar velikosti asi 12 mm, v centru hypersignální, na periferii hyposignální, bez expanzivního chování. V pravém temporálním laloku se v bílé hmotě sbíhají žilní struktury v obraze caput medusae se silnou odvodnou žilou do povrchového žilního systému. V zadní jámě v dolních částech obou mozečkových hemisfér jsou vidět nepravidelné hypersignální defekty velikosti asi 24 × 17 mm vlevo a asi 10 mm vpravo jako ischemická ložiska po intervenční angiografii. Jinak je mozková tkáň homogenní, bez známek infiltrace, vnitřní i vnější likvorové prostory jsou symetrické a nerozšířené. Závěr: kavernom se stopami po krvácení vlevo frontálně, venózní angiom vpravo temporálně stav po iatrogenních ischemizacích v bazálních partiích obou mozečkových hemisfér.“
Terapie: Protože v době hospitalizace jsme neměli schválenou úhradu zvažovaného thalidomidu, tak byl podán interferon α, který byl pro tuto indikaci schválen revizními lékaři. Pacientka však dostala pouze 3 injekce interferonu α v dávce 1,5 milionů jednotek 3krát týdně. V plánu bylo při dobré toleranci tuto dávku zvýšit na 3 miliony jednotek 3krát týdně. Jenže v průběhu prvních 3 aplikací interferonu α se dostavil velmi brzy dřeňový útlum se závažným poklesem počtu neutrofilů a trombocytů. Vyšetření kostní dřeně po ukončení podávání interferonu α popsalo výrazný deficit železa, sideroblasty byly sníženy na 4 % a zásobní železo nebylo přítomno. Při cytologickém hodnocení nebyly přítomny dysplastické rysy. Histologické hodnocení trepanobioptického válečku popsalo kostní dřeň s útlumem v granulopoéze a potvrdilo absenci barvitelného stromálního železa.
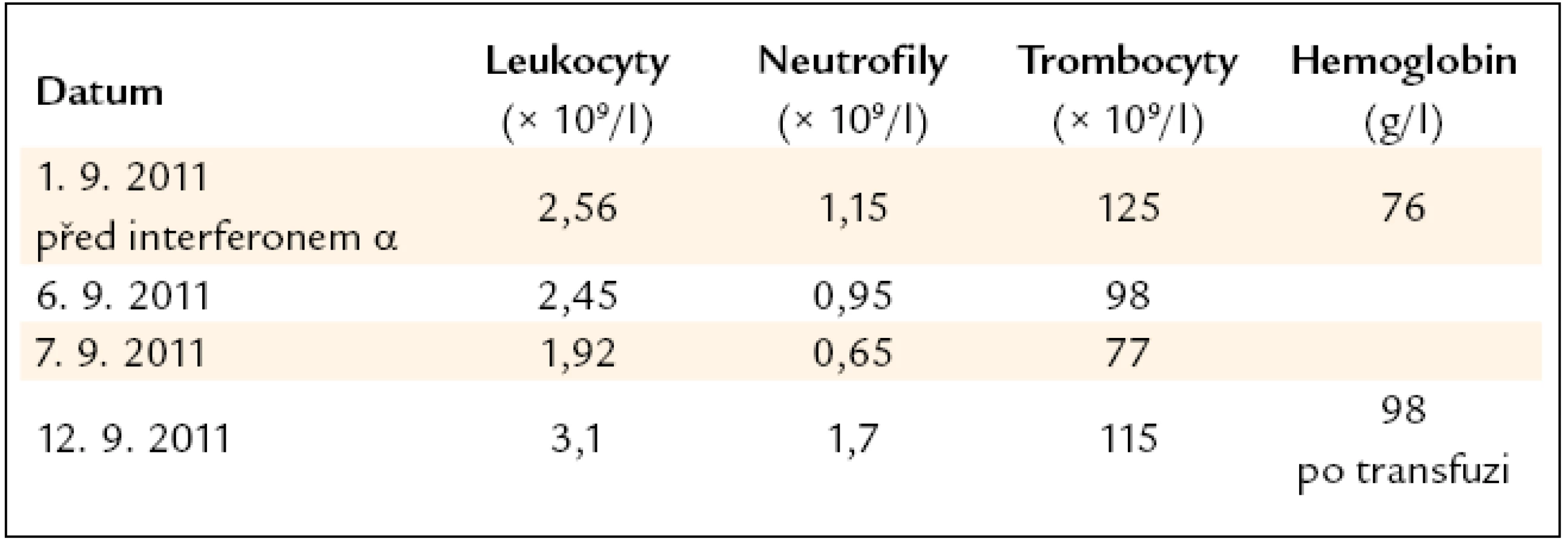
Léčba interferonem byla pro neutropenii okamžitě ukončena. Dále pokračovala pouze substituce železa. A protože i při této substituci pacientka anemizovala a anémie již měla klinické projevy, bylo nutné podání transfuzí erytrocytů. Vývoj počtu krvinek v 1. týdnu léčby interferonem α ukazuje tab. 2.
Po ukončení léčby interferonem α měla pacientka dále trvalou substituci železa a při klinických projevech anémie dostává ještě transfuze erytrocytů. Nyní po schválení preparátu thalidomidu (Myrin) revizními lékaři jsme zahájili pravidelné podávání thalidomidu v dávce 100 mg denně. Pacientka říká, že při léčbě thalidomidem (Myrinem) v dávce 100 mg užívané večer nepozoruje žádné nežádoucí účinky. Ihned v 1. měsíci léčby se zmenšila frekvence epistaxí. Dále pacientka udává, že pokud k epistaxi dojde, jsou krevní ztráty menší, než tomu bylo před léčbou.
Po měsíci léčby jsme však opět zjistili neutropenii s poklesem absolutního počtu neutrofilů až na 0,6 × 109/l. Z tohoto důvodu jsme dávku snížili na 100 mg ob den.
Diskuze
Charakteristika nemoci
Nemoc zvaná Osler-Weber-Rendu syndrom neboli též hereditární hemoragická teleangiektazie je vrozená choroba s autozomálně dominantní dědičností. Její prevalence se udává 1/10 000 obyvatel [1]. Pro ČR by to znamenalo 100 případů/10 milionů obyvatel. Jde však o celkovou prevalenci, tedy jak o mladé pacienty s počínajícím rozvojem nemoci – s občasnými epistaxemi, tak i pacienty s rozvinutou chorobou, ztrátami do zažívacího traktu a závažnými cévními arteriovenózními malformacemi. Dejme tomu, že těch závažným případů s rozvinutou chorobou bude jen 1/4, tedy 0,25/100 000, což odpovídá 25 na 10 000 000 obyvatel ČR. Takže vzhledem k vzácnosti této nemoci by tato diagnóza neměla být ekonomickou zátěží zdravotních pojišťoven.
V české a slovenské medicínské literatuře, citované v databázi MEDLINE--PUBMED, jsme našli pouze 6 publikací, které tuto nemoc zmiňují [2–7]. Vzácnost kliniky závažných forem této nemoci je zřejmě důvodem toho, proč lze v celosvětové zdravotnické literatuře registrované v databázi MEDLINE-PUBMED nalézt pouze 2 publikace charakteru doporučení pro léčbu (guidelines) [8,9]. Proto chceme následujícím popisem případu upozornit na tuto nemoc, předložit čtenáři přehled léčby a poukázat na nové léčebné možnosti thalidomidem či bevacizumabem.
Hereditární hemoragická teleangiektazie je charakterizována postupnou tvorbou nových teleangiektazií, které postihují kůži, slizniční povrchy, plíce, mozek, celou trávicí trubici (gastrointestinální trakt) a játra [8,9].
Teleangiektazie vznikají z dilatovaných postkapilárních venul, které se rozšiřují a anastomozují se sousedícími arteriolami, takže nakonec krevní tok neteče přes kapiláry, ale rovnou přes nově vytvořenou arteriovenózní komunikaci [1]. Tyto morfy mohou tvořit diskrétní izolované arteriovenózní malformace nebo se mohou slévat do difuzních arteriovenózních malformací či ploch s difuzními teleangiektaziemi.
Prvním příznakem nemoci obvykle bývá epistaxe. První ataky epistaxe přicházejí u postižených osob po 12. roce života a ve 40 letech již trpí epistaxí 100 % postižených. Interval od 1. epistaxe do výsevu viditelných teleangiektazií v obličeji, v ústech a na končetinách se pohybuje mezi 5–30 roky. Pacienti často udávají, že viditelných teleangiektazií si všimli obvykle až ve 3. dekádě života [10].
Pro stanovení diagnózy byla přijata v roce 2000 klinická kritéria uvedená v tab. 3 [8,11].
![Diagnostická kritéria hereditární hemoragické teleangiektazie [8,11].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/c0f0bfc34242db2e6d570ff3a24e151a.png)
Pokud jsou přítomny 3 či více příznaků, je diagnóza jednoznačná. Pokud jsou přítomna jen 2 kritéria, je diagnóza pravděpodobná. Nemoc lze však prokázat také vyšetřením přítomnosti či nepřítomnosti genetické informace pro tuto chorobu.
Arteriovenózní malformace postihují individuálně v různé míře všechny orgány. Plicní parenchym bývá postižen u 33 % pacientů, trávicí trubice (jícen a gastrointestinální trakt) u 44 % a játra u 17 % nemocných. Postižení mozku bývá diagnostikováno až u 15–23 % nemocných [12–19].
Etiopatogeneze hemoragické hereditární teleangiektazie
Hereditární hemoragická teleangiektazie (HHT) je způsobena mutací jednoho ze 2 genů, což se odráží v subklasifikaci na typ I a typ II. Druhý typ je častější. Oba geny kódují transmembránové proteiny, které jsou součástí signální cesty takzvaného „transforming growth factor (TGF)-β“ a které jsou exprimovány převážně na cévních endotelech [21].
U hereditární hemoragické teleangiektazie 1. typu byla prokázána mutace genu pro „endoglin“ na chromozomu 9q34 [10] a u 2. typu mutace genu pro „activin receptor-like kinase 1 (ALK-1)“ na chromozomu 12q13 [11].
Dále byl ojediněle popsán 3. typ u rodiny s hemoragickou hereditární teleangiektazií postihující plíce. U těchto pacientů byla prokázána mutace genu pro endoglin, ALK-1 a také Smad4. U této rodiny byly prokázány změny na chromozomu 5 [22].
Sadick et al [23] prokázali zvýšenou plazmatickou hladinu vaskulárního endoteliálního růstového faktoru (vascular endothelial growth factor – VEGF) a transforming growth factor-β – TGF-β – u pacientů s hemoragickou hereditární teleangiektazií. Dnes jsou již genetické změny u této nemoci natolik dobře prozkoumány, že jejich průkaz lze použít také jako genetické kritérium přítomnosti této nemoci [24–26].
Klinické příznaky v jednotlivých orgánech a jejich lokální léčba
Krvácející teleangiektazie v nosní sliznici
Nemoc obvykle začíná krvácením z nosu, které je způsobeno postižením nosní sliznice teleangiektaziemi. Krvácení, zpočátku nenápadné, se stále opakuje ve větší a větší intenzitě a v pokročilých stadiích nemoci může být tak intenzivní, že ohrozí život nemocného vykrvácením. Lokální léčba v ORL oblasti je zmíněna v odstavci věnovaném léčbě.
Cerebrovaskulární a spinální A-V malformace
V oblasti mozku a míchy bývají u pacientů s dlouhodobějším průběhem HHT detekovány arteriovenózní (A-V) cévní malformace až ve 23 %. Proto se doporučuje provádět u pacientů s klinickými příznaky této nemoci screeningové vyšetření, které může odhalit potenciálně léčitelné A-V malformace v CNS, které by při spontánním krvácení mohly poškodit svého nositele (způsobit krvácení či kompresi nervové tkáně, což způsobí bolest hlavy anebo další neurologické příznaky) [15]. Riziko krvácení z mozkových A-V malformací se u těchto pacientů uvádí 0,5 % za rok [8]. Standardním postupem pro diagnostiku je stále angiografie, stejně však lze použít MR angiografii s aplikací kontrastní látky (gadolinia).
Pokud se prokáže cévní malformace v mozku, která by mohla ohrozit svého nositele, pak je na zvážení lokální léčba, embolizace, chirurgický výkon, stereotaktická radioterapie nebo kombinace těchto postupů [27,28].
U naší pacientky byl zjištěn kavernom se stopami krvácení vlevo frontálně. Pacientka nyní zvažuje, zda přijme nabídku neurochirurgů na operační řešení.
Plicní A-V malformace
Plicní arteriovenózní malformace se objevují asi u 15–50 % pacientů s HHT [8,9]. Cévní malformace v plicním parenchymu mohou způsobit 2 typy komplikací. Tím, že cévní malformace umožní průtok krve přímo z arterioly do plicní žíly, bez nutnosti průchodu plicními kapilárami, tak se naruší filtrační efekt plicního kapilárního řečiště. Bakterie, ale i tromby, mohou projít A-V malformací v plicním řečišti do systémového oběhu, aniž by uvízly v plicní kapilární síti. Bakterie, tromby i infikované mikrotromby se tak mohou jako emboly dostat do systémového krevního oběhu (tedy i do mozkového oběhu) a způsobit závažné komplikace. K těm typickým patří mozkové abscesy či uzávěr mozkových tepen s odpovídajícími neurologickými příznaky. Z tohoto důvodu se doporučuje u těchto pacientů antibiotická profylaxe při všech výkonech, kdy by mohla vzniknout bakteriemie [29,30].
Další komplikací, kterou mohou plicní A-V malformace způsobit, je hemoptýza. V případě umístnění A-V malformace v blízkosti pleurální dutiny může vzniknout hemothorax [31].
Cévní malformace mohou také vést k plicní hypertenzi, kterou lze jen obtížně řešit [32,33].
Proto se má provést u pacientů s HHT vhodný dostupný screening s cílem detekovat plicní A-V malformace, a pokud jsou prokázány a pokud je průměr přívodné arterie do aneuryzmatu nejméně 3 mm, tak zvážit lokální (embolizační) léčbu [8,9].
Kardiální problémy způsobené A-V malformacemi
Arteriovenózní spojky zkracují, a tedy zrychlují krevní oběh. Hyperkinetický oběh může vést k srdečnímu selhání [34,35]. Khalid et al uvádí, že výrazné zintenzivňování epistaxí může ohlašovat progresi nemoci, která způsobí selhání hyperkinetického krevního oběhu [36].
Gastrointestinální krvácení
Asi 80 % pacientů s HHT má teleangiektazie v žaludku a v tenkém střevě. Jsou dobře viditelné při endoskopickém vyšetření či při vyšetření polknutou videokapslí. Teleangiektazie v žaludku, duodenu a v tenkém střevě jsou častější než teleangiektazie ve střevě tlustém. Symptomatické krvácení ze zažívacího traktu se však udává jen u 30 % pacientů, a to obvykle až po začátku 5. životní dekády. Před 40. rokem bývá krvácení do trávicího traktu vzácné, není však vyloučené [8–10]. Ztráty krve do trávicí trubice obvykle bývají malé, takže na prstu lékaře vyšetřujícího per rectum obvykle nezůstává stolice charakteru melény. Ztráty jsou však trvalé, a proto jsou příčinou hypochromní anémie.
Krvácení do trávicí trubice z teleangiektazií obvykle nezpůsobuje jiné symptomy než hypochromní anémii. Zpočátku je anémie řešitelná substitucí železem, ať již p.o., nebo i.v. cestou. Pokud ale ztráty krve přesáhnou kompenzační schopnosti kostní dřeně, tak je nutné řešit tuto ztrátovou anémii krevními transfuzemi. Výjimečné je masivní krvácení do trávicí trubice, které může být příčinou smrti [13–15,18].
Vzhledem k rozsahu postižení trávicí trubice nelze pro léčbu mnohočetných žaludečních a střevním teleangiektazií použít embolizační léčbu.
Jaterní cévní malformace HHT
Zajímavé je, že játra bývají nejčastěji postižena vaskulárními malformacemi, které mohou způsobovat portovenózní, arteriovenózní a arterioportální zkratový krevní oběh (shunting). Postižení jater se popisuje u 32–78 % pacientů s HHT [8–12].
Cévní malformace v játrech mohou způsobit následující příznaky:
- selhání srdce při hyperkinetickém oběhu neboli „hight output cardic failure“,
- portální hypertenzi,
- biliární nekrózu.
Nejčastější klinickou manifestací jaterních A-V malformací je kardiální selhání způsobené hyperkinetickým krevním oběhem. Klinickými důsledky je jako u každého kardiálního selhání: dušnost a retence tekutin.
Ascites, a případně krvácení z portokaválních varixů, může být důsledkem portální hypertenze způsobené arterioportálním zkratem. Naproti tomu arteriovenózní zkraty mohou způsobit ischemii biliárních cest s následnou nekrózou a sklerotizující cholangitidou [37–43].
Cévní malformace lze znázornit sonograficky, metodou CT zobrazení s aplikací kontrastní látky či selektivní angiografií.
Pro léčbu jaterních A-V malformací lze použít angiografie a selektivní embolizace. Tato léčebná metoda má však nepřehlédnutelné komplikace. Jako nejčastější z nich se popisuje ischemické poškození jater. Komplikace embolizační léčby jaterních A-V malformací vedly asi u 30 % léčených k úmrtí či k nutnosti akutní transplantace jater. Literatura uvádí 2leté přežití u pacientů po embolizační léčbě jaterních A-V malformací 73 %, zatímco po transplantaci jater uvádí 5leté přežití 83 %. Transplantace jater je tedy úspěšnější, a tudíž vhodnější než embolizační léčba [44–47].
Klinické projevy této nemoci jsme rozvedli v úvodu. Naše pacientka již má pokročilé projevy nemoci, generalizované teleangiektazie na povrchu těla. Teleangiektazie má také na jazyku a stejným způsobem bude pravděpodobně postižena celá trávicí trubice. Z toho rezultuje anémie. Ztráty jsou tak velké, že vyžadují podávání transfuzí. A samozřejmě A-V malformace komplikují průběh.
Otázka je, jak ji dále léčit. Proto v diskuzi shrnujeme fakta publikovaná o léčbě v odborné literatuře, včetně citovaných mezinárodních doporučení publikovaných v roce 2011.
Lokální léčba
Léčba epistaxe
Léčebné postupy se odvíjí od fáze nemoci. V první fázi nemoci, kdy jsou dominujícím problémem ztráty krve epistaxí, je léčba lokální. Je tedy pevně v rukou ORL specialistů, kteří mohou krvácení zastavovat klasickou mastnou tamponádou a následně elektrokauterizací krvácejících cévek. Další možností je ev. použití laseru, embolizace nazální arterie. Při refrakterních epistaxích lze aplikovat do nosních průchodů balónkovou tamponádu, výjimečně se provádí i ligatura a. carotis externa, a to nad odstupem a. thyroidea superior. Dalším operačním řešením je náhrada fragilní nazální mukózy bukální sliznicí (dermoplastika). O tom, jaká forma lokální léčby bude zvolena, rozhoduje ORL specialista dle toho, jaké prostředky má k dispozici [8,9,48–50]. Novinkou pro řešení epistaxí je použití tkáňového lepidla [51].
Léčba cévních A-V malformací
Pro lokální léčbu A-V malformací pacientů s HHT se používají embolizační techniky, lze použít ale také operační řešení či radioterapii, pokud je ložisko dobře radioterapeuticky ošetřitelné.
Endoskopické zákroky
U pacientů s HHT jsou často prováděny endoskopie s diagnostickým cílem, ale i s cílem léčebným – endoskopicky zastavit krvácení. Ale podobně jako u krvácení z nosu ani tyto endoskopické či operační výkony neřeší nic s definitivní platností.
Podávání léků s cílem snížit krevní ztráty
U pacientů s hemoragickou teleangiektazií se podávají následující léky s cílem snížit krevní ztráty.
Venofarmaka
Rutin
Bioflavonoid je důležitý pro normální funkci kapilár. Je obsažen v mnoha kompozitních preparátech, z nichž nejznámější je asi Ascorutin.
Kyselina askorbová
Vitamin C je rozpustný ve vodě. Je důležitý pro normální permeabilitu kapilár. Obě látky, rutin a vitamin C, účinně upravují zvýšenou fragilitu a permeabilitu kapilár. V případě krvácivých potíží lze podávat preparát Ascorutin v dávce až 3krát denně 2 tbl., toto lze i dlouhodobě. Mimo krvácení je podávána udržovací dávka 2–3krát denně 1 tbl.
Hemostatika
Etamsylát (Dicynone)
Je syntetická látka s antihemoragickým a angioprotektivním účinkem, zvyšuje adhezivitu trombocytů a zkracuje dobu krvácení. Nemá vazokonstrikční účinek. V případě krvácení podáváme 2 amp. nitrožilně po 4–6 hod, v případě perorálního podání 1 tobolku po 500 mg po 4–6 hod, případně v rámci udržovací léčby 1 tobolku 2–3krát denně.
Antifibrinolytika
Kyselina aminometylbenzoová (Pamba)
Antihemoragický účinek kyseliny aminometylbenzoové spočívá ve specifické blokádě plazminu na fibrin. Obvyklá dávka k zástavě krvácení je 1 tbl. po 6 hod, v případě parenterálního podání 50–100 mg nitrožilně, případně intramuskulárně.
Kyselina tranexamová (Exacyl)
Antihemoragický účinek kyseliny tranexamové spočívá v inhibici fibrinolytické aktivity plazminu. Indikací podání jsou menometroragie, krvácení z gastrointestinálního traktu, dolních cest močových, ORL a stomatologické oblasti. K inhibici fibrinolytické aktivity jsou dostatečné dávky 2 g po 8 hod.
U obou antifibrinolytik je nutné brát na zřetel jejich protrombogenní potenciál, tedy u pacientů s anamnézou žilního tromboembolizmu musí být jejich podání jasně indikované. Které antifibrinolytikum zvolit, závisí na názoru hematologa [8,9].
Substituce železa
Pokud lokální léčba epistaxe přestane plnit svůj cíl, tak epistaxe způsobí hypochromní anémii. Příčinou hypochromní anémie jsou však také ztráty střevním traktem. Prvním léčebným krokem je perorální či nitrožilní substituce železa. A pokud podávání preparátů železa nestačí zabránit anemizaci, jsou další léčebnou možností transfuze erytrocytů.
Estrogeny a antiestrogeny
V předchozích desetiletích lékaři marně hledali léky, které by zpomalily progresi této nemoci a snížily krevní ztráty. První účinnou léčbou, dle limitovaných zkušeností staršího data, byla aplikace kombinované hormonální léčby estrogen-progesteron (etinylestradiol 0,05 mg + noretisteron 1 mg). Ve srovnání s placebem snížila tato hormonální léčba počet potřebných transfuzí v prospektivní randomizované kontrolované studii [52,53]. Hormonální léčba deriváty estrogenu a progesteronu však může způsobit komplikace, zvláště pokud je podávána mužům. Proto je tato hormonální léčba dnes považována za vhodnou pro ženy, které jsou po menopauze a po hysterektomii.
Hypotéza, že stimulace estrogenních a progesteronových receptorů může zmenšit míru krevních ztrát, je však stále živá. V posledních letech byl v této indikaci testován raloxifen [54] a dále se objevily výsledky prospektivní randomizované klinické studie, v níž byl se stejným cílem použit antiestrogen tamoxifem. Výsledkem této prospektivní studie bylo snížení krevních ztrát ve skupině dostávající pravidelně tamoxifen [55–57]. Preparáty ze skupiny antiestrogenů mají svoji slabou vnitřní aktivitu (intrinsic activity), a proto u žen, které po menopauze užívají tamoxifen, dochází k hypertrofii endometria, ale také ke zpomalení progrese HHT [55,56].
Thalidomid
Thalidomid je lék, který byl uveden na trh v roce 1960 pod názvem Contergan s cílem potlačit nevolnost u těhotných. Výsledkem použití tohoto léku u těhotných byly malformace plodu, nevyvinutí končetin. Lék byl sice testován u gravidních myší, ale zřejmě díky jejich krátké březosti tento vliv nebyl odhalen. Teprve později se přišlo na to, že thalidomid snižuje rychlost angiogeneze. A tím, že se nevytvářely nové cévy, které by zásobovaly krví vyvíjející se končetiny, došlo poruchám vývoje končetin [58–60].
Dnes je již dostatečně prokázáno, že thalidomid stimuluje maturaci cév a brzdí novotvorbu cév nových. Thalidomid ovlivňuje expresi jednotlivých komponent signálních cest angiogeneze, včetně tvorby vaskulárního endoteliálního růstového faktoru – VEGF.
Thalidomid velmi intenzivně působí na endoteliální buňky nezralých cév, indukuje jejich maturaci a tím zmenšuje jejich křehkost [61].
Thalidomid má však i jiné účinky než pouze vliv na angiogenezi, moduluje imunitní odpověď, a proto je dnes řazen do skupiny immunomodulatory drugs – IMIDS. Díky komplexnímu působení na organizmus je thalidomid účinným lékem pro pacienty s mnohočetným myelomem, plazmacelulárním typem Castlemannovy choroby, ale také pro pacienty s histiocytózou z Langerhansových buněk a pro další vzácné nemoci [62,63]. Jedním z problémových účinků thalidomidu je prokoagulační efekt, který zodpovídá za tromboembolické komplikace [64].
Díky svému inhibičnímu vlivu na angiogenezi a díky indukci maturace již vytvořených cévních struktur se thalidomid osvědčil u léčby cévních malformací. Trávicí trakt může být postižen drobnými angiodysplaziemi, které způsobují krvácení. Tyto střevní angiodysplazie mohou být projevem HHT, ale mohou vzniknout i bez genetického pokladu HHT jako vícečetné střevní angiodysplazie. Střevní angiodysplazie mohou být součástí poměrně agresivně se chovající angiomatózy.
A u všech těchto nemocných s angiodysplaziemi různé morfologické klasifikace byl použit thalidomid s pozitivním výsledkem [65–73]. Obvyklá dávka používaná pro léčbu pacientů s angiodysplaziemi, hemangiomy a angiomatózou je stejná jako dávka u mnohočetného myelomu, tedy 100–200 mg denně, tedy 1–2 tbl. večer.
Při pohledu do literatury je zřetelné, že thalidomid se používá u všech typů angiodysplazií: u hemangioblastomu, který komprimoval míchu [74], u intrakraniálních hemangiomů a hemangioblastomů [75,76]. Byl s úspěchem použit u dětské formy hemangiomatózy [77]. Byl také použit ke kontrole poradiační zánětlivé reakce s cévní novotvorbou [78].
Thalidomid byl také použit s úspěchem u epiteloidních hemangioendoteliomů [79,80].
Na našem pracovišti máme zkušenosti s aplikací thalidomidu pacientům s mnohočetným myelomem, ale použili jsme jej také s úspěchem u jednoho mladého muže s angiomatózou postihující kosti, mediastinum a břišní dutinu a dále u jednoho pacienta s četnými angiodysplaziemi v trávicí trubici. Tento pacient po zavedení léčby thalidomidem přestal potřebovat transfuze. Zmenšení cévní malformace, její zpevnění a minimalizaci spontánního krvácení jsme pozorovali u pacienta s velkým hemangiomem v obličeji [81].
U pacientky jsme pozorovali již v 1. měsíci léčby thalidomidem (Myrin 100 mg denně) zmenšení frekvence epistaxí. A pokud k epistaxi došlo, tak pacientka říkala, že krvácení bylo méně intenzivní a ztráty krve menší. Během 1. měsíce léčby thalidomidem nepotřebovala transfuzi krve. V průběhu léčby však došlo k poklesu neutrofilů. Myelosuprese či pouhý pokles neutrofilů není častým nežádoucím účinkem thalidomidu. Vzhledem k faktu, že i malé dávky interferonu α indukovaly neutropenii a stejně tak i nyní nízká dávka thalidomidu vedla k neutropenii, předpokládáme alteraci kostní dřeně a sníženou toleranci myelosupresivně působících léků oproti průměrné populaci.
Lenalidomid
Lenalidomid je novější derivát thalidomidu. Na rozdíl od thalidomidu nezpůsobuje neurotoxicitu, ale zato způsobuje myelosupresi. Antiangiogenní účinek si lenalidomid ponechává, udává se však, že v menší intenzitě než thalidomid. Zatím jsou s použitím lenalidomidu v této indikaci jenom malé zkušenosti. Lze z nich usoudit, že lenalidomid určitý klinicky zjevný antiangiogenní efekt má [82], a je možné jeho indikaci v těchto případech otestovat.
Bevacizumab
Bevacizumab je monoklonální protilátka namířená proti vaskulárnímu endoteliálnímu růstového faktoru – VEGF. Tento lék je pod názvem Avastin používán u některých karcinomů. Tím, že v nádoru zablokuje novotvorbu cév, zastaví růst nádoru.
Z cévních novotvarů jsou to právě malformace typu HHT, u nichž byl popsán pozitivní účinek bevacizumabu u poměrně velkého počtu pacientů. Bevacizumab lze použít ve formě lokální aplikace pro léčbu epistaxe [83,84]. Rohrmayer et al používali pro lokální aplikaci velmi nízké dávky 7,5 mg a i při této dávce popsali zlepšení všech sledovaných parametrů ve srovnání s kontrolní skupinou [85].
Při nitrožilní aplikaci se obvykle používal bevacizumab v dávce 5 mg/kg v i.v. infuzi v intervalu 2–3 týdnů. Autoři, kteří popisují léčbu HHT bevacizumabem, podávali tento lék obvykle jen po několik měsíců, do dosažení klinické léčebné odpovědi, a pak jeho aplikaci přerušili. V případně recidivy potíží se k léčbě bevacizumabem vrátili [86–94]. Důležité je, že v popisech případů HHT léčených bevacizumabem nejsou uváděny žádné nežádoucí účinky této léčby.
Přínos bevacizumabu u pacientů s HHT se však neomezuje pouze na zmenšení krevních ztrát. V průběhu léčby bevacizumabem došlo ke zmenšení průtoku krve cévními malformacemi v játrech, a zmenšila se tak intenzita hyperkinetického oběhu, který vedl k srdečnímu selhání. Léčba bevacizumabem v některých popisovaných případech odstranila nutnost transplantace jater [95,96]. Samozřejmě je možná synergická kombinace bevacizumabu a thalidomidu [97]. A podobně jako thalidomid je bevacizumab účinný také u lymfangioendoteliomatózy [98].
Interferon α
Interferon α je často používán pro léčbu hemangiomů [99], a když zadáme do MEDLINE-PUBMED klíčová slova interferone a haemangiom, objevíme podstatně více citací, než když hledáme počet publikací zmiňujících účinek interferonu u HHT. V celosvětovém písemnictví jsou pouze 2 publikace, které popisují pozitivní účinek interferonu α na HHT u pacientů, u nichž byl interferon podán z jiného důvodu [100,101]. S interferonem α je tedy u HHT podstatně méně zkušeností než s hormonální léčbou, thalidomidem a bevacizumabem. V našem případě interferon α v počáteční dávce 3krát 1,5 milionů jednotek týdně způsobil již po 3 injekcích závažnou neutropenii a trombocytopenii, takže bylo nutné léčbu ukončit. Interferon α jsme již v minulosti podávali pacientům s agresivně rostoucími hemangiomy a angiomatózou a tito pacienti jej, alespoň v prvních měsících, tolerovali v dávce 3krát 3 miliony jednotek celkem dobře, bez myelosuprese. Jeden z nich toleroval kupodivu i dávku 3krát 6 milionů jednotek bez nežádoucích účinků. Špatnou toleranci interferonu α v tomto případě vysvětlujeme tím, že interferon byl podán v situaci, kdy kostní dřeň svoji zvýšenou aktivitou již dlouhodobě kompenzuje trvalé ztráty krve. Je možné, že v této situaci je kostní dřeň na myelosupresivní působení interferonu α senzitivnější než v případě pacientů bez trvalých krevních ztrát.
Základní diagnostická vyšetření a sledování nemocných
Koagulační vyšetření
Každý pacient musí mít kromě základního krevního obrazu (trombocyty) s diferenciálním rozpočtem i podrobně vyšetřené koagulační parametry. Především predisponující další krvácivá porucha by mohla výrazně zhoršit krvácivé projevy při této závažné diagnóze.
Kromě základního koagulačního vyšetření, čímž rozumíme protrombinový čas, aktivovaný parciální tromboplastinový čas, trombinový čas a vyšetření koncentrace fibrinogenu, je doporučeno vyšetření samotné fibrinolýzy. V našich podmínkách vyšetřujeme euglobulinovou lýzu a retrakci koagula. Alternativou je vyšetření tromboelastografem. Došetřování koagulační aktivity jednotlivých koagulačních faktorů má význam v případě, že je prodloužený protrombinový nebo aktivovaný parciální tromboplastinový čas. V případě krvácivých komplikací má význam vyšetřování primární hemostázy – PFA 100, agregace trombocytů, vyloučení m. von Willebrand (vWF:RCO, AgvWF, F VIII), případně i F XIII. V případě patologické fibrinolýzy i vyšetření plazminogenu a α2-antiplazminu.
Vyhledávání arteriovenózních malformací
Zatímco teleangiektazie na kůži lze dobře diagnostikovat pohledem a v GIT pomocí endoskopických metod, případně pomocí videokapslí, k detekci A-V malformací ostatních orgánů potřebujeme jejich detailní zhodnocení pomocí zobrazovacích metod.
Zlatým standardem pro detekci plicních A-V malformací je v dnešní době použití počítačové tomografie. Klasická angiografie není v dnešní době prováděna jako primární diagnostické vyšetření. Některé malformace lze detekovat i na RTG hrudníku, tato metoda však nemá v této indikaci dostatečnou senzitivitu a specificitu. Jako screeningovou metodu lze použít kontrastní echokardiografii [102].
Jako metodu pro detekci jaterních A-V malformací lze provést ultrasonografii, která má dostatečně vysokou senzitivitu k detekci cévních lézí. Blíže můžeme cévní malformace posoudit pomocí kontrastního CT. Z dalších metod lze provést selektivní angiografii jaterní tepny, tato však není pro svou invazivitu metodou první volby [26].
Screening mozkových A-V malformací je diskutabilní, v některých zemích je prováděn, v jiných odmítán. Jako vyšetřovací modalitu můžeme zvolit CT mozkovou angiografii, lépe však kontrastní MR mozku s možným provedením MR angiografie, stejně tak lze provést i klasickou angiografii mozkových tepen [103].
Doporučení pro sledování
Po zjištění diagnózy je nutná dispenzarizace. Názory, kdy a co vyšetřit, se mohou lišit. Nicméně je třeba mít alespoň nějakou představu o tom, co se doporučuje v jiných zemích s vyspělým zdravotnictvím. Proto zde uvedeme doporučení Kanadské asociace lékařů pro pacienty s HHT. Dle kanadského doporučení by měla postižená osoba podstoupit alespoň:
- prohlídku u ošetřujícího lékaře 1krát ročně; cílem prohlídky je zdokumentovat rozvoj mukokutánních teleangiektazií, intenzitu epistaxe a krvácení do trávicího traku, příznaky plicního postižení (dušnost, snížení fyzické zdatnosti, hemoptýza) a přítomnost neurologických příznaků;
- laboratorní vyšetření minimálně 1krát ročně, jinak dle intenzity epistaxe s cílem včas diagnostikovat nastupující anémii (krevní obraz, Fe, Ferritin, saturace transferinu), a také průkaz okultního krvácení;
- měření saturace arteriální krve kyslíkem jednou za rok; cílem tohoto vyšetření je včas detekovat plicní A-V malformace se doporučuje; při poklesu pod 97 % provést echokardiografické vyšetření, CT hrudníku, případně kontrastní echokardiografické vyšetření;
- angiografii v případě větších plicních A-V malformací; při průměru přívodné artérie nad 3 mm se doporučuje pokusit se o účinnu embolizaci;
- opakované CT plic v 3–5letých intervalech v případně již dříve prokázáné plicní arteriovenózní malformace; opakováné CT plic má cíl však detekovat velkou A-V malformaci, která již vyžaduje embolizační léčbu;
- vyšetření jater pomocí CT s cílem je detekovat jaterní A-V malformace nemá přesně určené intervaly, protože léčba jaterních A-V malformací je obtížná a často neúspěšná;
- zobrazení mozku metodou MR s cílem detekovat mozkové A-V malformace se doporučuje při stanovení diagnózy a pak při jakýchkoliv neurologických příznacích [104].
Obecný problém špatné dostupnosti léků pro raritní choroby
V době, kdy jsem po ukončení studia nastoupil jako lékař na interní oddělení, jsme měli k dispozici určité množství léků, které jsme používali dle našeho nejlepšího vědomí a svědomí. Použití léku jsme zvažovali pouze na základě znalosti nemoci a znalosti léku a jeho vlastností a vše kontrolovalo bdělé oko našeho primáře. V té době nebylo použití léku omezováno dalšími nařízeními, která by ho limitovala jen pro určitou chorobu či stadium choroby.
V dnešní době je použití léků obestavěno kvantem administrativních předpisů. V současnosti, když nějaká farmaceutická firma vyvine lék s dobrou účinností, musí dát do chodu a zaplatit mašinerii registračních studií. V pozitivním případě nadnárodní schvalující instituce vydá potvrzení, že lék je vhodný pro léčbu té nemoci, která byla předmětem registračních studií. Pak firmy podstupují další jednání se zdravotními pojišťovnami, zda v té či oné zemi schválí úhradu léku pro konkrétní nemoc. Nemám vůbec představu o tom, kolik tyto kroky, které následují po ověření účinnosti léku v klinických zkouškách I. a II. fáze, stojí. Jisté je, že to jsou zřejmě velké peníze. A tyto velké peníze jsou důvodem, proč farmaceutická firma musí zvážit, zda její úsilí nebude ztrátové. A jedním z předpokladů, aby toto úsilí nebylo ztrátové, je, že daná choroba se vyskytuje v populaci přiměřeně často, aby spotřeba léku po omezenou dobu, než vyprší ochranná licence léku, navrátila zpět peníze, které stál vývoj léku.
A proto dnes žádná farmaceutická firma zřejmě nebude dělat registrační studie svého léku pro léčbu vzácných chorob, které se vyskytují s frekvencí nižší než třeba 0,5/100 000 obyvatel. A tak se naši spoluobčané, které postihne některá z vzácně se vyskytujících chorob, dostávají do svízelné situace, protože pro jejich nemoc nejsou registrovány žádné nové léky. Přitom z charakteru choroby a známého účinku některých léků by bylo možné usoudit, že ten či onen lék může u konkrétní vzácné nemoci pomoci. A toto rozhodnutí lze podpořit publikovanými zkušenostmi ze světové medicínské literatury, které sdělují, že při určité vzácné nemoci pomohl ten či onen lék. V současnosti lze pacienty se vzácnými chorobami léčit novými léky jedině v případech, kdy revizní lékaři zdravotních pojišťoven k tomu dají souhlas. A tak záleží jak na ošetřujícím lékaři, zda si dá práci a pokusí se podrobným rozborem případu, jako je tento, shrnout vše podstatné, a vstřícnosti revizního lékaře či přímo ředitele zdravotní pojišťovny.
Závěr pro praxi
- U pacientů s epistaxí je vhodné při prvním vyšetření zjistit, zda problémy s epistaxí mají rodinný výskyt, a pokud ano, pak vyšetřit rodinu s cílem potvrzení či vyloučení hereditární hemoragické teleangiektazie. V počátečních fázích se nemoc projevuje pouze epistaxí a v té době dominuje lokální léčba ORL specialisty.
- HHT je spojena s rozvojem A-V cévních malformací, které ohrožují život svého nositele. Proto je nutné po nich cíleně pátrat a při jejich průkazu zvážit další kroky. Pokud screeningové vyšetření A-V malformací neprovádí ošetřující ORL specialista, měl by pacienta předat na další pracoviště, kdy by se mu věnoval lékař znalý této nemoci, obvykle na oddělení klinické hematologie, proto druhým příznakem po epistaxi je anémie.
- Z medikamentů je u této nemoci používána substituce železem, antifibrinolytika, u vybraných pacientek lze zpomalit průběh nemoci estrogeny, případně antiestrogeny. Krevní ztráty krvácením z trávicího traktu, ale nejen z něho, lze zmenšit dlouhodobým podáváním thalidomidu, případně opakovaným podáváním bevacizumabu.
- U pacientů s HHT se nepoužívají glukokortikoidy, které se podávají u dětských hemangiomů. Důvod je jasný, anemičtí pacienti s HHT obvykle mají teleangiektazie v žaludku a proulcerózní efekt glukokortikoidů by zvyšoval riziko krvácení ze žaludečních teleangiektazií. A překvapivě málo je informací o přínosu interferonu u této nemoci, ačkoli u jiných typů cévních proliferací je interferon α doporučován.
Poděkování
Děkujeme řediteli Všeobecné zdravotní pojišťovny MUDr. Pavlu Horákovi, CSc., MBA, za vstřícný přístup k pacientům s řídce se vyskytujícími chorobami a podporu jejich léčby, v tomto případně za schválení léčby preparátem thalidomidu.
Práce byla vypracována v rámci aktivity následujících grantů: výzkumného záměru MZ ČR: FUNDIN MZ0MOU2005, výzkumného záměru MŠMT MSM0021622434, specifického výzkumu MUNI/A/0784/2011 a grantů IGA MZd NT11154, NT12130 a NT12215.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 16. 11. 2011
Přijato po recenzi: 25. 1. 2012
Zdroje
1. Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333: 918–924.
2. Ernest J, Krampolová Poláčková V, Charvát F. Oční komplikace po embolizaci v povodí arteria carotis interna – kazuistika. Cesk Slov Oftalmol 2008; 64: 202–206.
3. Takác M, Koval S, Klimcík J. Hereditární hemoragická telangiektasie a vznik artriovenózní fistuly po 24 letech. Vnitř Lék 1997; 43: 599–601.
4. Krahulec B, Jergus P, Bátovský M et al. Telangiektázie v žaludku u pacienta s Oslerovou chorobou. Bratisl Lek Listy 1988; 89: 11–13.
5. Štastný B, Kroslák M. Chirurgická léčba epistaxe u pacienta s morbus Osler-Rendu-Weber. Česk Otolaryngol 1981; 30: 53–56.
6. Izák M. Sturge-Weber-Krabbe syndrom. Česk Oftalmol 1971; 27: 267–272.
7. Chvojka J. Zástava epistaxe u pacienta s morbus Osler-Rendu-Weber. Česk Otolaryngol 1967; 16: 211–214.
8. Faughnan ME, Palda VA, Garcia-Tsao G et al. HHT Foundation International – Guidelines Working Group. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet 2011; 48: 73–87.
9. Trerotola SO, Pyeritz RE, White RI Jr et al. HHT Foundation. HHT Centers. 2009 Treatment guidelines for hereditary hemorrhagic telangiectasia. J Vasc Interv Radiol 2010; 21: 179.
10. Plauchu H, de Chadarévian JP, Bideau A et al. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32: 291–297.
11. Shovlin CL, Guttmacher AE, Buscarini E et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91: 66–67.
12. Haitjema T, Westermann CJJ, Overtoom TT et al. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu-disease): new insights in pathogenesis, complications, and treatment. Arch Intern Med 1996; 156: 714–719.
13. Reilly PJ, Nostrant TT. Clinical manifestations of hereditary hemorrhagic telangiectasia. Am J Gastroenterol 1984; 79: 363–367.
14. McDonald JE, Miller FJ, Hallam SE et al. Clinical manifestations in a large hereditary hemorrhagic telangiectasia (HHT) type 2 kindred. Am J Med Genet 2000; 93: 320–327.
15. Olitsky SE. Hereditary hemorrhagic telangiectasia: diagnosis and management. Am Fam Physician 2010; 82: 785–790.
16. Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev 2010; 24: 203–219.
17. Zarrabeitia R, Albiñana V, Salcedo M et al. A review on clinical management and pharmacological therapy on hereditary haemorrhagic telangiectasia (HHT). Curr Vasc Pharmacol 2010; 8: 473–481.
18. McDonald J, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia. Haematologica 2005; 90: 728–732.
19. Folz BJ, Zoll B, Alfke H et al. Manifestations of hereditary hemorrhagic telangiectasia in children and adolescents. Eur Arch Otorhinolaryngol 2006; 263: 53–61.
20. McAllister KA, Grogg KM, Johnson DW et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994; 8: 345–351.
21. Johnson DW, Berg JN, Baldwin MA et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996; 13: 189–195.
22. Cole SG, Begbie ME, Wallace GM et al. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet 2005; 42: 577–582.
23. Sadick H, Riedel F, Naim R et al. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica 2005; 90: 818–828.
24. Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol 2010; 20: 556–567.
25. Castonguay R, Werner ED, Matthews RG et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem 2011; 286: 30034–30046.
26. Dupuis-Girod S, Bailly S, Plauchu H. Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. J Thromb Haemost 2010; 8: 1447–1456.
27. Matsubara S, Mandzia JL, ter Brugge K et al. Angiographic and clinical characteristics of patients with cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia. AJNR Am J Neuroradiol 2000; 21: 1016–1020.
28. Drouet A, Le Moigne F, Donat A et al. Brain abscess as the first clinical manifestation of isolated pulmonary arteriovenous malformation without Rendu-Osler disease. Rev Neurol (Paris) 2011; 167: 29–34.
29. Shovlin C, Bamford K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205: 531–533.
30. Shovlin CL, Letarte M. Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54: 714–729.
31. Berg AM, Amirbekian S, Mojibian H et al. Hemothorax due to rupture of pulmonary arteriovenous malformation: an interventional emergency. Chest 2010; 137: 705–707.
32. Reichenberger F, Wehner LE, Breithecker A et al. Pulmonary hypertension in hereditary haemorrhagic teleangiectasia. Pneumologie 2009; 63: 669–674.
33. Chang SA, Jang SY, Ki CS et al. Successful bosentan therapy for pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Heart Vessels 2011; 26: 231–234.
34. Ruygrok M, Combs B, Campbell J et al. Heart failure, aneurysms and telangiectases, oh my! Am J Med 2011; 124: 605–607.
35. Koscielny A, Willinek WA, Hirner A et al. Treatment of high output cardiac failure by flow-adapted hepatic artery banding (FHAB) in patients with hereditary hemorrhagic telangiectasia. J Gastrointest Surg 2008; 12: 872–876.
36. Khalid SK, Pershbacher J, Makan M et al. Worsening of nose bleeding heralds high cardiac output state in hereditary hemorrhagic telangiectasia. Am J Med 2009; 122: 779. e1–e9.
37. Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol 2007; 46: 499–507.
38. Garcia-Tsao G, Korzenik JR, Young L et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343: 931–936.
39. Al-Saleh S, John PR, Letarte M et al. Symptomatic liver involvement in neonatal hereditary hemorrhagic telangiectasia. Pediatrics 2011; 127: e1615–e1620.
40. Buscarini E, Leandro G, Conte D et al. Natural history and outcome of hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic teleangiectasia. Dig Dis Sci 2011; 56: 2166–2178.
41. Henrion J, Deltenre P, Peny MO et al. Fatal hypoxic hepatitis in a patient with hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber’s disease). Acta Gastroenterol Belg 2010; 73: 61–64.
42. Khalid SK, Garcia-Tsao G. Hepatic vascular malformations in hereditary hemorrhagic telangiectasia. Semin Liver Dis 2008; 28: 247–258.
43. Sabbà C, Pompili M. Review article: the hepatic manifestations of hereditary haemorrhagic telangiectasia. Aliment Pharmacol Ther 2008; 28: 523–533.
44. Scelzo C, Greco S, Bonanni L et al. The role of liver transplantation in the treatment of hereditary hemorrhagic telangiectasia: a short literature review. Transplant Proc 2007; 39: 2045–2047.
45. Song X, Chen HQ, Chen YX et al. Individualized management of hepatic diseases in hereditary hemorrhagic telangiectasia. Am Surg 2011; 77: 281–285.
46. Lee M, Sze DY, Bonham CA et al. Hepatic arteriovenous malformations from hereditary hemorrhagic telangiectasia: treatment with liver transplantation. Dig Dis Sci 2010; 55: 3059–3062.
47. Lerut J, Orlando G, Adam R et al. Liver transplantation for hereditary hemorrhagic telangiectasia: report of the European liver transplant registry. Ann Surg 2006; 244: 854–864.
48. Chowdhry SA, Ponsky DC, Hsu DP. Treatment of a nasal vascular malformation in a patient with Osler-Weber-Rendu syndrome via percutaneous N-butyl 2-cyanoacrylate embolization: case report and review of the literature. J Otolaryngol Head Neck Surg 2011; 40: E11–E14.
49. Harvey RJ, Kanagalingam J, Lund VJ. The impact of septodermoplasty and potassium-titanyl-phosphate (KTP) laser therapy in the treatment of hereditary hemorrhagic telangiectasia-related epistaxis. Am J Rhinol 2008; 22: 182–187.
50. Lund VJ, Howard DJ. Closure of the nasal cavities in the treatment of refractory hereditary haemorrhagic telangiectasia. J Laryngol Otol 1997; 111: 30–33.
51. Richmon JD, Tian Y, Husseman J et al. Use of a sprayed fibrin hemostatic sealant after laser therapy for hereditary hemorrhagic telangiectasia epistaxis. Am J Rhinol 2007; 21: 187–191.
52. Flessa HC, Glueck HI. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease): management of epistaxis in nine patients using systemic hormone therapy. Arch Otolaryngol 1977; 103: 148–151.
53. van Cutsem E, Rutgeerts P, Vantrappen G. Treatment of bleeding gastrointestinal vascular malformations with oestrogen-progesterone. Lancet 1990; 335: 953–955.
54. Albiñana V, Bernabeu-Herrero ME, Zarrabeitia R et al. Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): Effects of raloxifene, on Endoglin and ALK1 expression in endothelial cells. Thromb Haemost 2010; 103(3): 525–534.
55. Yaniv E, Preis M, Hadar T et al. Antiestrogen therapy for hereditary hemorrhagic telangiectasia: a double-blind placebo-controlled clinical trial. Laryngoscope 2009; 119: 284–288.
56. Yaniv E, Preis M, Shevro J et al. Anti-estrogen therapy for hereditary hemorrhagic telangiectasia – a long-term clinical trial. Rhinology 2011; 49: 214–216.
57. Zheng JW, Zhou Q, Yang XJ et al. Anti-estrogenic agents might be more favorable option for treatment of hereditary hemorrhagic telangiectasia. Med Hypotheses 2009; 72: 230–231.
58. Melchert M, List A. The thalidomide saga. Int J Biochem Cell Biol 2007; 39: 1489–1499.
59. D’Amato RJ, Loughnan MS, Flynn E et al. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci 1994; 91: 4082–4085.
60. Therapontos C, Erskine L, Gardner ER et al. Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation. Proc Natl Acad Sci 2009; 106: 8573–8578.
61. Lebrin F, Srun S, Raymond K et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med 2010; 16: 420–428.
62. Nau JY. Thalidomide: new therapeutic indications? Rev Med Suisse 2010; 6: 846–847.
63. Akhurst RJ. Taking thalidomide out of rehab. Nat Med 2010; 16: 370–372.
64. Penaloza A, Vekemans MC, Lambert C et al. Deep vein thrombosis induced by thalidomide to control epistaxis secondary to hereditary haemorrhagic telangiectasia. Blood Coagul Fibrinolysis 2011; 22: 616–618.
65. Alberto SF, Felix J, de Deus J. Thalidomide for the treatment of severe intestinal bleeding. Endoscopy 2008; 40: 788–789.
66. Bauditz J, Lochs H. Angiogenesis and vascular malformations: antiangiogenic drugs for treatment of gastrointestinal bleeding. World J Gastroenterol 2007; 13: 5979–5984.
67. Bauditz J, Schachschal G, Wedel S et al. Thalidomide for treatment of severe intestinal bleeding. Gut 2004; 53: 609–612.
68. Bauditz J. Angiogenesis inhibitors for treatment of angiodysplasia-related gastrointestinal bleeding. Dtsch Med Wochenschr 2009; 134: 1893–1896.
69. Dabak V, Kuriakose P, Kamboj G et al. A pilot study of thalidomide in recurrent GI bleeding due to angiodysplasias. Dig Dis Sci 2008; 53: 1632–1635.
70. Chen CH, Hsu HH, Hu RH et al. Long term therapy with thalidomide in hereditary hemorrhagic telangiectasia: case report and literature review. J Clin Pharmacol 2011. In press.
71. Kamalaporn P, Saravanan R, Cirocco M et al. Thalidomide for the treatment of chronic gastrointestinal bleeding from angiodysplasias: a case series. Eur J Gastroenterol Hepatol 2009; 21: 1347–1350.
72. Kurstin R. Using thalidomide in a patient with epithelioid leiomyosarcoma and Osler-Weber-Rendu disease. Oncology 2002; 16: 21–24.
73. Pérez-Encinas M, Rabuñal Martínez MJ, Bello López JL. Is thalidomide effective for the treatment of gastrointestinal bleeding in hereditary hemorrhagic telangiectasia? Haematologica 2002; 87: ELT34.
74. Sardi I, Sanzo M, Giordano F et al. Monotherapy with thalidomide for treatment of spinal cord hemangioblastomas in a patient with von Hippel-Lindau disease. Pediatr Blood Cancer 2009; 53: 464–467.
75. Frei-Jones M, McKinstry RC, Perry A et al. Use of thalidomide to diminish growth velocity in a life-threatening congenital intracranial hemangioma. J Neurosurg Pediatr 2008; 2: 125–129.
76. Piribauer M, Czech T, Dieckmann K et al. Stabilization of a progressive hemangioblastoma under treatment with thalidomide. J Neurooncol 2004; 66: 295–299.
77. Jarvi K, Roebuck DJ, Sebire NJ et al. Successful treatment of extensive infantile hemangiomatosis of the small bowel in a 3-month-old with thalidomide and somatostatin analog. J Pediatr Gastroenterol Nutr 2008; 46: 593–597.
78. Tan DS, Evanson J, Plowman PN et al. Post-radiation inflammatory reaction controlled with thalidomide and rofecoxib. Clin Oncol (R Coll Radiol) 2004; 16: 585–586.
79. Bölke E, Gripp S, Peiper M et al. Multifocal epithelioid hemangioendothelioma: case report of a clinical chamaeleon. Eur J Med Res 2006; 11: 462–466.
80. Mascarenhas RC, Sanghvi AN, Friedlander L et al. Thalidomide inhibits the growth and progression of hepatic epithelioid hemangioendothelioma. Oncology 2004; 67: 471–475.
81. Adam Z, Pour L, Krejčí M et al. Úspěšná léčba angiomatózy thalidomidem a interferonem α. Popis 5 případů a přehled léčby angiomatózy a proliferujících hemangiomů. Vnitř Lék 2010; 56: 810–823.
82. Bowcock SJ, Patrick HE. Lenalidomide to control gastrointestinal bleeding in hereditary haemorrhagic telangiectasia: potential implications for angiodysplasias? Br J Haematol 2009; 146: 220–222.
83. Chen S 4th, Karnezis T, Davidson TM. Safety of intranasal Bevacizumab (Avastin) treatment in patients with hereditary hemorrhagic telangiectasia-associated epistaxis. Laryngoscope 2011; 121: 644–646.
84. Karnezis TT, Davidson TM. Efficacy of intranasal Bevacizumab (Avastin) treatment in patients with hereditary hemorrhagic telangiectasia--associated epistaxis. Laryngoscope 2011; 121: 636–638.
85. Rohrmeier C, Sachs HG, Kuehnel TS. A retrospective analysis of low dose, intranasal injected bevacizumab (Avastin) in hereditary haemorrhagic telangiectasia. Eur Arch Otorhinolaryngol 2012; 269: 531–536.
86. Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 2009; 360: 2143–2144.
87. Brinkerhoff BT, Poetker DM, Choong NW. Long-term therapy with bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 2011; 364: 688–689.
88. Cruikshank RP, Chern BW. Bevacizumab and hereditary haemorrhagic telangiectasia. Med J Aust 2011; 194: 324–325.
89. Davidson TM, Olitsky SE, Wei JL. Hereditary hemorrhagic telangiectasia/avastin. Laryngoscope 2010; 120: 432–435.
90. Flieger D, Hainke S, Fischbach W. Dramatic improvement in hereditary hemorrhagic telangiectasia after treatment with the vascular endothelial growth factor (VEGF) antagonist bevacizumab. Ann Hematol 2006; 85: 631–632.
91. Fodstad P, Dheyauldeen S, Rinde M et al. Anti-VEGF with 3-week intervals is effective on anemia in a patient with severe hereditary hemorrhagic telangiectasia. Ann Hematol 2011; 90: 611–612.
92. Oosting S, Nagengast W, de Vries E. More on bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med 2009; 361: 931–932.
93. Simonds J, Miller F, Mandel J et al. The effect of bevacizumab (Avastin) treatment on epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 2009; 119: 988–992.
94. Suppressa P, Liso A, Sabbà C. Low dose intravenous bevacizumab for the treatment of anaemia in hereditary haemorrhagic telangiectasia. Br J Haematol 2011; 152: 365.
95. Mitchell A, Adams LA, MacQuillan G et al. Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transpl 2008; 14: 210–213.
96. Buscarini E, Manfredi G, Zambelli A. Bevacizumab to treat complicated liver vascular malformations in hereditary hemorrhagic telangiectasia: a word of caution. Liver Transpl 2008; 14: 1685–1686.
97. Amanzada A, Töppler GJ, Cameron S et al. A case report of a patient with hereditary hemorrhagic telangiectasia treated successively with thalidomide and bevacizumab. Case Rep Oncol 2010; 3: 463–470.
98. Kline RM, Buck LM. Bevacizumab treatment in multifocal lymphangioendotheliomatosis with thrombocytopenia. Pediatr Blood Cancer 2009; 52: 534–536.
99. Lindner DJ. Interferons as antiangiogenic agents. Curr Oncol Rep 2002; 4: 510–514.
100. Wheatley-Price P, Shovlin C, Chao D. Interferon for metastatic renal cell cancer causing regression of hereditary hemorrhagic telangiectasia. J Clin Gastroenterol 2005; 39: 344–345.
101. Massoud OI, Youssef WI, Mullen KD. Resolution of hereditary hemorrhagic telangiectasia and anemia with prolonged alpha-interferon therapy for chronic hepatitis C. J Clin Gastroenterol 2004; 38: 377–379.
102. Faughnan ME, Granton JT, Young LH. The pulmonary vascular complications of hereditary haemorrhagic telangiectasia. Eur Respir J 2009; 33: 1186–1194.
103. Jaskolka J, Wu L, Chan RP et al. Imaging of hereditary hemorrhagic telangiectasia. AJR Am J Roentgenol 2004; 183: 307–314.
104. Grand’Maison A. Hereditary hemorrhagic telangiectasia. CMAJ 2009; 180: 833–835.
Internetové adresy s informacemi o HHT pro lékaře i pacienty
Stránky HHT Foundation international: www.hht.org,
Stránky Toronto HHT Centrum: www.hhttoronto.com
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2012 Číslo 6
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Význam hydratace při hojení ran
Najčítanejšie v tomto čísle
- Teleangiectasia hereditaria haemorrhagica – syndrom Osler-Weber-Rendu. Popis případu a zkušeností s léčbou
- Poruchy hemostázy u sepse
- Kladribin je velmi účinným lékem pro léčbu histiocytózy z Langerhansových buněk a vzácných histiocytárních nemocí ze skupiny juvenilního xantogranulomu
- Vliv kontinuální a intermitentní náhrady renálních funkcí na antibiotickou léčbu u kriticky nemocných v sepsi – praktický pohled na léčbu vankomycinem a gentamicinem