
Projekt MedPed – pacienti s familiární hypercholesterolemií ve středu pozornosti
Project MedPed – spotlight on patients with familial hypercholesterolaemia
Familial hypercholesterolaemia is an inborn defect of cholesterol metabolism, which seems to be considerably more prevalent than previously thought. Given the recently proposed prevalence of 1 : 250 in the general population, we estimate up to some 40,000 individuals are affected in the Czech Republic. FH patients should be diagnosed, adequately informed and treated as early as possible. Comprising of a number of specialised outpatient centres, the MedPed network has been successfully working to this objective for the last 17 years.
Keywords:
MedPed – familial hypercholesterolaemia – cascade screening – cardiovascular risk – mutation – ScreenPro FH
Authors:
M. Vaclová 1; M. Vráblík 1; T. Freiberger 2; R. Češka 1
Authors‘ workplace:
III. interní klinika 1. LF UK a VFN v Praze
1; Genetická laboratoř, Centrum kardiovaskulární a transplantační chirurgie, Brno
2
Published in:
Kardiol Rev Int Med 2016, 18(3): 203-207
Overview
Familiární hypercholesterolemie je vrozené onemocnění metabolizmu cholesterolu, které je mnohem častější, než se publikovalo dříve. Při nově stanovené frekvenci heterozygotů 1 : 250 je možné, že se v ČR jedná zhruba o 40 000 jedinců postižených tímto onemocněním. Je více než žádoucí, aby co největší část těchto jedinců byla diagnostikována, o svém onemocnění informována a adekvátně léčena. K tomuto účelu již více než 17 let slouží síť projektu MedPed a velké množství lékařů, kteří jsou do projektu zapojeni.
Klíčová slova:
MedPed – familiární hypercholesterolemie – kaskádový screening – kardiovaskulární riziko – mutace – ScreenPro FH
Úvod
Projekt MedPed (Make early diagnosis to Prevent early deaths) vznikl v roce 1994 a dosud se do něj zapojilo více než 30 zemí po celém světě. Cílem tohoto projektu je snížení rizika předčasného úmrtí u pacientů s familiární hypercholesterolemií (FH) [1]. Síť projektu MedPed v ČR je dynamická, vznikla v roce 1998 a od té doby se neustále zapojují nová pracoviště a někdy naopak některá končí. V tuto chvíli je síť projektu MedPed v ČR tvořena dvěma národními centry v Praze a v Brně, dále ji tvoří 15 regionálních center pro dospělé, 10 center pro děti a 18 specializovaných pracovišť pro dospělé a sedm pro děti, dále s projektem spolupracuje devět lékařů. Do projektu jsou zapojeni lékaři mnoha odborností, kteří přicházejí do styku s pacienty s FH, např. internisté, kardiologové, pediatři, biochemici a genetici. Projekt funguje pod záštitou České společnosti pro aterosklerózu a je založen na dobrovolné a bezplatné spolupráci zúčastněných lékařů. Cílem projektu je vyhledávání postižených jedinců, jejich diagnostika a léčba. Dále snaha o kontaktování a diagnostiku jejich dosud nediagnostikovaných příbuzných. Nejen k těmto aktivitám slouží celonárodní databáze MedPed, která umožňuje vyhledávání jednotlivých pacientů a event. jejich ošetřujících lékařů. Tím lze dosáhnout komplexní erudované péče i u rodinných příslušníků žijících v odlišných regionech. Lékaři tak mají přehled i o rodinných příslušnících svého pacienta vč. výsledků genetických vyšetření a nemusí se spoléhat na anamnestická data uváděná pacientem. Nedílnou součástí projektu MedPed jsou také vzdělávací aktivity jeho spolupracovníků a snaha udržet spolupracující lékaře na vysoké odborné úrovni. S tím souvisí i snahy o zlepšení povědomí o FH jak mezi laickou veřejností, tak i mezi lékaři 1. linie (praktickými, kožními či očními lékaři), kteří mohou pacienta s FH odhalit jako první. Projekt MedPed má v budoucnu ambice rozšířit svou působnost i na všechny těžké hyperlipoproteinemie a dyslipidemie. Všechny výše uvedené aktivity projektu MedPed vedou k jednomu cíli: zajistit diagnostiku a dlouhodobé sledování vč. nejlepší možné dostupné léčby co největšímu počtu pacientů (především s FH), a tím snížit riziko předčasného kardiovaskulárního (KV) úmrtí v této populaci.
Definice FH
FH je jedno z nejčastějších metabolicky podmíněných vrozených onemocnění člověka. Je podmíněna genetickou poruchou některého z genů uplatňujících se v metabolizmu LDL cholesterolu (LDL-c). Je to tedy monogenní autozomálně dominantní (výjimečně se může jednat i o recesivní) onemocnění, v jehož důsledku dochází od narození k hromadění LDL-c v cirkulaci a ve tkáních. Tak dochází k urychlení rozvoje aterosklerózy.
Epidemiologie FH
Práce Goldsteina et al ze 70. let minulého století uváděla prevalenci FH heterozygotů 1 : 500 a homozygotů 1 : 1 milionu obyvatel [2]. Studie z roku 2012 provedená u rozsáhlého vzorku více než 69 000 osob dánské populace však prokázala prevalenci mnohem vyšší [3]. Tato studie byla založena na stanovení diagnózy FH pomocí běžně užívaných kritérií (Dutch Lipid Network Criteria), zatímco nizozemská studie na molekulárně genetické analýze více než 104 000 osob [4]. Tyto nové studie prokázaly prevalenci FH s frekvencí přibližně 1 : 250 u heterozygotů. Homozygotní postižení bylo zjištěno s frekvencí 1 : 160 000–300 000 osob [4]. Tato čísla tedy prokazují přibližně 2–6× vyšší prevalenci tohoto onemocnění v běžné populaci než uváděl Goldstein et al. Takto frekventní výskyt se dříve připouštěl jen u populací zatížených efektem zakladatele rodu, jako je např. bělošská populace v Jihoafrické republice, populace francouzských Kanaďanů či populace v Libanonu [5–7]. Toto zjištění dělá z FH nejčastější vrozené metabolicky podmíněné onemocnění známé u člověka.
Fenotyp FH
Fenotypické známky tohoto onemocnění jsou známy a popsány mnoho desítek let. Patří mezi ně výskyt šlachových xantomů, které jsou pro FH patognomické. Nejčastěji bývá postižena Achillova šlacha a šlachy extenzorů především na hřbetech rukou. U Achillovy šlachy se také může jednat o její rupturu – traumatické změny kvalitativně poškozené šlachy. Xantelazma palpebrarum jsou méně specifické příznaky FH, ale jejich výskyt v mladších věkových kategoriích, obzvláště s recidivami i po plastických chirurgických výkonech, by měl na možnost FH upozornit. Zde jsou 1. linií kožní lékaři a specialisté v oboru plastické chirurgie, kteří by měli zajistit vyšetření lipidogramu k vyloučení či potvrzení suspekce na FH. Arcus lipoides corneae je příznak nespecifický, u pacientů ve vysokém věku se často vyskytuje i při nepříliš vysoké cholesterolemii, avšak ve věku do 50 let by měl opět vést k vyšetření lipidogramu. Klinické projevy závisí na tíži cholesterolemie a délce doby, po kterou je jí tělo vystaveno. Nepřekvapí tedy, že většina dětí a mladistvých klinické známky spíše nemá nebo je má pouze při velmi vysokých hodnotách lipidogramu, např. jedná-li se o homozygoty. Stejně tak při dobře vedené a dobře tolerované mnohaleté léčbě se některé klinické příznaky vytrácí. To je jistě jeden z důvodů, proč již těžké a jasné klinické příznaky FH – obzvláště šlachové xantomy – vidíme v klinické praxi spíše zřídka. Ta část pacientů s FH, která je mnoho let léčena i „symptomaticky“ statiny pro hypercholesterolemie bez potvrzení diagnózy FH, je např. ani nevyvine.
U homozygotů s FH se klinické příznaky objevují v mladším věku než u heterozygotů, často v mnohem větším rozsahu, a můžeme vidět i kožní xantomy.
Předčasný výskyt KV příhod v rodinné i osobní anamnéze je u heterozygotů s FH běžný. Za předčasný výskyt KV příhody považujeme věk u mužů do 55 let a u žen do 60 let. Výjimkou však není ani KV příhoda (často fatální) ve věku okolo 30 let. Zda a v kolika letech se u daného jedince KV onemocnění rozvine, záleží na řadě okolností, např. na typu mutace a jejím dopadu na metabolizmus cholesterolu nebo na koexistenci ostatních rizikových faktorů (např. mužské pohlaví, kouření, obezita apod.). U homozygotů se může manifestní KV onemocnění posunout až do prepubertálního věku (např. méně než 10 let). Takto postižení jedinci často umírají ve druhé či třetí dekádě svého života po četných recidivách KV příhod. U homozygotů je také častá akcelerovaná ateroskleróza kořene aorty a aterosklerotické postižení aortální chlopně. Pouze účinná a velmi časně zahájená léčba jim dává naději na prodloužení předpokládané doby dožití.
Předčasný rozvoj KV onemocnění se dá zvrátit či odsunout do vyšších věkových kategorií. Výsledek závisí na tíži postižení jednotlivce, na jeho ostatních rizikových i protektivních faktorech a na věku zahájení léčby (graf 1). Důležitou zůstává i strategie léčby a její dávkování. Farmakologická léčba by měla být vedena odborníkem, vždy nejpozději od věku 18 let (pokud není indikována léčba již v dětském či pubertálním věku). Cílem léčby u pacienta s FH v primární prevenci by měla být hodnota LDL-c pod 2,5 mmol/l. Cílem léčby u pacientů s FH v sekundární prevenci je hodnota LDL-c pod 1,8 mmol/l. Pokud těchto cílů nelze dosáhnout ani při maximální farmakologické léčbě, pak usilujeme o dosažení sekundárního léčebného cíle, tj. snížení hladin LDL-c o nejméně 50 %. Každé snížení LDL-c o 1 mmol/l i u běžné populace je spojeno s redukcí KV mortality o 22 % a celkové mortality o 12 % za pět let [8].
![Kumulativní zátěž LDL cholesterolem (LDL-c) u osob s a bez FH se vztahem k době zahájení léčby statiny [8].](https://pl-master.mdcdn.cz/media/image/865107cae3649eb5dd897206d195fc2c.png?version=1537795035)
Diagnostická kritéria FH
Zda bychom o FH měli začít uvažovat, nám na první pohled ozřejmí koncentrace celkového a LDL-c bez léčby. Podmínkou jsou nejméně dvě měření lipidogramu s časovým odstupem alespoň jeden měsíc po poučení a dodržování diety ke snížení hladin cholesterolu. Diagnózu FH připouští hodnoty LDL-c vyšší než 95. percentil pro danou populaci, věk a pohlaví. Podmínkou je normotriglyceridemie nebo hypertriglyceridemie nepřekračující hodnoty 3 mmol/l a vyloučení sekundární příčiny hypercholesterolemie (např. hypotyreózy, Cushingova syndromu, užívání hormonální antikoncepce, mentální anorexie, cholestázy nebo nefropatie).
Orientačně bychom o FH měli začít uvažovat při překročení 8 mmol/l u celkového cholesterolu a zároveň překročení 5 mmol/l u LDL-c nebo při koncentraci LDL-c nad 6 mmol/l.
Laboratorní kritéria projektu MedPed byla vypočtena pro pacienty, u kterých je již známý příbuzný s tímto onemocněním, a u kterých je tedy jejich vlastní riziko přítomnosti FH větší než u obecné populace. Tito pacienti mají odlišné hranice LDL-c rozhodné pro diagnózu FH, a to v závislosti na svém stupni příbuznosti k nemocnému s prokázanou FH a v závislosti na věku, ve kterém byl odběr krve bez léčby proveden (tab. 1) [9].
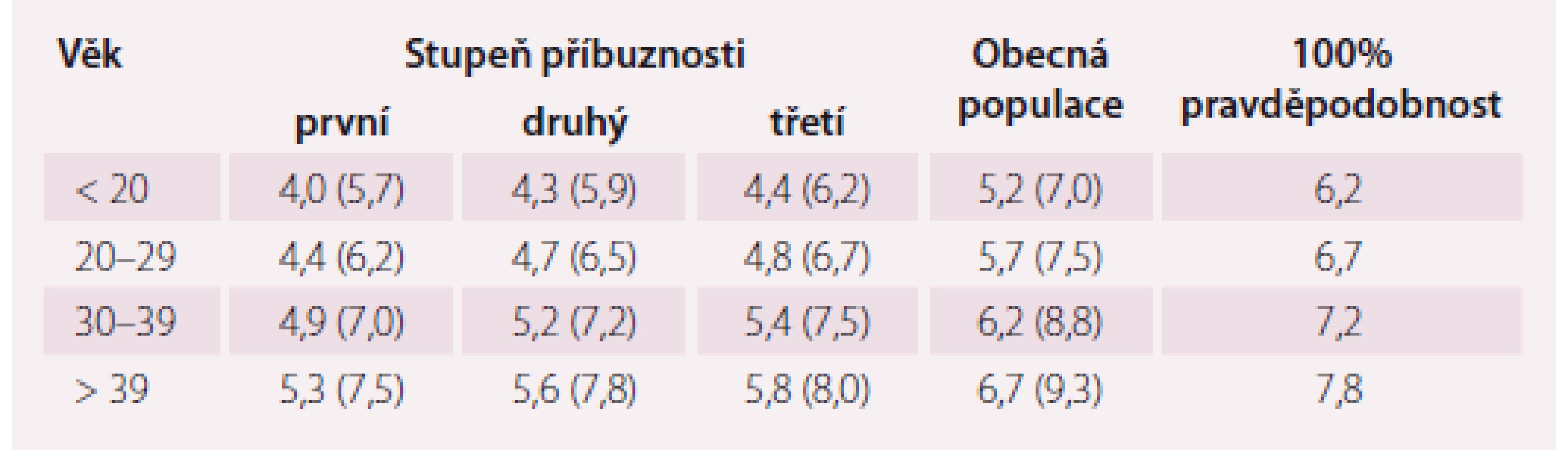
Nejpřesnějším se ale jeví využití komplexních diagnostických kritérií, která v sobě zahrnují i osobní, rodinnou anamnézu, výsledek genetického testování apod. Jedněmi z prvních komplexních kritérií byla britská, založená na datech ze Simone Broome registru (tab. 2) [10].
![Diagnostická kritéria FH dle Simone Broome registru pro probandy [10].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/05bd9fb3cd2503fbf05bb9ce60ceded4.png)
Nejrozšířenější je však nejspíše nizozemský systém skórování (Dutch Lipid Network Criteria – DLNC) (tab. 3) [11]. Tento systém se jeví nejkomplexnější, zahrnuje informace z rodinné i osobní anamnézy jedince, jeho fyzikální i laboratorní vyšetření a výsledek DNA analýzy, je-li k dispozici.
![Dutch Lipid Network Criteria pro diagnózu familiární hypercholesterolémie [11].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/1ead3457902e35257df8a1c20b8c1de9.png)
Genetický podklad FH
Mnoho let byla FH považována za jasný příklad onemocnění děděného autozomálně dominantním typem dědičnosti. Rozlišujeme tři základní geny, ve kterých může být přítomna mutace podmiňující FH. Jedná se o gen pro LDL receptor. Zde je známo více než 1 800 různých mutací. Dále je to gen pro apolipoprotein B-100 (FDB). Zde je dominantně se vyskytující mutací způsobující fenotyp FH aminokyselinová záměna v pozici 3 527 peptidové sekvence (p.Arg3527Gln). Dále mohou být zjištěny i tzv. gain-of-function mutace genu pro PCSK9 (proprotein konvertázu subtilizin/kexin typu 9) (zvyšují transkripci genu), těch bylo popsáno již více než 20 [8]. S postupujícími možnostmi genetického vyšetřování a technologickým pokrokem bylo zjištěno, že se velmi výjimečně může jednat i o onemocnění recesivní (mutace obou alel genu pro LDL receptor adaptorový protein).
Naprostá většina případů FH je ovšem děděna autozomálně dominantně. Důkazem toho jsou v praxi sestavené rodokmeny, kde by měl být postižen vždy jeden z rodičů a průměrně polovina sourozenců a potomků. Proto byl ustanoven pojem autozomálně dominantní hypercholesterolemie (ADH) [12]. Dle genetické analýzy pak toto onemocnění můžeme dělit na:
- tzv. klasickou FH podmíněnou mutacemi v genu pro LDL receptor,
- familiární defekt apolipoproteinu B-100 – zapříčiněný výše zmíněnou mutací v genu pro Apo B,
- tzv. non-FH/non-FDB ADH – někdy také označovanou jako FH3 či ADH3, která je způsobena mutací v genu PCSK9 nebo mutacemi jiných genů, které nejsou dosud známy.
U dvou heterozygotních rodičů je 25% riziko, že se jim narodí potomek s homozygotní konstitucí mutace, a 50% riziko narození potomka s heterozygotní konstitucí. V tomto případě průměrně pouze 25 % potomků nebude nositelem FH.
Jak již bylo zmíněno výše, FH může být i onemocněním způsobeným recesivní dědičností. Příčinou je v tomto případě defekt genu LDLRAP1 (LDL receptor adaptor protein 1). Toto onemocnění se pak nazývá autozomálně recesivní hypercholesterolemie (ARH). Toto onemocnění je velmi vzácné a klinicky může odpovídat homozygotní formě ADH. V rodokmenu nenacházíme typickou přítomnost onemocnění ve všech generacích, naopak rodiče fenotypově zdraví mají 25 % potomků s těžkou hypercholesterolemií [13].
Výsledek DNA analýzy musí být vždy hodnocen v kontextu klinických informací – precizně vypracovaného rodokmenu, informací z rodinné anamnézy, klinického stavu a laboratorních výsledků. DNA analýza také může mnohdy osvětlit situaci u klinicky a laboratorně nejasných případů.
Důležitou součástí genetického vyšetřování je i kaskádový screening. Pokud je v rodině znám proband a jeho konkrétní mutace, lze velmi snadno a relativně rychle vyšetřit jeho suspektní rodinné příslušníky, a tak se rychle a jednoduše dobrat odpovědi, kteří rodinní příslušníci jsou nositeli FH a kteří nikoli.
Projekt MedPed v ČR
Jak bylo řečeno výše, projekt MedPed je na světě od roku 1994 a od roku 1998 je v ČR funkční síť jeho center. K polovině roku 2016 je v projektu zadáno 12 246 záznamů jednotlivých pacientů. Z toho je pacientů s FH 6 889 (z toho dětí do 19 let v době diagnózy je 669 a dětí do 19 let dosud je 427). V databázi je zadáno celkem 5 181 probandů s FH, jejich příbuzných s FH 1 670. Dále je tam zadáno 1 356 zdravých příbuzných. Tato čísla řadí ČR na první místa v Evropě i ve světě. Při původně odhadované frekvenci heterozygotů 1 : 500 by mělo být v ČR přítomno cca 20 000 pacientů s FH. Uvedených 6 889 pacientů s FH tedy představuje 34 % odhadovaného celkového počtu. Proto lze ČR hodnotit jako třetí nejlepší zemi ve vyhledávání pacientů s FH v Evropě i na světě za Nizozemskem (se 71 %) a Norskem (se 43 %) (obr. 2) [8].
![Odhad počtu diagnostikovaných FH na světě v jednotlivých státech.
Převzato a upraveno dle [8].](https://pl-master.mdcdn.cz/media/image/bee171dca034389138fa13c215c35780.png?version=1537796338)
Závěr
FH je metabolické vrozené onemocnění, které je mnohem častější, než se původně myslelo. Při nově stanovené frekvenci heterozygotů 1 : 250 je možné, že se v ČR jedná zhruba o 40 000 jedinců postižených tímto onemocněním. Je více než žádoucí, aby co největší část těchto osob byla diagnostikována, o svém onemocnění informována a adekvátně léčena. K tomuto účelu již přes17 let slouží síť projektu MedPed a velké množství lékařů, kteří v rámci projektu dobrovolně a bezplatně spolupracují.
Děkujeme všem centrům a spolupracujícím lékařům projektu MedPed v ČR. Projekt MedPed v ČR podporují společnosti Amgen, AOP Orphan, KRKA, MSD, Pfizer a Sanofi.
Práce byla podpořena grantem AZV č. 15-28277A.
Doručeno do redakce: 18. 8. 2016
Přijato po recenzi: 31. 8. 2016
MUDr. Martina Vaclová, Ph.D.
www.vfn.cz
vaclova.martina@seznam.cz
Sources
1. World Health Organization: Familial hypercholesterolemia – report of a second WHO Consultation. Geneva, Switzerland: World Health Or - ganization 1999 (WHO publication No. WHO/HGN/ /FH/CONS/99.2). Available from: http: //apps.who.int/iris/bitstream/10665/66346/1/ WHO_HGN_ FH_CONS_99.2.pdf.
2. Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-metylglutaryl coenzyme A reductase aktivity associated with overproduction of cholesterol. Proc Natl Acad Sci USA 1973; 70 : 2804–2808.
3. Benn M, Watts GF, Tybjaerg-Hansen A et al. Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab 2012; 97 : 3956–3964. doi: 10.1210/jc.2012–1563.
4. Sjouke B, Kusters DM, Kindt I et al. Homozygous autosomal dominant hypercholesterolemia in the Netherlands: prevalence, genotype-fenotype relationship, and clinical outcome. Eur Heart J 2015; 36 : 560–565. doi: 10.1093/eurheartj/ehu058.
5. Delport R. Familial hypercholesterolaemia in South Africans: tracking findings and developments over time – with reference to: prevalence of hypercholesterolaemia in young Afrikaners with myocardial infarction. Ischaemic heart disease risk factors. Cardiovasc J Afr 2009; 20 : 18–22.
6. Simard LR, Viel J, Lambert M et al. The Delta >15 Kb deletion French Canadian founder mutation in familial hypercholesterolemia: rapid polymerase chain reaction-based diagnostic assay and prevalence in Quebec. Clin Genet 2004; 65 : 202–208.
7. Fahed AC, Safa RM, Haddad FF et al. Homozygous familial hypercholesterolemia in Lebanon: a genotype/phenotype correlation. Mol Genet Metab 2011; 102 : 181–188. doi: 10.1016/j.ymgme.2010.11.006.
8. Nordestgaard BG, Chapman MJ, Humphries SE et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013; 34 : 3478–3490. doi: 10.1093/eurheartj/eht273.
9. Williams RR, Hunt SC, Schumacher MC at al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol 1993, 72 : 171–176.
10. Identification and Management of Familial Hypercholesterolaemia (FH). NICE Clinical Guidelines, No. 71. National Collaborating Centre for Primary Care (UK). London: Royal College of General Practitioners (UK) 2008.
11. van Aalst-Cohen ES, Jansen AC, Tanck MW et al. Diagnosing familial hypercholesterolaemia: the relevance of genetic testing. Eur Heart J 2006; 27 : 2240–2246. doi: 10.1093/eurheartj/ehl113.
12. Varret M, Abifadel M, Rabès JP et al. Genetic heterogeneity of autosomal dominant hypercholsterolemia. Clin Genet 2008; 73 : 1–13. doi: 10.1111/j.1399-0004.2007.00915.x
13. Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest 2003; 111 : 1795–1803. doi: 10.1172/JCI20031892.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2016 Issue 3
Most read in this issue
- Betablokátory, které nejméně negativně ovlivňují kardiorespirační zdatnost u zdravých osob
- Endovaskulární léčba ileofemorální hluboké žilní trombózy
- Možnosti endovaskulární léčby akutní končetinové ischemie
- Disekce aorty typu B s těžkou viscerální a končetinovou ischemií řešená kompletní endovaskulární revaskularizací – kazuistika