
Idiopatická plicní fibróza – změny v diagnostice a léčbě
Idiopathic pulmonary fibrosis – changes in diagnostics and treatment
Idiopathic pulmonary fibrosis (IPF) belongs to the most difficult-to-treat and most serious of lung diseases, with prognosis similar to lung cancer. Epidemiologic data are highly probably underestimated in many countries since IPF used to be frequently misdiagnosed for other diagnoses or not diagnosed at all. In the last century, when described for the first time, IPF was an extremely rare diagnosis. Nowadays, the incidence of IPF is undoubtedly rising, which is also supported by improved diagnostic opportunities. Previously, IPF was considered an untreatable disease; however, this has changed in the last eight years as antifibrotic therapy has emerged. Due to this fact, IPF patients have a better chance of survival, although the disease is still uncurable.
Keywords:
idiopathic pulmonary fibrosis – diagnosis
Authors:
Vašáková M.
Authors‘ workplace:
Pneumologická klinika 1. LF UK a Thomayerovy nemocnice, Praha
Published in:
Kardiol Rev Int Med 2019, 21(3): 135-141
Idiopatická plicní fibróza (IPF) patří mezi nejobtížněji léčitelné a zároveň nejzávažnější plicní nemoci s prognózou podobnou rakovině plic. Velmi pravděpodobně jsou epidemiologická data v řadě zemí podhodnocena, neboť IPF bývá diagnosticky zaměňována s jinými diagnózami nebo není diagnostikována vůbec. V minulém století, kdy byla poprvé poznána, se jednalo o diagnózu raritní, nyní je nepochybně incidence IPF na vzestupu, na čemž se jistě podílí i zlepšená diagnostika. Původně se jednalo o nemoc neléčitelnou, což se změnilo až v posledních 8 letech s nástupem antifibrotických léků. Díky tomu je dána nemocným šance na lepší přežití, i když zcela vyléčit IPF stále nedovedeme.
Overview
Idiopatická plicní fibróza (IPF) patří mezi nejobtížněji léčitelné a zároveň nejzávažnější plicní nemoci s prognózou podobnou rakovině plic. Velmi pravděpodobně jsou epidemiologická data v řadě zemí podhodnocena, neboť IPF bývá diagnosticky zaměňována s jinými diagnózami nebo není diagnostikována vůbec. V minulém století, kdy byla poprvé poznána, se jednalo o diagnózu raritní, nyní je nepochybně incidence IPF na vzestupu, na čemž se jistě podílí i zlepšená diagnostika. Původně se jednalo o nemoc neléčitelnou, což se změnilo až v posledních 8 letech s nástupem antifibrotických léků. Díky tomu je dána nemocným šance na lepší přežití, i když zcela vyléčit IPF stále nedovedeme.
Klíčová slova:
idiopatická plicní fibróza – diagnóza
Úvod
Idiopatická plicní fibróza (IPF) je smrtícím onemocněním plic s prognózou stejnou jako bronchogenní karcinom. Jde o progredující idiopatickou intersticiální pneumonii (IIP), která je charakterizována radiologickým a histopatologickým obrazem obvyklé intersticiální pneumonie (usual interstitial pneumonia – UIP). Poznání morfologického, a tím i radiologického obrazu IPF, se vyvíjí v čase ruku v ruce s poznáním patogeneze této nemoci a hlavně přesných zobrazovacích metod jako je počítačová tomografie hrudníku s vysokou rozlišovací schopností (HRCT). Dříve v podstatě jakákoli fibróza plic, kde nebyla zjištěna jiná příčina, byla hodnocena jako IPF a plicní biopsie byla považována za zlatý standard diagnostiky. Názor se začal měnit v roce 2000, kdy se oddělila od IPF nová podjednotka IIP a bylo zároveň zjišťováno, že obraz UIP může mít více nemocí, nejen IPF, a že tedy histopatologická diagnóza pravděpodobně není zlatým grálem. V roce 2000 pak bylo publikováno doporučení ATS/ ERS/ JRS/ ALAT, které stanovilo kombinaci velkých a malých kritérií pro diagnózu IPF [1]. Stále více se začíná hovořit o multidisciplinárním teamu (MDT) a jeho roli pro definitivní diagnózu této nemoci. V roce 2011 vychází nové doporučení respiračních společností, které stanovuje histopatologická kritéria pro definitivní a možnou diagnózu IPF. Na základě kombinací kritérií pak určuje pravděpodobnost diagnózy IPF. Také vylučuje bronchoalveolární laváž (BAL) z diagnostických vyšetření jako nevhodnou a nepřínosnou a stanovuje, že pacienti s typickým klinickým obrazem IPF a radiologickým obrazem definitivní UIP nemusí podstoupit plicní biopsii. Radiologický nález se tak stává dominantním pro diagnózu [2]. Nicméně stále neexistuje léčba na základě evidence, používá se stále ještě trojkombinace kortikosteroidů, N-acetylcysteinu a azathioprinu na základě studie IFIGENIA s velmi slabými důkazy [3]. Již se ale objevují první pozitivní studie s pirfenidonem, které zcela mění situaci na poli léčby IPF. Trojkombinace padá na základě studie PANTHER a je prakticky odstavena jako silně nedoporučená. Po další úspěšné studii s antifibrotikem, tentokrát nintedanibem, se v roce 2015 objevuje první evidence-based léčebné doporučení pro IPF [4–7]. Nedokonalost doporučeného postupu diagnostiky a léčby IPF je čím dál více evidentní, a proto vychází bezprostředně po novém návrhu klasifikace IPF radiologické Fleischnerovy společnosti doposud poslední mezinárodní dokument v roce 2018, který vyděluje z možné radiologické UIP ještě podskupinu pravděpodobné, u které nemusí být za určitých podmínek biopsie prováděna vůbec, a navíc se vrací do diagnostiky i BAL [8,9].
Definice IPF
V novém doporučeném postupu pro diagnózu je IPF definována jako specifická forma chronické progredující fibrotizující intersticiální pneumonie nejasné etiologie. Objevuje se primárně u dospělých jedinců, postihuje pouze plíce a IPF je spojená s histopatologickým a/ nebo radiologickým obrazem UIP. Diagnóza IPF vyžaduje vyloučení jiných forem intersticiálních pneumonií, zvláště ostatní idiopatické intersticiální pneumonie, systémové nemoci pojiva (SNP) a intersticiální plicní procesy (IPP) vzniklé v příčinné souvislosti s expozicí vlivům prostředí [9].
Epidemiologie IPF
Incidence IPF je odhadována mezi 6,8 a 16,3/ 100 000 (v USA). Prevalence této nemoci je celosvětově udávána v rozmezí 2–29/ 100 000, předpoklad prevalence vycházející z údajů databáze zdravotního pojištění v USA je však ještě vyšší (14–42,7/ 100 000) [10]. IPF postihuje tedy asi 5 milionů lidí na celém světě. Epidemiologické studie, které se uskutečnili v ČR, odhadují incidenci IPF maximálně na 1/ 100 000, odhadovaná prevalence se dle údajů z registru IPF EMPIRE v ČR pohybuje mezi 5–6/ 100 000 [11,12]. IPF nemá dle provedených výzkumů žádnou jednoznačnou geografickou, sociální a rasovou distribuci. Častěji postihuje muže než ženy. Obvykle se vyskytuje sporadicky, familiární případy jsou vzácné (5 %) [13]. Mortalita IPF je vysoká a kupříkladu v USA dosahuje 61,2 úmrtí / 1 000 000 obyvatel u mužů a 54,5/ 1 000 000 u žen. Příčinou smrti je nejčastěji progrese IPF (60 %), z dalších příčin je pak zaznamenána nemoc koronárních tepen, plicní embolie a rakovina plic [14].
Etiopatogeneze IPF
Poznání etiopatogeneze IPF se dynamicky vyvíjí a spolu s tím se objevují stále nové terapeutické cíle. Pravděpodobné je, že podkladem nemoci je uniformní patologická odpověď plicní tkáně na různá infekční i neinfekční agens, která způsobují opakované poškození výstelky plicních sklípků, vyúsťující v nekontrolovatelné a progredující jizvení. Z vyvolávajících faktorů, které způsobují léze alveolární výstelky, je důležité zmínit hlavně virové infekce, inhalaci organických a anorganických prachů, kouření a také reflux, který je v subklinické podobě přítomen až u 90 % pacientů s IPF. Ke spuštění fibroproliferativního hojení je pravděpodobně nezbytný genetický podklad (multifaktoriální s genetickými polymorfizmy genu pro mucin, surfaktantové proteiny, telomerázy aj.) a senescence, neboť IPF nepozorujeme u jedinců mladších 40 let [15–17].
Klinický obraz IPF
Na IPF musíme myslet v případě nově detegovaného IPP nejasného původu, u pacienta typicky staršího 60 let s poslechovým nálezem inspiračních chrůpků bilaterálně bazálně, s radiologickým obrazem (na skiagramu hrudníku a/ nebo HRCT) plicní fibrózy oboustranně. U mladších pacientů (40–60 let) se může obdobný klinicko-radiologický obraz objevit též, pak se může jednat i o familiární IPF. Typickým znakem IPF je progredující dušnost a u některých pacientů i kašel, pacienti ale mohou být v době diagnózy i asymptomatičtí, zvláště pak, je-li diagnóza stanovena časně. I když je pro IPF typický pozvolný a plíživý nástup dušnosti a její pozvolná progrese, u některých pacientů se vyskytnou epizody tzv. akutní exacerbace IPF charakterizované náhlým výrazným zhoršením dušnosti [7,9].
Fyzikální vyšetření
U 75 % pacientů se vyskytují fenotypové projevy jako jsou paličkovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomén krepitu slyšitelný nad plicními bazemi.
Funkční vyšetření
Základem je vyšetření ventilace metodou spirometrie a bodypletysmografie, difuzní kapacity a vyšetření krevních plynů. V počátku může být u IPF ventilace i difuze v normě, postupem času při fibrotizaci plicní tkáně dochází k poklesu difuzní kapacity, vitální kapacity (VC) a totální kapacity (TLC). Vzniká restrikční ventilační porucha a objevuje se obvykle hypoxemická respirační insuficience, zprvu pouze při zátěži, pak i klidová. Vyšetření plicních funkcí má význam hlavně pro sledování dynamiky poklesu VC a TLC pro stanovení prognózy nemoci a posouzení indikace transplantace plic a úhrad antifibrotické léčby. Vyšetření krevních plynů má zejména význam v rámci kyslíkového testu pro indikaci dlouhodobé domácí oxygenoterapie (viz doporučené postupy na www.pneumologie.cz) [18]. Při indikaci mobilního kyslíkového přístroje je pak nutné doplnit šestiminutový test chůzí.
Radiologické vyšetření
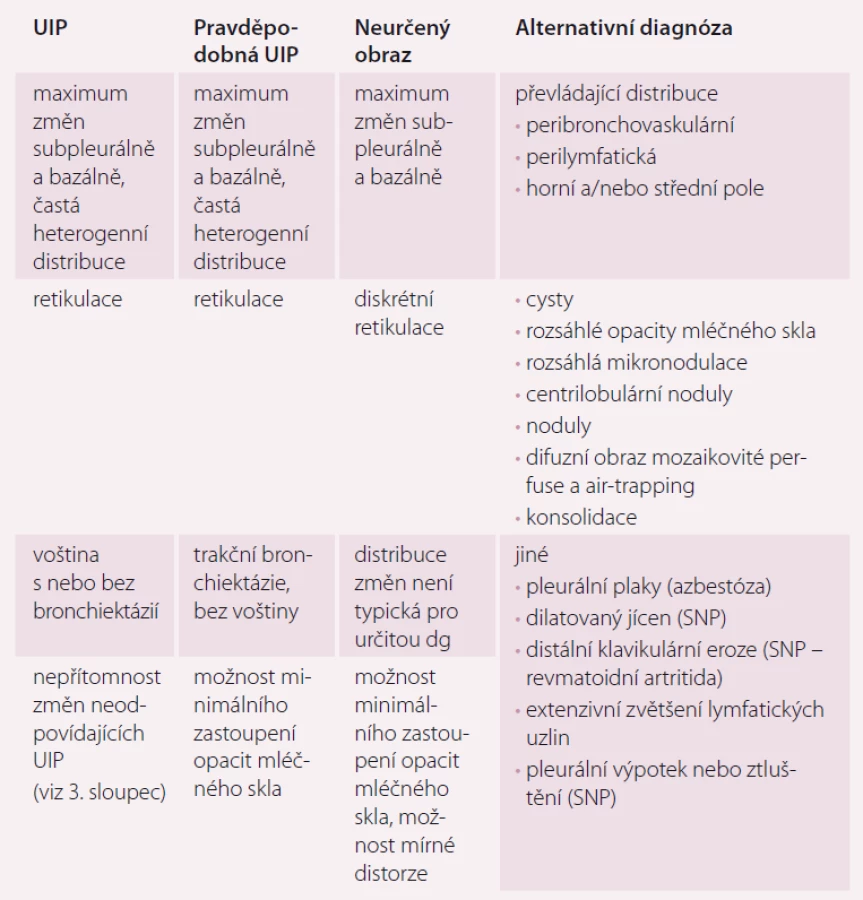
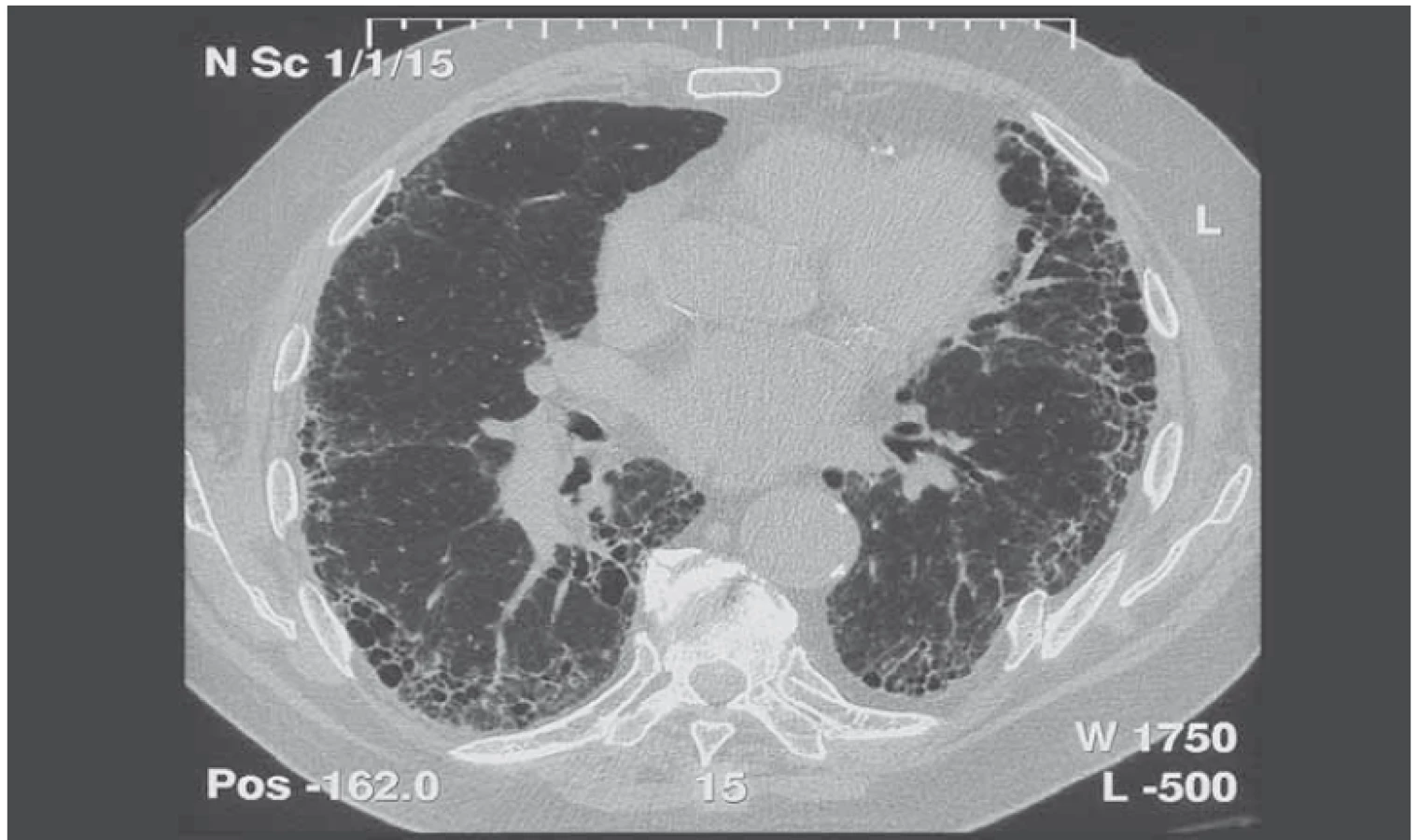
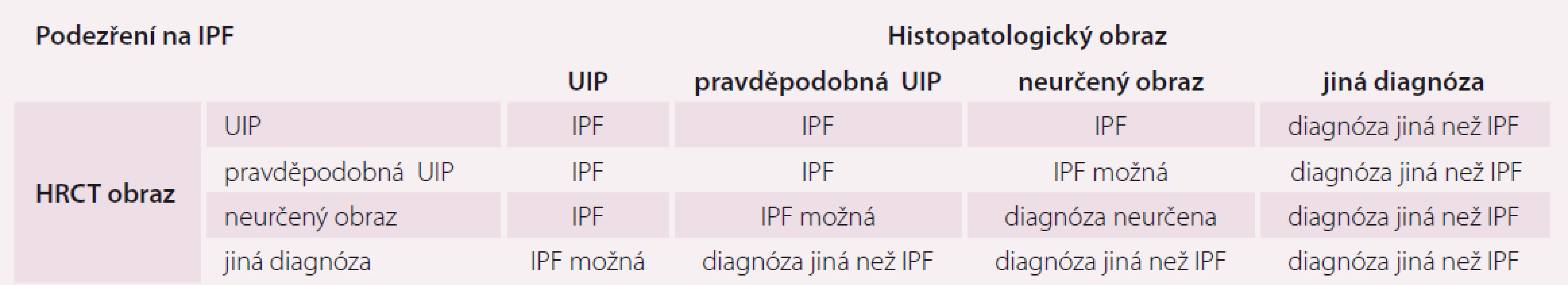
Vnímání radiologického obrazu IPF, tedy UIP, se změnilo a tyto změny se odrazily jak v publikovaném vyjádření Fleischnerovy společnosti, tak i v diagnostickém doporučení ATS/ ERS/ JRS/ ALAT z roku 2018. Nové rozdělení rozlišuje radiologické obrazy potenciálně kompatibilní s IPF na UIP, pravděpodobnou UIP, neurčený obraz a alternativní diagnózu (tab. 1 a obr. 1) [4].
Bronchoalveolární laváž
BAL je cenným pomocným diferenciálně diagnostickým nástrojem. Provedení BAL v době diagnózy je indikované u každého pacienta s podezřením na IPF, který nemá obecné kontraindikace BAL, případně neodmítá BAL. Pro IPF je typické zmnožení granulocytů obvykle s malou příměsí eozinofilů v tekutině získané BAL (BALTe), lymfocyty bývají zvýšeny minimálně. Pokud jsou lymfocyty v BALTe zvýšeny nad 20 %, je nutné uvažovat o exogenním původu IPP, případně o IPP v rámci SNP, a to i pokud by radiologický obraz odpovídal UIP.
Plicní biopsie
U pacientů s nově diagnostikovaným IPP s klinickým obrazem kompatibilním s IPF a typickým radiologickým obrazem UIP, bez nálezu jiné etiologie IPP není plicní biopsie doporučena. U pacientů s radiologickým obrazem pravděpodobné UIP s typickým klinickým obrazem a při vyloučení jiné etiologie onemocnění nepovažujeme též plicní biopsii za nutnou [8,9]. Plicní biopsie je doporučena v případě neurčeného obrazu či pokud radiologický obraz odpovídá spíše alternativní diagnóze. Plicní biopsii zvažujeme u každého pacienta individuálně, i s ohledem na celkový stav pacienta a jeho plicní funkce. Biopsie lze provést chirurgicky nebo cestou transbronchiální kryobiopsie (transbronchial lung cryobiopsy – TBLC) [19,20]. Kryobiopsii jako alternativa chirurgické biopsie je doporučeno provádět pouze v centrech, která mají příslušné personální i technické vybavení a zkušenosti s prováděním TBLC a s řešením případných komplikací.
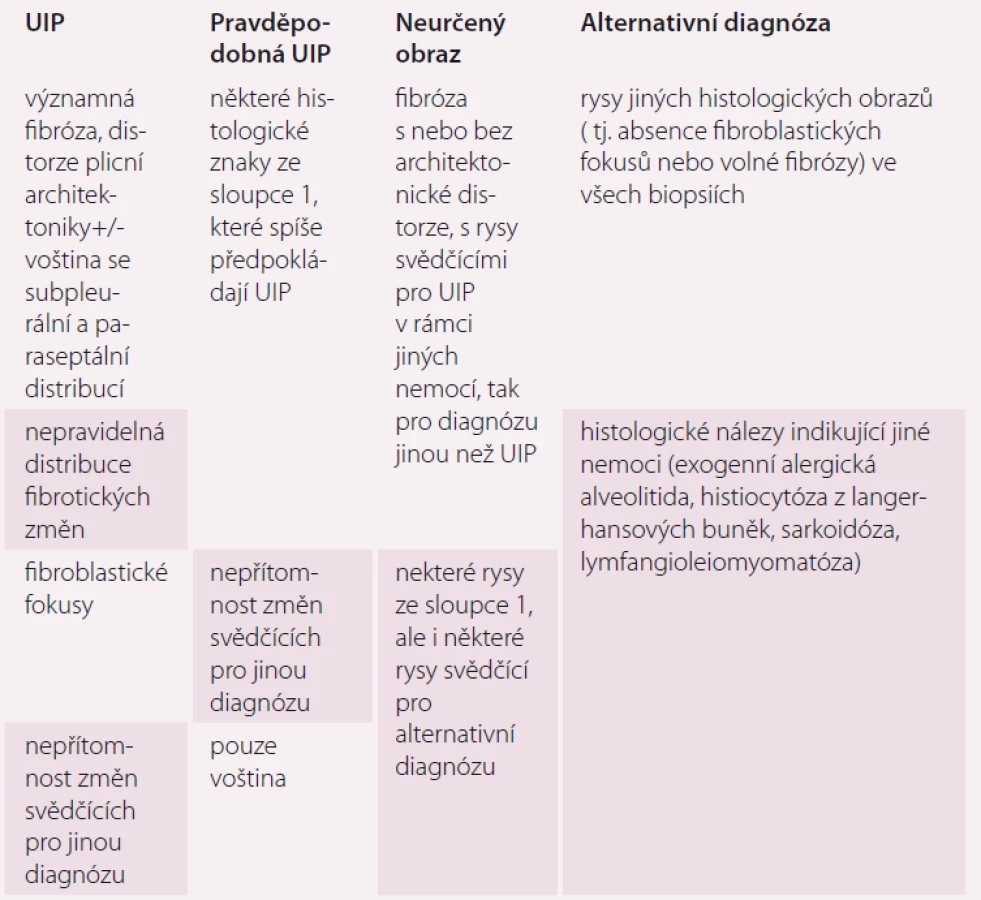
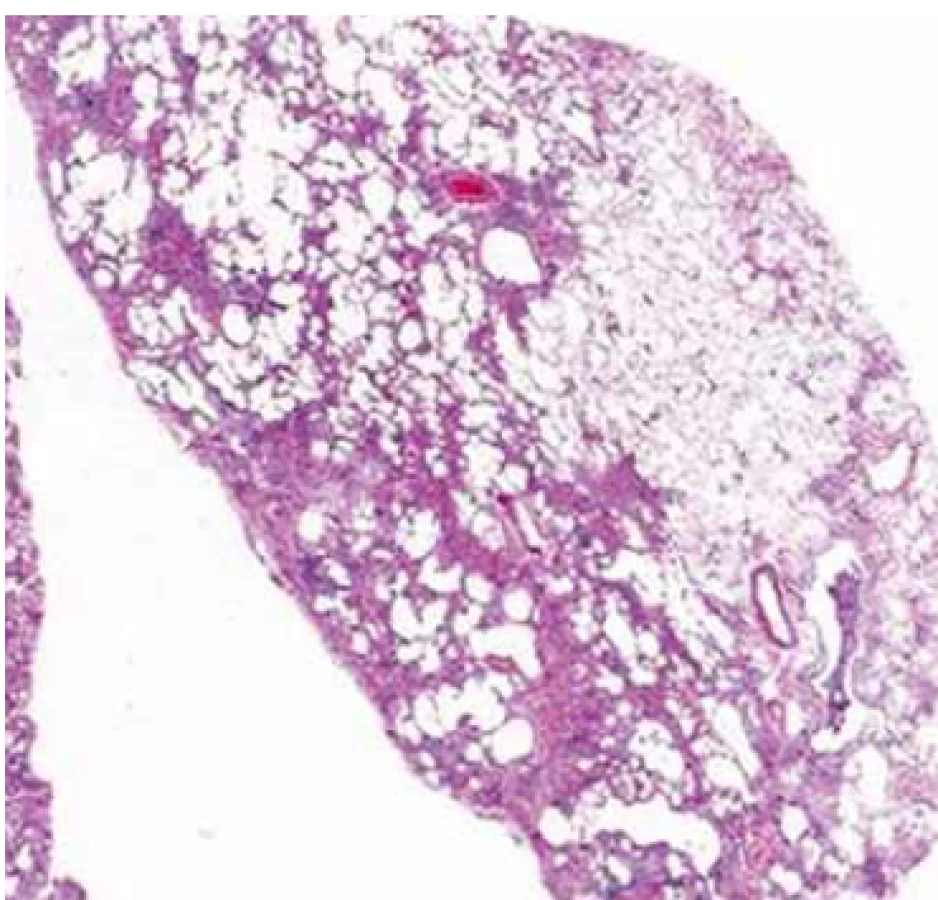
Hlavním histopatologickým znakem UIP je časově heterogenní vzhled s obrazem ložisek fibrózy a voštiny střídajícím obraz méně postižené nebo normální tkáně. Změny obvykle postihují subpleurální oblasti a paraseptální parenchym. Zánět je většinou minimální [9]. Diferenciální diagnostika není široká a zahrnuje hlavně exogenní alergickou alveolitidu (EAA), SNP a pneumokoniózy. Některé biopsie mohou vykazovat obraz tzv. neklasifikovatelné fibrózy, pokud však neobsahují změny typické pro jinou diagnózu (sarkoidózu, EAA, jiné), může být v případě typických klinických a radiologických změn po konzultaci MDT nález hodnocen jako IPF (tab. 1 a obr. 2). Někdy nemusí být diagnóza zcela jednoznačná a může být kupříkladu v plicních biopsiích z rozdílných plicních laloků nalezen obraz UIP v jednom laloku a fibrotické nespecifické intersticiální pneumonie (NSIP) v druhém laloku. V tomto případě se jedná o diskordantní UIP, na rozdíl od konkordantní UIP, kde všechny vzorky odpovídají UIP. Klinický obraz i průběh jsou u obou těchto variant stejné a pokud je odpovídající klinický obraz, jedná se o IPF.
Multidisciplinární tým
Pacienty s podezřením na IPF je doporučeno projednat v rámci MDT, a to optimálně před indikací plicní biopsie. Je možné, že i u pacientů s pravděpodobnou UIP či neurčeným obrazem bude možno takto rozhodnout o finální diagnóze, což může hrát výzanmnou úlohu zejména v individuálních situacích, kdy nelze provést plicní biopsii (pokročilý věk, špatné plicní funkce, komorbidity). Případně MDT rozhodne o tom, zda a jaká plicní biopsie má být provedena. S výsledkem biopsie je pak doporučeno pacienta probrat ještě jednou v rámci MDT, hlavně když diagnóza není definitivní ani s přihlédnutím k histopatologickému obrazu.
Diagnostická kritéria IPF
- Vyloučení jiných příčin intersticiálních plicních procesů (domácí a profesní expozice, systémové nemoci pojiva, léková toxicita).
- Přítomnost HRCT vzoru UIP u pacientů bez plicní biopsie (tab. 1).
- Specifické kombinace HRCT a histopatologického UIP vzorce u pacientů s plicní biopsií (tab. 3).
Diagnostika a léčba IPF v ČR má probíhat dle doporučení Sekce IPP ČPFS v Centrech pro diagnostiku a léčbu intersticiálních plicních procesů (Centra IPP), která jsou určena ČPFS a uznána i zdravotními pojišťovnami. Znamená to nejenom záruku kvality diagnostiky a péče, ale zároveň i možnost preskribce antifibrotik, která jsou vázána na Centra IPP. Pacient se suspektní IPF by měl tedy projít některým z Center IPP, aby byla určena správně diagnóza a doporučen další léčebný postup.
Farmakologická léčba
Základem farmakologické léčby je antifibrotická léčba.
Pirfenidon snižuje proliferaci fibroblastů a produkci s fibrózou asociovaných proteinů a cytokinů pravděpodobně inhibicí TGF-beta a destičkového růstového faktoru (platelet-derived growth factor – PDGF), a to cestou blokády nukleární translokace proteinu SMAD. Klinická účinnost léčby pirfenidonem byla prokázána v několika studiích, společné analýze multinárodních studií CAPACITY 004 a 006, japonské studii Shionogi (SP3) a následně ve studii ASCEND [5,6]. Pirfenidon významně snižuje rychlost poklesu plicních funkcí u pacientů s IPF, navíc i prodlužuje vzdálenost ušlou při šestiminutovém testu chůze (6MWD) a snižuje mortalitu ze všech příčin. Pirfenidon je hrazen z prostředků zdravotního pojištění pro nekouřící pacienty s IPF s VC mezi 50–90 % náležitých hodnot a TLC vyšší než 30 % náležitých hodnot, a to v Centrech pro diagnostiku a léčbu intersticiálních plicních procesů ČPFS (uvedeny na www.pneumologie.cz). Bohužel trvá omezení úhrady týkající se pacientů s poklesem plicních funkcí v 6měsíčních intervalech mezi jednotlivými měřením, kdy z úhrady vypadávají jedinci s poklesem VC > 10 % a TLC > 15 % oproti předchozím hodnotám. Toto úhradové omezení odporuje medicíně založené na důkazech, neboť pacienti, jejichž plicní funkce klesají rychleji, se mohou po vysazení antifibrotik ještě výrazněji zhoršit. Pacienti indikovaní k léčbě pirfenidonem zahajují 1.–7. den 3 × 1 kapslí po 267 mg, 8.–15. den pak zvyšují dávku na 3 × 2 kapsle a od 16. dne pak užívají 3 × 3 kapsle (2 403 mg pirfenidonu/ den), při dobré toleranci je možný přechod na variantu 801 mg 1-1-1. Pacienti, kteří mají výrazné nežádoucí gastrointestinální účinky (obvykle nauzeu a nechutenství) mohou mít redukovanou dávku pirfenidonu na nejvyšší tolerovanou (kupříkladu 3 × 2 nebo pouze 3 × 1 kapsle po 267 mg/ den). Také v případě fotosenzitivní reakce či významné elevace transamináz je zpravidla nutná přechodná redukce dávky léku či jeho úplné přechodné vysazení. Pacient musí být při zahájení léčby seznámen s rizikem fototoxicity léku a s nutností soustavné ochrany proti slunečnímu záření (celoročně krémy s vysokým ochranným faktorem, tzv. sun-blockery na nechráněná místa kůže, a pokrývka hlavy a ochrana těla oděvem při riziku oslunění). Také musí vědět o gastrointestinálních nežádoucích účincích, hlavně o nevolnosti a nechuti k jídlu a o možnosti snížit dávku léku při jejich výskytu. Musí být poučeni i o možnosti poruchy funkce jater při léčbě pirfenidonem a nutnosti opakovaných odběrů krve v průběhu léčby s kontrolou jaterních testů. Vzhledem k tomu, že tabákový kouř je induktorem CYP1A2, čímž ovlivňuje významně farmakokinetiku, a tím biologickou dostupnost (expozici) pirfenidonu, pacienti léčení pirfenidonem by neměli kouřit. Navíc nekouření je samo o sobě jednou z podmínek úhrady pirfenidonu v ČR (viz výše). Zvýšené opatrnosti je potřeba při současné léčbě léky snižujícími aktivitu CYP1A2, kdy může dojít až k významné toxicitě pirfenidonu (např. fluvoxamin, který je při léčbě pirfenidonem kontraindikován, chinolony). Naopak léky zvyšující aktivitu CYP1A2 mohou snižovat účinnost léčby pirfenidonem (rifampicin, omeprazol).
Nintedanib je dalším lékem, který je dostupný pro léčbu IPF v ČR. Jedná se o trikinázový inhibitor – inhibice receptoru pro růstový faktor fibroblastů (fibroblast growth factor – FGF), vaskulární endoteliální růstový faktor (vascular endothelial growth factor – VEGF) a od destiček odvozený růstový faktor (platelet derived growth factor – PDGF), který v dávce 150 mg 2× denně perorálně prokazatelně snižuje pokles plicních funkcí u pacientů s IPF. Efektivita a bezpečnost nintedanibu v léčbě IPF byly prokázány ve dvou velkých studiích fáze 3 INPULSIS-1 a INPULSIS-2, do kterých bylo zařazeno 1 066 pacientů s IPF [21]. Nintedanib prokázal v obou těchto studiích efektivitu vyjádřenou významnou redukcí poklesu VC a navíc v INPULSIS-2 i benefit ve smyslu redukce počtu a doby do výskytu akutních exacerbací IPF. Nintedanib je v ČR hrazen u pacientů s VC 50–90 % NH (normální hodnota) a TLC vyšší než 30 % NH.I pro nintedanib platí ukončení úhrady léku při poklesu plicních funkcí tak jako pro pirfenidon. Lék je pro perorální užití, ve formě kapslí, užívá se 2 × 1 kapsle po 150 mg denně (přibližně v intervalu 12 hod). Při intoleranci je možné dávku snížit na 2 × 100 mg. Nejčastějšími nežádoucími účinky léčby jsou průjmy a elevace jaterních enzymů. Frekvenci stolic lze snížit podáním loperamidu. Při elevaci transamináz lze při významném vzestupu přechodně přerušit léčbu, častěji však postačí většinou přechodné snížení dávky na 2 × 100 mg denně.
Klinické studie
Je doporučeno zařazovat pacienty s IPF do klinických studií s novými léky, a to zejména ty, kteří nedosáhnou na kritéria úhrady antifibrotické léčby, a i ty, kteří jsou již na léčbě antifibrotiky a studijní lék je podáván jako lék přidaný do kombinace.
Další léky
Ne všichni pacienti s IPF dosáhnou na antifibrotickou léčbu dle kritérií úhrady, případně ji netolerují. I s ohledem na tento fakt se členové Sekce IPP dohodli na základě důkazů ze studie s N-acetylcysteinem, že je možné podávat N-acetylcystein nemocným s IPF v dávce 1 800 mg/ den p.o. v případě, že jsou vyšetřeny polymorfizmy TOLLIP a nemocný má přítomen polymorfizmus, který predikuje pozitivní efekt léku na pokles plicních funkcí [22].
Z léků je dále možno použít v rámci symptomatické léčby dušnosti a kašle u IPF opiáty, systémové perorální kortikosteroid v nízké dávce a antitusika v obvyklých dávkách. Stran léčby inhibitory protonové pumpy, nebyla prokázána jejich efektivita na pokles plicních funkcí u IPF, a proto je tato léčba doporučena pouze u pacientů se symptomatickou refluxní chorobou.
Nefarmakologická léčba
Dlouhodobá domácí oxygenoterapie je doporučena pro všechny pacienty s IPF s klidovou hypoxemií, kteří splní kritéria ČPFS po přidělení přístroje.
Transplantace plic
Transplantace je pro vhodně vybrané pacienty s IPF rozhodně doporučena. Pětileté přežití po transplantaci plic u pacientů s IPF je odhadováno na 50–56 %. Transplantace plic u adekvátně indikovaných pacientů s IPF snižuje významně riziko úmrtí v 5. roce po transplantaci. Navíc pacienti s IPF mají lepší dlouhodobé přežití po transplantaci plic než ti transplantovaní pro jinou diagnózu. Je nutné pacienta s IPF zvážit pro transplantační léčbu včas, aby byl dostatek času pro vyšetření před zařazením na čekací listinu.
Umělá plicní ventilace
Pacienti s IPF by při respiračním selhání způsobeném IPF neměli být paušálně invazivně uměle ventilováni, ale tento způsob léčby může být vhodný pro některé selektované jedince. Mortalita při umělé plicní ventilaci (UPV) u pacientů s IPF je 96 %. Většinou pouze ti pacienti, kteří mohou být transplantováni z ventilátoru, mají z UPV profit. V praxi to znamená, že by invazivní UPV měla být indikována pro zhoršení IPF s respiračním selháním pouze v případě nemocných, kteří jsou již zařazeni na čekací listině transplantace plic, nebo u kterých existuje šance, že se podaří je urgentně zařadit k transplantaci plic. I tak je pravděpodobnost úspěšné transplantace, a tudíž přežití těchto pacientů, velmi malá.
Rehabilitace
Pacienti s IPF by měli být indikováni k plicní rehabilitaci ve většině případů. Jedna z možností, jak zlepšit kvalitu života pacienta, je zlepšení pacientovy funkční výkonnosti a zmírnění dušnosti. Rehabilitace musí být v případě IPF komplexní, zahrnující učení, poradu a behaviorální techniky ke zlepšení sebeobsluhy, redukci symptomů a optimalizaci funkční kapacity [23].
Léčba komplikací a komorbidit
Léčba akutní exacerbace IPP
Většina pacientů by měla být v době exacerbace léčena systémovými kortikosteroidy a antibiotiky (podrobněji viz standard Akutní exacerbace IPP v Sekci pro intenzivní pneumologii ČPFS) [24].
Léčba plicní hypertenze
U pacientů s IPF by plicní hypertenze neměla být paušálně léčena, nicméně u některých je vhodné tuto léčbu zvážit. Jedná se hlavně o pacienty s dysproporční plicní hypertenzí, která svou závažností neodpovídá rozsahu plicního postižení při IPF. Základním screeningem plicní hypertenze u IPF je echokardiografie. Podezření na plicní hypertenzi však může vzejít již z funkčního vyšetření plic (poměr VC%NH/ TLC%NH > 1,8 – vysoká pravděpodobnost postižení plicní cirkulace), případně z vyšetření spiroergometrie a laboratorního vyšetření (mozkový natriuretický peptid – BNP). Definitivně pak stanoví plicní arteriální hypertenzi pravostranná srdeční katetrizace.
Monitorace klinického průběhu onemocnění a sledování efektu léčby
K monitorování klinického průběhu je doporučováno sledování symptomů, vyšetření plicních funkcí a sledování progrese fibrózy na HRCT.
Pokud není známa jiná příčina, která by vedla ke zhoršení stavu, je nutno jakoukoliv příhodu z níže uvedených brát jako progresi IPF:
- progredující dušnost,
- progredující pokles VC oproti výchozí hodnotě,
- progredující pokles transfer faktoru oproti výchozí hodnotě,
- progrese plicní fibrózy na HRCT oproti výchozímu stavu,
- akutní exacerbace,
- smrt na respirační selhání.
V současné době je jako progrese onemocnění označován pokles celkového objemu vzduchu, který umí pacient vydechnout (FVC) o 10 % absolutních hodnot a pokles transfer faktoru o 15 % absolutních hodnot. Interval sledování je doporučen 3–6měsíční, spočívá ve funkčním (spirometrie, transfer faktor, krevní plyny) a klinickém vyšetření pacienta, HRCT hrudníku není třeba zhotovovat při každé z těchto kontrol, pokud nedojde k významné změně klinického obrazu a funkčních parametrů. Pokud máme však podezření na tzv. akutní exacerbaci IPF, je provedení HRCT hrudníku na prvním místě. V případě podezření na infekci či nádor v terénu IPF jsou další vyšetřovací postupy včetně radiologického vyšetření samozřejmostí.
Staging a prognóza
Rozsah nemoci a závažnost funkčního postižení v době diagnózy je mezi jednotlivými pacienty s IPF značně variabilní. Je to dáno jednak odlišným stupněm vnímání obtíží u jednotlivých nemocných a pak také stupněm povědomí lékařské veřejnosti o této nemoci. Stran prognózy je nutné identifikovat pacienty s vysokým rizikem úmrtí v následujících 2 letech, aby mohlo být u těchto nemocných včas zváženo zařazení na čekací listinu transplantace plic (tab. 4).

Kompozitní skórovací systémy
Pro určení závažnosti postižení při IPF byl vytvořen Kompozitní fyziologický index (Composite Physiologic Index), který využívá hodnot FVC, FEV1 (objem úsilného výdechu za 1 s) a transfer faktoru k předpovědi rozsahu nemoci na HRCT. Je silnějším prediktivním faktorem než jednotlivé funkční parametry a dokonce než jiné složené indexy, jako je CRP (clinical-radiographic-physiological scoring system), vč. jeho nových variant. Klinické užití kompozitních systémů však není rozšířeno a jejich užitečnost pro klinickou praxi je zatím nejasná [25].
Biomarkery v séru a BALTe
Zatím neexistuje dostatek údajů pro užitečnost biomarkerů v klinickém sledování IPF, nicméně některá pracoviště již biomarkery používají, hlavně pro předpověď prognózy pacienta a monitoraci rizika vzniku akutního zhoršení.
Krebs von den Lungen-6 (KL-6) patřící mezi muciny je produkován regenerujícími pneumocyty II. typu a je zvýšen v séru a BALTe u pacientů s IPF. Jeho hodnota koreluje s mortalitou pacientů s IPF. Také sérové hladiny surfaktantových proteinů A a C (SP-A,C), chemokin CCL-18,BNP a matrix metaloproteináza 7 (MMP-7) mohou mít prediktivní hodnotu pro přežití u těchto pacientů. Z buněčných parametrů mohou být cirkulující fibrocyty v periferní krvi negativním prognostickým faktorem pro krátkodobé přežití u pacientů s IPF. Nicméně rutinně tyto markery nelze doporučit pro sledování a léčebná rozhodnutí u pacientů s IPF [26].
Doručeno do redakce: 2. 8. 2019
Přijato po recenzi: 14. 8. 2019
prof. MU Dr. Martina Vašáková, Ph.D
Sources
1. American Thoracic Society, European Respiratory SocietyAmerican Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med 2000; 161(2 Pt 1): 646–664. doi: 10.1164/ ajrccm.161.2.ats3-00.
2. Raghu G, Collard HR, Egan JJ et al. ATS/ ERS/ JRS/ ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ ERS/ JRS/ ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6): 788–824. doi: 10.1164/ rccm.2009-040GL.
3. Demedts M, Behr J, Buhl R et al. IFIGENIA Study Group. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005; 353(21): 2229–2242. doi: 10.1056/ NEJMoa042976.
4. McGrath EE, Millar AB. Hot off the breath: triple therapy for idiopathic pulmonary fibrosis-hear the PANTHER roar. Thorax 2012; 67(2): 97–98. doi: 10.1136/ thoraxjnl-2011-201398.
5. Noble PW, Albera C, Bradford WZ et al. CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779): 1760–1769. doi: 10.1016/ S0140-6736(11)60405-4.
6. King TE Jr, Bradforf WB, CastroBernardini S et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2083–2092. doi: 10.1056/ NEJMoa1402582.
7. Raghu G, Rochwerg B, Zhang Y et al. American Thoracic Society; European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association. An Official ATS/ ERS/ JRS/ ALAT Clinical Practice Guideline: treatment of idiopathic pulmonary fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015; 192(2): e3–e19. doi: 10.1164/ rccm.201506-1063ST.
8. Lynch DA, Sverzellati N, Travis WD et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med 2018; 6(2): 138–153. doi: 10.1016/ S2213-2600(17)30433-2.
9. Raghu G, Remy-Jardin M, Myers JL et al. American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ ERS/ JRS/ ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2018; 198(5): e44–e68. doi: 10.1164/ rccm.201807-1255ST.
10. Raghu G, Chen SY, Hou Q et al. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J 2016; 48(1): 179–186. doi: 10.1183/ 13993003.01653-2015.
11. Homolka J, Altmann V, Votava V. Increasing prevalence of idiopathic pulmonary fibrosis in the Czech Republic. Chest 1999; 116 (Suppl 2): 155.
12. European MultiPartner IPF Registry. Available at: http:/ / empire.registry.cz/ index-en.php.
13. Ravaglia C, Tomassetti S, Gurioli C et al. Features and outcome of familial idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2014; 31(1): 28–36.
14. Dove EP, Olson AL, Glassberg MK. Trends in IPF-related mortality in the United States: 2000–2017. Am J Respir Crit Care Med 2019. doi: 10.1164/ rccm.201905-0958LE.
15. Kropski JA, Blackwell TS, Loyd JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J 2015; 45(6): 1717–1727. doi: 10.1183/ 09031936.00163814.
16. Peljto AL, Zhang Y, Fingerlin TE et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013; 309(21): 2232–2239. doi: 10.1001/ jama.2013.5827.
17. Wolters PJ, Blackwell TS, Eickelberg O et al. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir Med 2018; 6(2): 154–160. doi: 10.1016/ S2213-2600(18)30007-9.
18. Doporučené postupy. Dostupné na: www.pneumologie.cz.
19. Lentz RJ, Argento AC, Colby TV et al. Transbronchial cryobiopsy for diffuse parenchymal lung disease: a state-of-the-art review of procedural techniques, current evidence, and future challenges. J Thorac Dis 2017; 9(7): 2186–2203. doi: 10.21037/ jtd.2017.06.96.
20. Iftikhar IH, Alghothani L, Sardi A et al. Transbronchial lung cryobiopsy and video-assisted thoracoscopic lung biopsy in the diagnosis of diffuse parenchymal lung disease. A Meta-analysis of Diagnostic Test Accuracy. Ann Am Thorac Soc 2017; 14(7): 1197–1211. doi: 10.1513/ AnnalsATS.201701-086SR.
21. Richeldi L, du Bois RM, Raghu G et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2071–2082. doi: 10.1056/ NEJMoa1402584.
22. Oldham JM, Ma SF, Martinez FJ et al. IPFnet Investigators.TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2015; 192(12): 1475–1482. doi: 10.1164/ rccm.201505-1010OC.
23. Swigris JJ, Brown KK, Make BJ et al. Pulmonary rehabilitation in idiopathic pulmonary fibrosis: a call for continued investigation. Respir Med 2008; 102(12): 1675–1680. doi: 10.1016/ j.rmed.2008.08.014.
24. Standard Akutní exacerbace IPP v Sekci pro intenzivní pneumologii ČPFS. Dostupné na: http: / / www.pneumologie.cz/ stranka/ 58/ sekce-pro-intenzivni-pneumologii.
25. Tomassetti S, Ryu JH, Poletti V. Staging systems and disease severity assessment in interstitial lung diseases. Curr Opin Pulm Med 2015; 21(5): 463–439. doi: 10.1097/ MCP.0000000000000198.
26. Organ LA, Duggan AR, Oballa E et al. Biomarkers of collagen synthesis predict progression in the PROFILE idiopathic pulmonary fibrosis cohort. Respir Res 2019; 20(1): 148. doi: 10.1186/ s12931-019-1118-7.
Labels
Paediatric cardiology Internal medicine Cardiac surgery Cardiology Pneumology and ftiseology General practitioner for adults RadiodiagnosticsArticle was published in
Cardiology Review

2019 Issue 3
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
Most read in this issue
- Kardiovaskulární působení rekreačních drog (kokain, marihuana, metamfetamin)
- Sarkoidóza – aktuální pohled na patogenezi, diagnostiku a léčbu
- Epidemiologie tuberkulózy
- PCSK9 inhibitory v klinické praxi – update