
Genetika kardiomyopatií
Genetics of cardiomyopathies
Cardiomyopathies are a heterogeneous group of diseases with structural and/ or functional myocardial impairment in the absence of ischemic or hemodynamic conditions capable of causing such impairment. Some cardiomyopathies are familial diseases with genetic and phenotypic variability. The heritability of cardiomyopathies became an integral part of new classification schemes of myocardial diseases. Genetic testing is especially beneficial in selected groups of patients and in the management of relatives. New methods of genetic testing will provide diagnosis for more patients and new data regarding genetic variants in patients with cardiomyopathies.
Keywords:
cardiomyopathies – hypertrophic cardiomyopathy – mutation – phenotype – genetic testing
Authors:
P. Tomašov
Authors‘ workplace:
Kardiologická klinika 2. LF UK a FN v Motole, Praha
Published in:
Kardiol Rev Int Med 2015, 17(1): 15-19
Category:
Cardiology Review
Overview
Kardiomyopatie jsou heterogenní skupinou onemocnění se strukturálním anebo funkčním postižením srdečního svalu při absenci ischemických anebo hemodynamických podmínek schopných způsobit toto postižení. Část kardiomyopatií má familiární výskyt s velkou genotypovou i fenotypovou variabilitou. Dědičnost kardiomyopatií je zmíněna i v nových klasifikačních schématech onemocnění myokardu. Genetické testování má význam zejména u selektovaných skupin pacientů a pro diagnostiku příbuzných. Nové metody genetického vyšetření zpřístupní diagnostiku více pacientům a přinesou nová data o genetických variantách u nemocných s kardiomyopatiemi.
Klíčová slova:
kardiomyopatie – hypertrofická kardiomyopatie – mutace – genotyp – genetické testování
Úvod
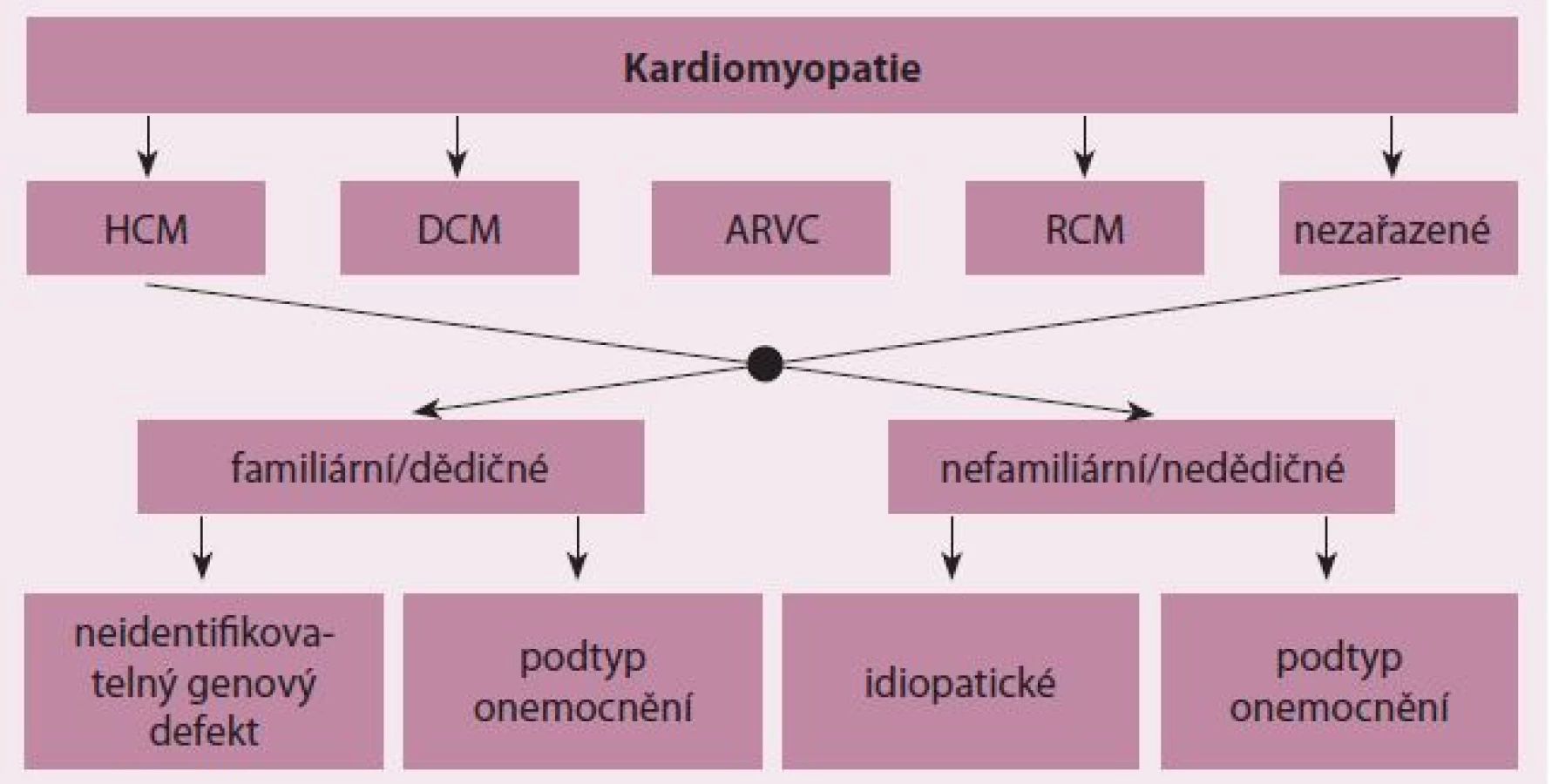
Kardiomyopatie jsou skupinou onemocnění srdečního svalu s poruchou struktury a/ nebo funkce myokardu různé etiologie [1]. Mezi kardiomyopatie nepatří stavy s dysfunkcí myokardu v důsledku hemodynamických efektů vrozených a chlopenních vad a arteriální hypertenze a postižení koronárních tepen při ischemické chorobě srdeční [1]. Významná část kardiomyopatií jsou monogenně podmíněná onemocnění s velkou genetickou variabilitou. Mutace způsobující jednotlivé formy kardiomyopatií byly nalezeny v mnoha genech pro důležité proteiny kardiomyocytů, přičemž seznam těchto genů se pro jednotlivé formy kardiomyopatií částečně překrývá [2]. Zejména geny pro sarkomerické proteiny mohou být postiženy u více typů onemocnění. Familiární formy kardiomyopatií jsou zahrnuty i v aktuálních klasifikačních schématech těchto onemocnění, včetně klasifikace Evropské kardiologické společnosti (ESC), která dělí kardiomyopatie do jedné ze tříd podle morfologického a funkčního obrazu postižení a následně rozlišuje u každé kardiomyopatie dědičné a nedědičné formy (obr. 1) [1].
Hypertrofická kardiomyopatie
Hypertrofická kardiomyopatie (HCM) je onemocnění charakterizované přítomností zvětšené tloušťky a/ nebo masy myokardu levé komory srdeční při absenci arteriální hypertenze nebo vrozených a chlopenních vad schopných zapříčinit danou míru hypertrofie [1]. HCM je nejčastější monogenně dědičné onemocnění srdce s prevalencí 1 : 500 v obecné populaci a je významnou příčinou náhlé srdeční smrti u mladých lidí a morbidity v důsledku srdečního selhání a cévních mozkových příhod [3,4]. HCM má autozomálně dominantní způsob přenosu a je zapříčiněna mutacemi zejména v genech pro sarkomerické proteiny [3,4]. Jako první byla identifikována mutace v genu pro těžký řetězec β myozinu, následně bylo nalezeno velké množství dalších mutací [3,5,6]. V současnosti je se vznikem HCM asociováno více než 1 400 mutací v alespoň 20 různých genech (tab. 1) [2,7]. Většina pacientů s pozitivním genetickým vyšetřením má mutaci v jednom ze dvou genů (gen pro těžký řetězec β myozinu a gen pro myozin vazebný protein C), zastoupení ostatních genů je výrazně nižší. Značná část geneticky vyšetřených pacientů (kolem 40 %) nemá žádnou nalezenou mutaci. Pozitivní výsledek genetického vyšetření je častější u mladších pacientů, u pacientů s pozitivní rodinnou anamnézou HCM a u pacientů s reverzní morfologií mezikomorového septa (septum konvexně vyklenuté do dutiny levé komory) [8]. Naopak nejnižší záchyt mutace je u starších pacientů se septum sigmoideum (konkávní do dutiny levé komory) [8]. Kromě postižení genů pro proteiny sarkomery dochází ke vzniku fenotypu HCM i u dalších genetických poruch (metabolická a střádavá onemocnění a mitochondriopatie, někdy se specifickými fenotypovými rysy a odlišným typem dědičnosti (tab. 2) [7]. V minulosti byly tyto nemoci označovány jako fenokopie HCM, ale podle aktuální klasifikace ESC splňují definici HCM [1]. Velká genetická variabilita HCM je pravděpodobně jedním z důvodů velké variability fenotypu a klinických projevů onemocnění. Nalezení vztahů konkrétních mutací a fenotypových znaků nebo prognózy HCM je obtížné pro velké množství mutací (některé z nich jsou identifikovány pouze v jedné konkrétní rodině), chybějící potvrzení již publikovaných asociací a ovlivnění fenotypu HCM i dalšími genetickými variantami kromě příčinné mutace. Mutace v genech pro srdeční troponiny byly v minulosti označeny za rizikové a mutace v genu pro myozin vazebný protein naopak za benigní stran náhlé srdeční smrti, neexistují ale přesvědčivé důkazy o vlivu konkrétních mutací na vznik náhlé srdeční smrti [9]. Porovnání skupin pacientů s mutací v genech pro tenká a tlustá filamenta (dva nejčastěji zastoupené geny pro těžký řetězec β myozinu a pro myozin vazebný protein C patří mezi tlustá filamenta) v nedávné práci ukázalo vyšší pravděpodobnost vývoje systolické dysfunkce i těžké diastolické dysfunkce u pacientů s mutacemi v genech pro tenká filamenta [10]. Naproti tomu arytmické příhody včetně náhlé smrti nebyly u pacientů s postižením tenkých filament (kam patří i srdeční troponiny) častější [10].
![Geny asociované se vznikem HCM a četnost záchytu mutací v jednotlivých genech – upraveno podle [7].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/12b540de012190badd444ea2636323a0.jpg)
![Geny s mutacemi způsobující metabolická a střádavá onemocnění a mitochondriopatie s fenotypem HCM – upraveno podle [7].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/2b9b2e428e5c55a8dded2b1df47e0060.jpg)
Dilatační kardiomyopatie
U dilatační kardiomyopatie (DCM) dochází k dilataci a systolické dysfunkci levé komory srdeční při nepřítomnosti hemodynamické anebo ischemické příčiny dostatečné pro vznik globální poruchy funkce [1]. Prevalence DCM v dospělé populaci se odhaduje na 1 : 2 500 [1]. Přibližně 25 – 40 % případů má familiární výskyt, většina případů onemocnění má získané příčiny. Familiární DCM může být způsobena širokou škálou genetických defektů srdečních bílkovin (až 40 genů) [1,11,12]. Většina mutací je přenášena jako autozomálně dominantní znak, možný je ale i autozomálně recesivní a gonozomálně recesivní přenos. První byla objevena mutace v genu pro dystrofin, základní součást dystroglykanového komplexu, který propojuje extracelulární matrix a cytoskeleton myocytů (i kosterních svalů) [11]. Onemocnění označované jako X ‑ vázaná DCM je alelickou variantou svalových dystrofií Duchennova a Beckerova typu s akcentovaným srdečním a potlačeným svalovým fenotypem [11]. Typ přenosu je v tomto případě samozřejmě gonozomální. Příčinné mutace DCM byly objeveny i u dalších komponent dystroglykanového komplexu (δ ‑ sarkoglykan, laminin α4, integrinová kináza) [11]. Některé mutace desminu, klíčového proteinu intermediárních filament, udržujícího správné uspořádání myofibril mohou způsobit kardiální poškození s obrazem DCM [11]. Postižení genů pro bílkoviny jaderného obalu, lamin A/ C a emerin způsobuje autozomálně dominantě vázanou a X ‑ vázanou formu Emeryho ‑ Dreifussovy muskulární dystrofie s různě vyjádřeným srdečním postižením vyskytujícím se zpravidla společně s převodními poruchami [11,13]. Mutace v genu pro lamin A/ C jsou asociovány s vyšším rizikem náhlé srdeční smrti [13]. Sarkomerické proteiny hrají rovněž roli v etiologii DCM [11].
Restriktivní kardiomyopatie
Restriktivní kardiomyopatie (RCM) je stav s restriktivní poruchou komorového plnění, bez zvětšení komor a s normální tloušťkou srdeční stěny [1]. Zařazují se zde ale i stavy s jistou mírou hypertrofie srdeční stěny (srdeční amyloidóza) [1]. Část pacientů může mít rovněž familiární výskyt onemocnění, dalšími příčinami jsou systémová onemocnění, toxické a poradiační postižení a patologie endokardu [1]. U familiární RCM byly identifikovány mutace v genu pro troponin I, který svým inhibičním účinkem brání interakci aktinu a myozinu v situaci, kdy nejsou na troponin C navázány ionty kalcia [14]. Porucha této inhibice může vést k diastolické dysfunkci až obrazu RCM. Další sarkomerické proteiny, které mohou být porušeny u RCM, jsou troponin T, esenciální lehký řetězec myozinu a těžký řetězec β ‑ myozinu [14]. Protein intermediárních filament desmin a jemu příbuzné proteiny, například alfa‑B ‑ krystalin a plektin, jsou rovněž dávány do souvislosti s RCM [9,14]. Dědičná jsou samozřejmě i některá systémová onemocnění s kardiálním postižením splňujícím definici RCM, například hereditární formy amyloidózy (mutace v genu pro prealbumin), infiltrativní a střádavá onemocnění jako Gaucherova nemoc, hemochromatóza a v některých případech Fabryho nemoc [1,14].
Arytmogenní kardiomyopatie (pravé) komory
U arytmogenní kardiomyopatie (ARVC) dochází k nahrazení myokardu pravé komory tukovou a fibrózní tkání a ke vzniku elektrokardiografických abnormalit [1]. U některých pacientů ovšem dominuje postižení levé komory [15].Většina případů je dědičná, existují i oblasti s endemickým výskytem (Itálie) [1]. ARVC má většinou autozomálně dominantní přenos s mutacemi v genech kódujících nejčastěji desmozomální proteiny plakophilin‑2, desmoglein‑2, desmocollin‑2 [15 – 17]. Existují i varianty s autozomálně recesivním přenosem – Naxos disease, u které byla prokázána mutace v genu pro plakoglobin a Carvajal syndrom s mutací v genu pro desmoplakin [15]. Postižením struktury desmozomů dochází k porušení elektrického i mechanického propojení kardiomyocytů, což vede ke vzniku arytmií a poruchám kinetiky. Další genetické poruchy vedoucí ke vzniku ARVC byly identifikovány mimo oblast desmozomu. Jsou to mutace genu pro transformační růstový faktor ‑ 3, genu pro srdeční ryanodinový receptor a genu pro transmembránový protein 43 [15,17,18].
Nonkompaktní kardiomyopatie
Nonkompaktní kardiomyopatie (LVNC), v ESC klasifikaci zařazená mezi neklasifikované kardiomyopatie, je stav, kdy dochází k výrazné trabekularizaci části levé komory (nejčastěji apikální) a ztenčení kompaktní subepikardiální vrstvy myokardu s alespoň částečně familiárním výskytem (převládá autozomálně dominantní přenos) [1]. Nepanuje jednoznačný názor, zda je LVNC samostatná kardiomyopatie nebo znak sdílený s ostatními kardiomyopatiemi [19]. Nonkompaktní část levé komory se může vyskytovat u jakékoli jiné kardiomyopatie, některé mutace ale prokazatelně způsobují trabekularizaci myokardu [19]. LVNC navíc může vzniknout i při zvýšení preloadu (například u sportovců a během těhotenství). U LVNC se podařilo identifikovat mutace v genech některých sarkomerických proteinů (těžký řetězec β ‑ myozinu, aktin, troponin T), dále v genu pro lamin A/ C (protein jaderného obalu), Cypher/ ZASP (strukturální protein Z disku sarkomery) nebo protein tafazzin s předpokládanou úlohou v mitochondriálním metabolizmu fosfolipidů [19 – 20]. Mutace v posledně jmenovaném genu se přenáší jako X ‑ vázaný znak a u kojenců způsobují Barthův syndrom s dilatovanou, dysfunkční a trabekularizovanou levou komorou (kombinace fenotypu LVNC a DCM) [19].
Genetické vyšetření pacientů s kardiomyopatiemi
Přes velkou genetickou heterogenitu familiárních forem kardiomyopatií umožňuje znalost základů jejich dědičnosti správně indikovat vyšetřování příbuzných a do jisté míry i zhodnotit prognózu pacienta nebo ovlivnit terapii. Genetické testování pacientů je při velkém množství potenciálně postižených genů náročné vyšetření, které se ale postupně stává součástí klinické praxe. ESC vydala v roce 2010 stanovisko zabývající se genetickým testováním u pacientů s kardiomyopatiemi a jeho přínosem pro sledování pacientů a jejich příbuzných [12]. Evropské i americké doporučené postupy pro diagnostiku a léčbu HCM rovněž doporučují genetické vyšetření pacientů k umožnění diagnostiky příbuzných [21,22]. Součástí péče o rodiny pacientů s familiárními formami kardiomyopatií je i genetické poradenství [12]. Genetické vyšetření je zvlášť výhodné při neobvyklých fenotypových rysech, které umožňují zúžit rozsah zkoumaných genů, představují vysoké riziko anebo mají dopad na léčbu pacienta [12]. Příkladem může být Fabryho nemoc s možností specifické terapie, nutnost implantace kardiostimulátoru u nemocných s převodními poruchami u DCM a HCM a ovlivnění indikace implantace ICD u pacientů s DCM (mutace v genu pro lamin A/ C) [12]. Pozitivní genetické vyšetření ale zásadně mění strategii sledování příbuzných (obr. 2) [12]. Při identifikaci příčinné mutace v rodině je rychlá a přesná DNA diagnostika u příbuzných vysoce efektivní. Při pozitivním výsledku by příbuzní měli podstoupit stejná vyšetření jako pacienti s danou kardiomyopatií, včetně vyšetření jejich příbuzných prvního stupně (kaskádový přístup), při negativním výsledku již nepotřebují další kardiologické sledování (obr. 2) [12]. Příbuzné prvního stupně pacientů bez genetického vyšetření anebo s negativním výsledkem vyšetření je nutné kardiologicky sledovat [12]. Většina kardiomyopatií má totiž s věkem stoupající penetranci (fenotyp onemocnění se u nositelů mutace může vyvinout až do věku kolem 60 let) [12,21]. Moderní metody genetického vyšetření (NGS – next generation sequencing) umožňují rychlou analýzu velkého množství DNA, případně i celého genomu [23]. U pacientů s kardiomyopatiemi tak zvyšují časovou i ekonomickou efektivitu vyšetření. Identifikované varianty DNA se klasifikují podle úrovně důkazů kauzality k vývoji onemocnění na patogenní, pravděpodobně patogenní a varianty nejasného významu [21].
![Sledování příbuzných pacientů s kardiomyopatiemi v závislosti na výsledku genetického vyšetření [12].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/7ef7a29e2cee3112019f8456508a47f1.jpg)
Závěr
Jedním z projevů různorodosti kardiomyopatií je genetická variabilita jejich familiárních forem. Velké množství objevených mutací v různých genech vysvětluje etiologii jenom části dědičných kardiomyopatií. Chybí detailní vysvětlení patofyziologických pochodů vedoucích od konkrétních mutací k rozvoji fenotypu a omezené jsou naše poznatky o korelaci mutací a prognóze onemocnění, nejvíc dat je v tomto ohledu dostupných u HCM. Aktuální klinický přínos genetického vyšetření je zejména v diagnostice příbuzných a u pacientů se zvláštními fenotypovými rysy. Rozšíření vysokokapacitních metod genetického testování (NGS) zpřístupní DNA diagnostiku více pacientům a poskytne velké množství dat o genetických variantách u pacientů s kardiomyopatiemi.
Doručeno do redakce: 10. 1. 2015
Přijato po recenzi: 29. 1. 2015
MUDr. Pavol Tomašov
www.fnmotol.cz
pavol.tomasov@fnmotol.cz
Sources
1. Elliott P, Andersson B, Arbustini E et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29 : 270 – 276.
2. Perrot A, Dietz R, Osterziel KJ. Is there a common genetic basis for all familial cardiomyopathies? Eur J Heart Fail 2007; 9 : 4 – 6.
3. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet 2013; 381 : 242 – 255. doi: 10.1016/ S0140 ‑ 6736(12)60397 ‑ 3.
4. Maron BJ, Gardin JM, Flack JM et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92 : 785 – 789.
5. Jarcho JA, McKenna W, Pare JA et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med 1989; 321 : 1372 – 1378.
6. Geisterfer ‑ Lowrance AA, Kass S, Tanigawa G et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990; 62 : 999 – 1006.
7. Yingchoncharoen T, Tang WW. Recent advances in hypertrophic cardiomyopathy. F1000Prime Rep 2014; 6 : 12. doi: 10.12703/ P6 – 12.
8. Binder J, Ommen SR, Gersh BJ et al. Echocardiography ‑ guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc 2006; 81 : 459 – 467.
9. Ashrafian H, Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol 2007; 49 : 1251 – 1264.
10. Coppini R, Ho CY, Ashley E et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin‑filament gene mutations. J Am Coll Cardiol 2014; 64 : 2589 – 2600. doi: 10.1016/ j.jacc.2014.09.059.
11. Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2005; 45 : 969 – 981.
12. Charron P, Arad M, Arbustini E et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010; 31 : 2715 – 2726. doi: 10.1093/ eurheartj/ ehq271.
13. Malhotra R, Mason PK. Lamin A/ C deficiency as a cause of familial dilated cardiomyopathy. Curr Opin Cardiol 2009; 24 : 203 – 208. doi: 10.1097/ HCO.0b013e32832a11c6.
14. Willott RH, Gomes AV, Chang AN et al. Mutations in Troponin that cause HCM, DCM and RCM: what can we learn about thin filament function? J Mol Cell Cardiol 2010; 48 : 882 – 892. doi: 10.1016/ j.yjmcc.2009.10.031.
15. Marcus FI, McKenna WJ, Sherrill D et al. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/ Dysplasia (ARVC/ D). Circulation 2010; 121 : 1533 – 1541. doi: 10.1161/ CIRCULATION AHA.108.840827.
16. Dalal D, James C, Devanagondi R et al. Penetrance of mutations in plakophilin‑2 among families with arrhythmogenic right ventricular dysplasia/ cardiomyopathy. J Am Coll Cardiol 2006; 48 : 1416 – 1424.
17. Basso C, Corrado D, Marcus F et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009; 373 : 1289 – 1300. doi: 10.1016/ S0140 ‑ 6736(09)60256 ‑ 7.
18. Vatta M, Marcus F, Towbin JA. Arrhythmogenic right ventricular cardiomyopathy: a ‘final common pathway‘ that defines clinical phenotype. Eur Heart J 2007; 28 : 529 – 530.
19. Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol 2014; 64 : 1840 – 1850. doi: 10.1016/ j.jacc.2014.08.030.
20. Klaassen S, Probst S, Oechslin E et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008; 117 : 2893 – 2901. doi: 10.1161/ CIRCULATIONAHA.107.746164.
21. Gersh BJ, Maron BJ, Bonow RO et al. 2011 ACCF/ AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines. Circulation 2011; 124 : 783 – 831. doi: 10.1161/ CIR.0b013e318223e2bd.
22. Elliott PM, Anastasakis A, Borger MA et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35 : 2733 – 2779. doi: 10.1093/ eurheartj/ ehu284.
23. Faita F, Vecoli C, Foffa I et al. Next generation sequencing in cardiovascular diseases. World J Cardiol 2012; 4 : 288 – 295. doi: 10.4330/ wjc.v4.i10.288.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2015 Issue 1
Most read in this issue
- TDM digoxinu v klinické praxi
- Lékové interakce a současná klinická praxe
- Srdeční resynchronizační terapie – kdy a u koho ji v současnosti indikovat?
- TDM antibiotik v klinické praxi