
Hsp90 – cíl protinádorové terapie
Hsp90 – a Target for Anticancer Therapy
Molecular chaperones help other proteins to achieve and maintain their proper conformation. Chaperones bind to newly synthesized or unfolded polypeptide chains, actively modify their conformation and participate on their transport or degradation. Chaperones play an important role in cancer cell, where their increased activity enables stabilization of many mutant proteins and overcoming the stress generated by genetic instability. Hsp90 represents a key chaperone in cancer cells. Growth factor receptors, steroid hormone receptors and signal proteins are among its substrates, so-called client proteins; many of them being targets for anti-cancer therapy. Adverse conditions of the tumor microenvironment, such as hypoxia and nutrient deficiency, contribute to destabilization of proteins and further escalate dependence on chaperones. This is why molecular chaperones, in particular Hsp90, may represent a promising target for anticancer therapy. Importantly also, tumour-based Hsp90 has a significantly higher sensitivity to inhibitors than that in normal cells, and Hsp90 activity inhibition in tumours leads to a suppression of cellular signaling in many different oncogenic pathways. Several inhibitors of Hsp90 are currently undergoing clinical evaluation and new agents with different mechanisms of action are continually being identified.
Key words:
molecular chaperones – Hsp90 – cancer – therapy
This work was supported by grants of MZCR NS/9812-4 and RECAMO CZ.1.05/2.1.00/03.0101.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
18. 4. 2011
Accepted:
25. 5. 2011
Authors:
E. Růčková; P. Müller; B. Vojtěšek
Authors‘ workplace:
Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2011; 24(5): 329-337
Category:
Reviews
Overview
Molekulární chaperony jsou proteiny, které se podílejí na vytváření a udržování správné konformace ostatních proteinů v buňce. Vážou se na nově syntetizované nebo denaturované polypeptidové řetězce, aktivně mění jejich konformaci a podílejí se na jejich transportu nebo degradaci. Chaperony hrají velmi důležitou roli v nádorové buňce, kde jejich zvýšená aktivita umožňuje stabilizovat řadu mutantních proteinů a překonávat stres vzniklý genetickou nestabilitou. Klíčovým chaperonem nádorových buněk je Hsp90, mezi jehož klienty náleží receptory růstových faktorů, steroidních hormonů a signální proteiny, z nichž řada patří mezi cíle protinádorové terapie. Nepříznivé podmínky mikroprostředí nádoru, jako jsou hypoxie a nedostatek živin, přispívají ke zvýšené destabilizaci proteinů, čímž závislost na chaperonech ještě dále prohlubují. Z těchto důvodů představují molekulární chaperony, a zvláště Hsp90, nadějný cíl protinádorové terapie. Hsp90 je dále výjimečný tím, že v nádorových buňkách vykazuje výrazně vyšší citlivost k inhibitorům oproti normálním buňkám a inhibice Hsp90 v nádorech vede k paralelnímu potlačení různých drah onkogenní signalizace. V současné době probíhají klinické testy několika inhibitorů Hsp90 a jsou stále identifikovány nové látky s různým mechanizmem účinku.
Klíčová slova:
molekulární chaperony – Hsp90 – nádorová onemocnění – terapie
Úvod
Přestože je konformace proteinů dána pořadím aminokyselin a je utvářena podle zákonů termodynamiky, je v intracelulárním prostředí pro vytvoření správné konformace vyžadována spoluúčast specializovaných komplexů, molekulárních chaperonů. Je to způsobeno tím, že koncentrace proteinů v savčích buňkách dosahuje velmi vysokých hodnot, a neustále tak dochází k nežádoucím interakcím polárních řetězců peptidových vazeb a hydrofobních aminokyselinových zbytků nově syntetizovaných i částečně denaturovaných proteinů. Tím by mohlo docházet k jejich nesprávnému sbalování či spojování do nefunkčních agregátů. Přechodná vazba molekulárních chaperonů tedy poskytuje vznikajícím proteinům ochranu a umožňuje sbalení do příslušné terciární struktury [1]. Molekulární chaperony také úzce spolupracují s ubikvitin-proteazomovým systémem, který denaturované proteiny odbourává, stejně jako se systémem transportujícím proteiny na místo určení. Dohromady tento aparát zajišťující homeostázu proteinů v buňce tvoří rozsáhlou a strukturně různorodou skupinu proteinů. Exprese řady molekulárních chaperonů je výrazně zvýšena působením vyšších teplot. Proto jsou nazývány proteiny tepelného šoku (Heat Shock Proteins – Hsp) a jejich označení je často tvořeno zkratkou Hsp a číslem udávajícím molekulovou hmotnost. K indukci exprese chaperonů však dochází také působením nepříznivých vlivů vnějšího i vnitřního prostředí [2].
Aktivita většiny chaperonů závisí na hydrolýze ATP, přičemž jejich struktura a mechanizmus aktivity se mezi jednotlivými třídami výrazně liší. Chaperony se dělí do čtyř tříd. Třída chaperoninů zahrnuje oligomerní molekuly GroEL a TRiC (TCP1 Ring Complex) vytvářející velké soudkovité útvary, uvnitř kterých jsou uzavřeny celé proteiny nebo jejich domény, a je jim tak poskytnuta ochrana pro vytvoření správné konformace [3]. Chaperony třídy Hsp70 jsou monomerní a vyžadují spolupráci ko-chaperonů třídy Hsp40. Své klientní proteiny rozpoznávají podle krátkých lineárních hydrofobních úseků. Hsp70 slouží jako chaperon velkému množství klientních proteinů, některé z nich jsou však poté předány k dalšímu zpracování chaperonům třídy Hsp90 [4]. Chaperony třídy malých Hsp tvoří oligomerní komplexy, které se vážou na denaturované proteiny a zabraňují jejich nevratné agregaci. Malé Hsp interagují se svými substráty bez účasti ATP, ale opětovné vytvoření správné konformace vyžaduje zapojení dalších chaperonů [5]. Kromě výše zmíněných chaperonů se širokým spektrem klientních proteinů existují i specializované chaperony podílející se na utváření konformace jediného specifického proteinu nebo multi-proteinových komplexů. Byly popsány chaperony účastnící se sestavení nukleozomu [6] nebo proteazomu [7]. Mezi nejvýznamnější chaperony patří bezesporu Hsp90. Za normálních nestresových podmínek tvoří 1–2 % celkových buněčných proteinů a v případě stresu jeho exprese dále prudce narůstá. Hsp90 je nejen nezbytnou složkou obrany před poškozením buňky teplotním stresem, ale i za fyziologických podmínek se účastní sbalování řady proteinů do funkční konformace. Poprvé byla interakce Hsp90 s proteiny objevena během afinitní purifikace prvního molekulárně charakterizovaného onkogenu, kinázy v-Src (Viral-Sarcoma), kdy Hsp90 precipitoval společně s v-Src z kuřecích buněk nakažených virem Rousova sarkomu [8]. Následně byly popsány dvě hlavní třídy klientních proteinů Hsp90, proteinkinázy a receptory steroidních hormonů a řada dalších klientních proteinů s funkcí transkripčních faktorů, chromatin remodelujících proteinů a polymeráz [2].
Struktura a funkce Hsp90
Hsp90 je evolučně konzervovaný, přičemž jeden nebo více genů kódujících Hsp90 nese ve svém genomu všechny organizmy od bakterií až po savce. Hsp90 se nachází téměř ve všech kompartmentech eukaryotických buněk a u většiny eukaryot navíc existují příbuzné chaperony specifické pro dané organely. Jedná se např. o protein GRP94 (Glucose Related Protein 94) exprimovaný v endoplazmatickém retikulu nebo TRAP1 (TNF Receptor Associated Protein 1) v mitochondriích. Nejvíce poznatků bylo získáno o proteinech Hsp90 lokalizovaných v cytosolu. U obratlovců byly nalezeny dvě izoformy, Hsp90α a Hsp90β, které se navzájem liší funkcí, buněčnou lokalizací i mírou inducibility [9]. Izoforma Hsp90α se nachází i na buněčném povrchu a je také secernována do extracelulárního prostoru [10]. O úloze extracelulárního Hsp90 zatím není získáno mnoho poznatků, ale předpokládá se, že plní funkci molekulárního chaperonu i vně buňky. Na buněčném povrchu byl Hsp90 nalezen v komplexu s MMP2 (Matrix Metalloproteinase 2), která se podílí na degradaci extracelulární matrix. Působením inhibitoru Hsp90 modifikovaného tak, aby nepronikal dovnitř buňky, nebo inaktivací Hsp90 neutralizující protilátkou bylo pozorováno snížení aktivity MMP2 a invazivnosti nádorových buněk [11].
Hsp90 se nachází v cytoplazmě ve formě dimeru, přičemž každý monomer je složen ze tří funkčních domén: N-koncové, centrální a C-koncové. N-koncová doména obsahuje adenosintrifosfát (ATP) vazebnou kapsu. Motiv vazebné kapsy vykazuje strukturní homologii s ATPázovými doménami DNA-gyrázy, histidin kinázy a proteinu systému reparace DNA MutL. Tento motiv není příbuzný s ATP-vazebnou doménou proteinkináz ani Hsp70, což otvírá možnost využití vysoce specifických inhibitorů [12]. N-koncová doména je s centrální doménou flexibilně spojena, což umožňuje konformační změny proteinu po vazbě ATP a jeho hydrolýze na ADP. Ty mají zásadní význam pro aktivitu dimeru Hsp90 jako molekulárního chaperonu [13]. Hlavní úlohou centrální domény je zřejmě rozlišení typů klientních proteinů [14]. C-koncová doména je především zodpovědná za dimerizaci [15] a nachází se zde také konzervovaný motiv MEEVD (tvořený aminokyselinami Met-Glu-Glu-Val-Asp), který zprostředkovává interakce s ko-chaperony obsahujícími TPR (Tetratricopeptide Repeat) doménu [16].
Samostatná ATPázová aktivita Hsp90 je pouze slabá a je ovlivněna vazbou klientních proteinů a interakcemi s určitými ko-chaperony. U eukaryot bylo identifikováno více než 20 ko-chaperonů, ale jejich funkce je v mnoha případech dosud neznámá. Ko-chaperony modulují aktivitu Hsp90 čtyřmi způsoby:
- a) koordinují interakce s dalšími chaperonovými systémy,
- b) stimulují nebo inhibují ATPázovou aktivitu,
- c) zprostředkovávají interakce s různými typy klientních proteinů,
- d) ovlivňují svou enzymatickou aktivitou další aspekty chaperonového cyklu.
Největší skupinou jsou ko-chaperony s TPR doménou, která interaguje s MEEVD motivem v C-koncové doméně Hsp90. Patří mezi ně např. ko-chaperony HOP (Hsp90/Hsp70 Organizing Protein), CHIP (C-terminus of Hsc70-Interacting Protein), FKBPs (FK506-Binding Proteins), CyP40 (Cyclophilin40) a PP5 (Protein Phosphatase 5). S centrální doménou Hsp90 interaguje ko-chaperon AHA1 (Activator of Hsp90 ATPase homologue 1) a na N-koncovou doménu se vážou např. Cdc37 (Cell Division Cycle 37), p23 a SGT 1 [2]. Interakce některých ko-chaperonů s Hsp90 jsou schematicky znázorněny na obr. 1.
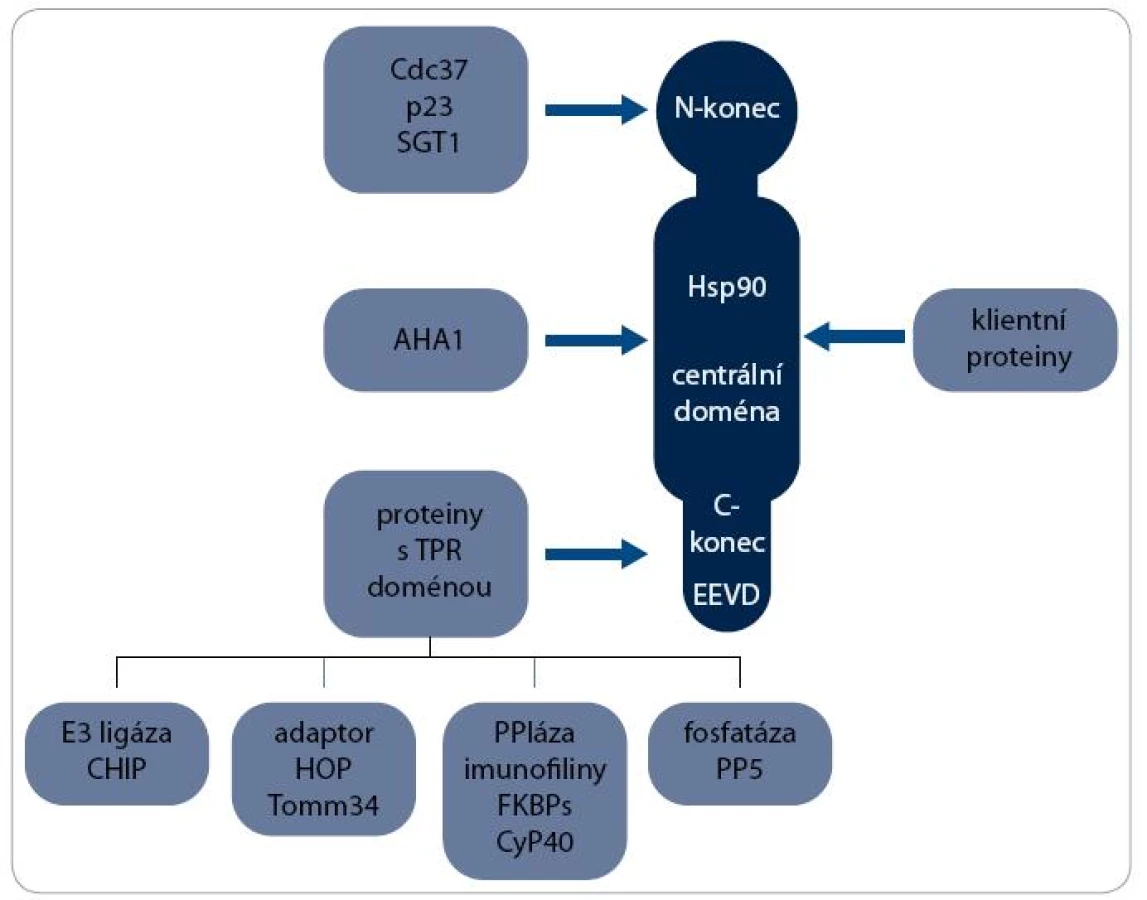
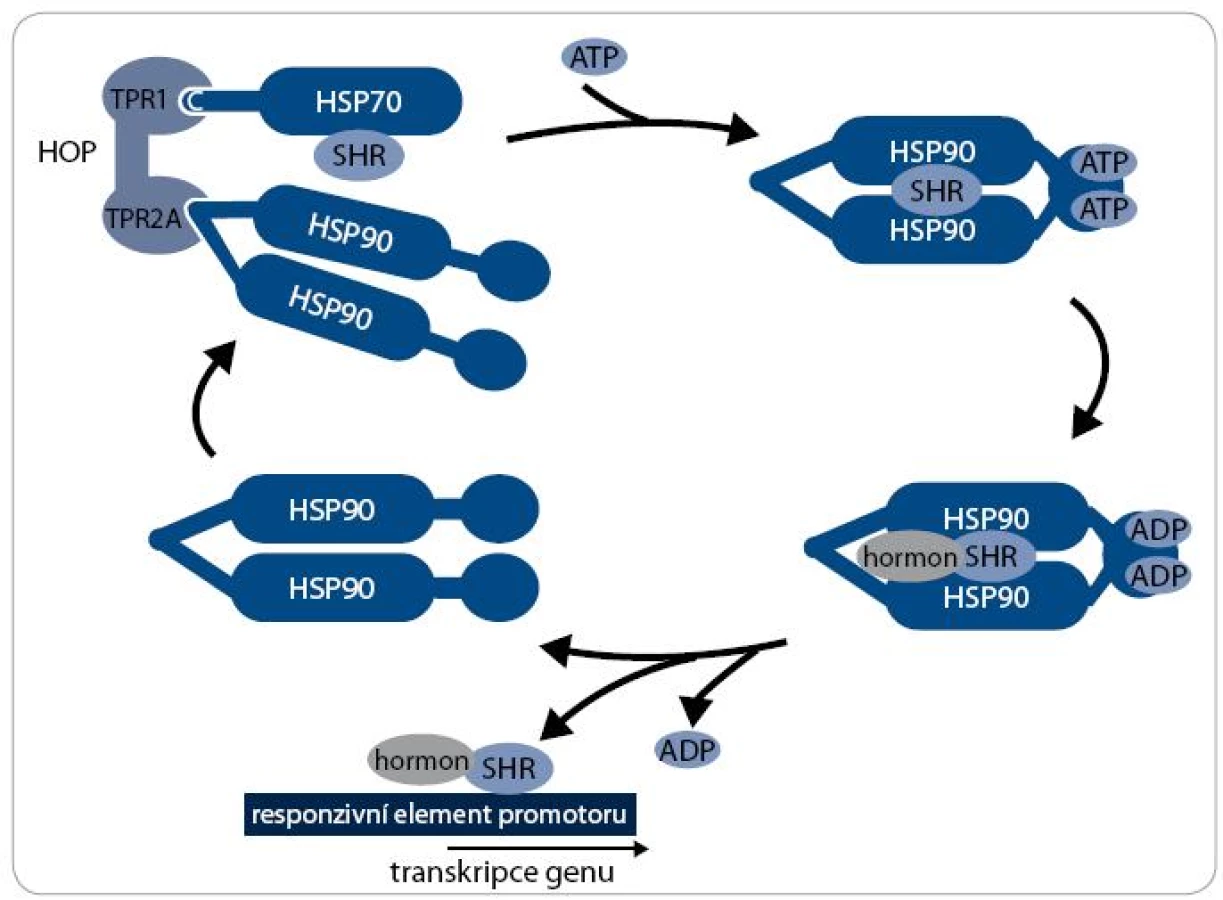
Mechanizmus aktivity multi-chaperonového systému Hsp90 je nejlépe prostudován při procesu maturace receptorů steroidních hormonů (SHR) – viz obr. 2. Klientní protein je vázán pouze na dimer Hsp90 v otevřeném stavu, kdy jsou jeho N-koncové domény separovány. Chaperonový cyklus začíná navázáním nově syntetizovaného nebo denaturovaného polypeptidového řetězce na komplex chaperonu Hsp70 a jeho ko-chaperonu Hsp40. Za účasti adaptorového proteinu HOP, který interaguje současně s Hsp70 i Hsp90, je poté klientní protein připojen k dimeru Hsp90. Vazba ATP způsobí uzavření víčka ATP-vazebné kapsy a ovinutí N-koncových domén kolem sebe, čímž vznikne kompaktní prstencová konformace dimeru Hsp90, v níž je klientní protein sevřen. Po vazbě ATP jsou dále Hsp70 a HOP nahrazeny ko-chaperony p23 a FKBP51 a vzniká zralý komplex. SHR je v tomto stavu aktivní, je připraven vázat steroidní hormony a být následně transportován do jádra [17]. Hydrolýza ATP je stimulována aktivitou FKBP52 a dalšího ko-chaperonu AHA1 [18]. Po hydrolýze ATP se dimer Hsp90 navrací do otevřeného stavu a klientní protein, v tomto případě SHR, je disociován [19]. Signální proteinkinázy jsou největší třídou klientních proteinů Hsp90. Mechanizmus jejich aktivace chaperonem Hsp90 je podobný jako u steroidních receptorů, v procesu jsou však zahrnuty odlišné ko-chaperony. Nově syntetizovaný polypeptidový řetězec kinázy je připraven komplexem Hsp70/Hsp40 k interakci s ko-chaperonem Cdc37 [20]. Cdc37 je ko-chaperon specifický pro proteinkinázy a společně s proteinem HOP zprostředkovává jejich vazbu k chaperonovému komplexu Hsp90. Cdc37 inhibuje ATPázovou aktivitu, a proto musí poté dojít k jejich disociaci [21]. Další ko-chaperony jako p23 a AHA1 se procesu pravděpodobně také účastní, ale detailnější informace o mechanizmu zrání proteinkináz zatím nejsou známy [22].
Význam Hsp90 v kancerogenezi
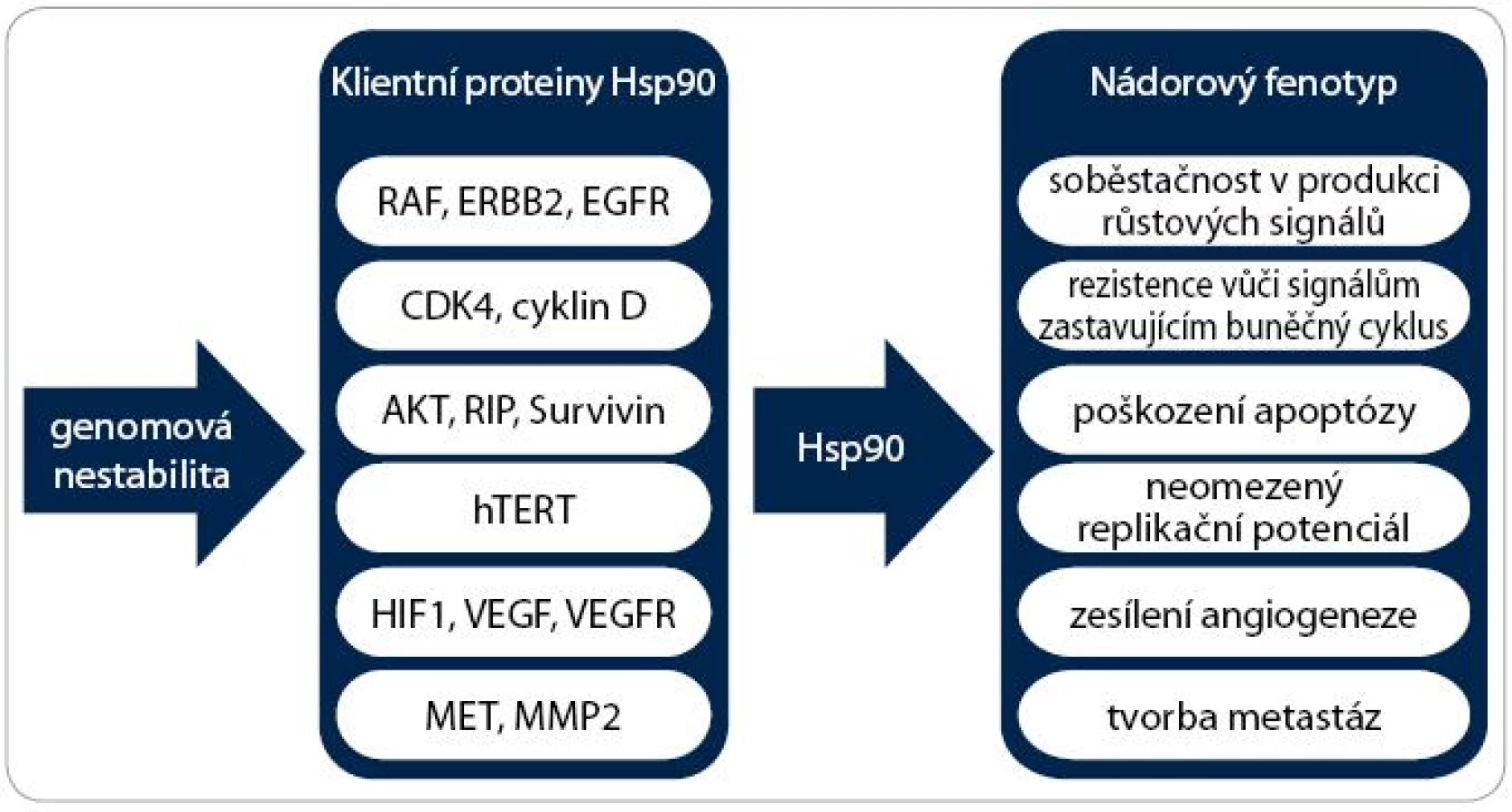
Zvýšená exprese a aktivita jednoho nebo více stresových proteinů je typickým znakem nádorů a předpokládá se jejich prognostický význam u řady malignit. Mnoho klientních proteinů Hsp90 jsou onkogeny. Příkladem mohou být proteinkinázy (Her-2 (Human Epidermal growth factor Receptor 2), Cdk4 (Cyclin-Dependent Kinase 4), Bcr-abl, AKT, c-Raf, POLO-1 a MET), transkripční faktory (estrogenový a androgenový receptor, mutantní p53 a HIF-1α (Hypoxia Inducible Factor 1α)) a další proteiny, jako je katalytická podjednotka telomerázy TERT (Telomerase Reverse Transcriptase) nebo eNOS (Endothelial Nitric Oxide Synthase) [23]. Tyto proteiny se podílejí na získání a udržení všech šesti charakteristických znaků nádorů:
- a) soběstačnosti v produkci růstových signálů,
- b) necitlivosti k signálům zastavujícím buněčný cyklus,
- c) poškození apoptózy,
- d) neomezeného replikačního potenciálu,
- e) posílení angiogeneze,
- f) tvorby metastáz [24].
V neposlední řadě se Hsp90 na vzniku onkogenních vlastností nepřímo podílí tím, že umožňuje existenci genomové nestability (obr. 3). Vzhledem k faktu, že inhibice Hsp90 vede k degradaci různorodých onkogenních proteinů, dochází k současné a kombinované supresi signálních drah způsobujících maligní transformaci a modulaci všech zmíněných znaků nádorové buňky [23].
Pohled na maligní progresi z hlediska evoluce nádoru ukazuje, že terapie ovlivňující klíčové faktory určující schopnost adaptace transformované buňky je efektivnější než inhibice jednotlivých onkogenních signálních drah, při nichž může dojít k vytvoření rezistence. Většina solidních nádorů má již v době diagnózy dostatečnou genetickou diverzitu pro vznik rezistence vůči terapii zaměřené pouze na jeden molekulární cíl [25]. Velmi často je např. pozorována rezistence vůči inhibitoru tyrozinkinázy EGFR (Epidermal Growth Factor Receptor), gefitinibu [26], která je potlačena právě kombinací gefitinibu a inhibitoru Hsp90, 17AAG [27]. Hsp90 může také maskovat fenotypový projev genetické variability, která je typická pro většinu nádorů [28]. Díky své funkci molekulárního chaperonu umožňuje Hsp90 akumulovat polymorfní varianty genů v klíčových signálních drahách za současného zachování standardního fenotypu. Tím je zajištěno přežití buňky i při extrémně vysokém výskytu mutací. Za stresových podmínek vyskytujících se v nádoru se zvyšují požadavky na aktivitu Hsp90 a po vyčerpání jeho stabilizační kapacity se začnou projevovat rozmanité genotypově-specifické fenotypy. Takto se může zrychlit vývoj invazivních, metastatických a rezistentních nádorů [29]. Zpomalení evoluce a adaptace nádorové buňky by mohlo být z terapeutického hlediska výhodné, může se však vyskytnout nebezpečí, že inhibice aktivity Hsp90 odhalí již existující genotypovou diverzitu a nepředvídatelně urychlí proces maligní progrese [30].
Mechanizmus inhibice Hsp90 a vývoj nových inhibitorů
Chaperon Hsp90 je nezbytný pro existenci eukaryotické buňky, a proto nebyl dlouhou dobu považován za potenciální cíl protinádorové terapie. Teprve objevem silných protinádorových účinků antibiotika geldanamycinu způsobených právě inhibicí molekuly Hsp90 [31] došlo k intenzivnímu výzkumu vztahu stresových proteinů a terapie nádorů, což vedlo k identifikaci a syntéze řady nových inhibitorů chaperonu Hsp90. Z hlediska klinického využití inhibitorů Hsp90 je nezbytným předpokladem výrazná citlivost nádorových buněk k inhibici tohoto stresového proteinu ve srovnání s buňkami normálními. Tato selektivita účinku může být vysvětlena řadou faktorů specifických právě pro nádorové buňky. V nádorových buňkách byl pozorován zvýšený podíl Hsp90 v aktivovaném stavu, tedy v multi-chaperonových komplexech. Takto má Hsp90 zvýšenou afinitu k ATP, a tudíž i k inhibitorům [32]. Nádorové buňky jsou závislé na aktivitě onkoproteinů, často exprimovaných jako mutantní varianty, které vyžadují stabilizaci pomocí Hsp90 ve větší míře oproti standardním formám. Jejich inaktivace tedy postihne zejména buňky nádorové. Dalším faktorem je nepříznivé prostředí uvnitř nádoru, kde buňky trpí nedostatkem kyslíku, živin a acidózou, což zvyšuje nároky na účast chaperonů [33].
ATP-vazebná kapsa na N-konci proteinu Hsp90 je místem vazby strukturně nepříbuzných přírodních látek, jako jsou geldanamycin a radicicol, jejich semi-syntetických derivátů a malých syntetických molekul. Tyto látky mají vyšší vazebnou afinitu než přirozené nukleotidy ATP, a inhibují tak funkci chaperonu zamezením cyklických změn konformace navozených vazbou a hydrolýzou ATP. Klientní proteiny proto nemohou být uvnitř dimeru Hsp90 zpracovány, což vede k jejich ubikvitinaci a následné degradaci [34]. Nejznámějším inhibitorem Hsp90 je bezesporu geldanamycin (GA), antibiotikum ze skupiny ansamycinů, izolované z kultury Streptomyces hygroscopicus v roce 1970 [35]. Jedná se také o první látku, u které byla objevena schopnost inhibovat Hsp90 [31]. Přestože GA vykazuje in vitro velmi silné protinádorové účinky, jeho klinický potenciál je bohužel nízký z důvodu špatné rozpustnosti ve vodných roztocích a hepatotoxicity [36]. Z výše uvedených důvodů byla syntetizována řada derivátů GA, které si zachovaly srovnatelné protinádorové účinky, ale mají lepší toxikologické vlastnosti. Do klinického testování doposud vstoupily deriváty 17AAG (17-allylamino-17-demethoxygeldanamycin, tanespimycin), 17DMAG (17-dimethylaminoethylamino-17-demethoxy-geldanamycin, alvespimycin), IPI-504 (retaspimycin hydrochlorid) a nejnověji pak 17AG (IPI-493) [37]. Sloučenina 17AAG se ze všech derivátů dostala až do třetí fáze klinického hodnocení.
Další intenzivně studovanou látkou je radicicol, makrocyklické přírodní antibiotikum izolované z houby Monosporium bonorden. Radicicol kompetuje, stejně jako GA, s ATP o vazebné místo v N-koncové doméně Hsp90 [38]. Radicicol vykazuje protinádorové účinky pouze in vitro, nikoliv in vivo, a to z důvodu jeho chemické a metabolické nestability. Naproti tomu nově vytvořené stabilnější oximové deriváty a cykloproparadicicol již u zvířecích modelů protinádorovou aktivitu s tolerovatelnou toxicitou prokázaly [39].
Kromě hledání přirozených inhibitorů Hsp90 se výzkum protinádorových látek zaměřuje i na syntézu malých molekul, které mohou potenciálně vykazovat větší specificitu i lepší farmakologické vlastnosti. Tento přístup využívá predikci molekulární struktury inhibitoru podle vlastností vazebného místa v N-koncové doméně. První syntetizovaný inhibitor PU3 je založen na kostře purinu, čímž napodobuje přirozené nukleotidy ATP [40]. Následně byl vytvořen panel různě chemicky modifikovaných derivátů PU3, z nichž N9-benzyl derivát CNF-2024 (BIIB021) byl vybrán pro klinické testování. Tyto látky mají podobné biologické účinky jako GA, ale vyznačují se lepší rozpustností ve vodných roztocích, možností orálního podání a metabolickou stabilitou [41]. Další skupinou syntetických molekul jsou pyrazoly. Molekula 3,4-diaryl pyrazolu (CCT018159) se váže do N-koncové ATP-vazebné kapsy Hsp90 a má podobné účinky jako GA, stejně jako nově vytvořené účinnější amidy pyrazolu a strukturně podobný isoxazol [41,42].
Aminokumarinové antibiotikum novobiocin izolované ze Streptomyces spheroides se váže v C-koncové doméně Hsp90. Novobiocin inhibuje interakce s některými ko-chaperony a vazbu ATP do potenciálního druhého ATP-vazebného místa Hsp90 [43]. Protože je vazebná afinita novobiocinu k C-konci Hsp90 velmi nízká, není klinicky využitelný. Byla však syntetizována řada analogů novobiocinu se srovnatelným inhibičním účinkem na proliferaci nádorových buněk, vykazujících minimální toxicitu při in vivo testech. Významným zjištěním je, že použití C-terminálních inhibitorů vede na rozdíl od N-terminálních k degradaci HSF-1, Hsp27 a Hsp70. Tyto látky proto mohou mít lepší terapeutické vlastnosti [44].
Nejnověji se výzkum inhibitorů zaměřuje na modulaci interakcí Hsp90 s jeho ko-chaperony. Tento přístup by oproti inhibici vazby ATP mohl mít výhodu ve větší specificitě účinku. Nejvíce poznatků bylo zatím získáno o inhibici ko-chaperonu Cdc37, který je nezbytný pro maturaci proteinkináz tím, že umožňuje jejich rozpoznání a připojení ke komplexu Hsp90. Odstranění Cdc37 metodou RNA interference vede ke snížení hladiny klientních proteinkináz, včetně Her-2, Raf-1, Cdk4 a Akt, a následně i k redukci buněčné proliferace. Kombinace posttranslačního umlčení Cdc37 a inhibice Hsp90 pomocí 17AAG pak snižuje hladinu klientních kináz a podporuje apoptózu ještě účinněji [45]. Tyto výsledky naznačují terapeutický potenciál inhibice interakce mezi Cdc37 a Hsp90. Zajímavým inhibitorem je celastrol, látka izolovaná z rostliny Tripterygium wilfordii, která reguluje dráhu Hsp90 a způsobuje stresovou odpověď podobně jako jiné inhibitory [46]. Pomocí počítačového modelování a biochemických metod bylo prokázáno, že celastrol nemá vliv na vazbu ATP, ale narušuje právě interakci Cdc37 s Hsp90. Jeho protinádorové účinky byly pozorovány in vitro i in vivo [47].
Klinické studie inhibitorů Hsp90
Aktuální údaje o probíhajících klinických studiích inhibitorů Hsp90 jsou přehledně shrnuty v nedávné publikaci, která popisuje 40 ukončených a 23 aktivních onkologických klinických studií [48]. Nejdéle známý inhibitor Hsp90, 17AAG (tanespimycin), vstoupil do klinického testování v roce 1999 a byl testován v nejrůznějších dávkovacích plánech a způsobech podání u pacientů s různými diagnózami, a to formou mono-terapie i kombinované terapie [48]. Největším nedostatkem 17AAG je jeho nízká rozpustnost ve vodných roztocích. Je tedy nutná aplikace v roztocích organických látek, jako je DMSO nebo Cremophor EL. DMSO se vyznačuje systémovou toxicitou a Cremophor vyvolává hypersenzitivní reakci organizmu, čímž oba ještě více podporují negativní vedlejší účinky samotného 17AAG. Proto jsou vyvíjeny nové formy nosičů 17AAG, jako jsou suspenze polysorbátu 80, lecitinu a sacharózy [49], stejně jako nano-vektory ve formě polymerních micel různého chemického složení, které zajišťují plynulejší uvolňování léčiva [50,51].
Během první fáze klinického hodnocení bylo zjištěno, že toxicita 17AAG závisí na četnosti dávkování. Např. při podávání po dobu pěti dnů ve třítýdenních intervalech byla maximální tolerovaná dávka (MTD) 40–56 mg/m2 [52], na rozdíl od 450 mg/m2 při podávání jednou týdně [53]. Z pozorovaných vedlejších účinků byla nejzávažnější hepatotoxicita. Studie druhé fáze klinického hodnocení vycházely z dávkovacího schématu jedna nebo dvě dávky týdně, ale nepřinesly ani v jednom případě klinicky významné odpovědi. Nedostatečná odpověď může být vysvětlena krátkodobým účinkem 17AAG v organizmu, který vyplývá z farmakodynamických údajů dvou klinických studií [53,54]. V rámci studie první fáze klinického hodnocení u pacientů s pokročilými malignitami byly odebrány vzorky tkáně před léčbou a dále jeden a pět dní po podání první dávky 320–450 mg/m2 17AAG v týdenních intervalech. Suprese klientních proteinů Hsp90 c-Raf a Cdk4 a indukce Hsp70 byla po jednom dni prokazatelná, zatímco po pěti dnech nebyly změny hladin těchto proteinů jednoznačně interpretovatelné [53]. Ve studii druhé fáze klinického hodnocení u pacientů s metastazujícím melanomem byly provedeny biopsie před léčbou a 18–50 hodin (medián 44 hodin) po podání první dávky. U vzorků tkání byla stanovena hladina proteinů Raf a ERK (Extracellular signal-Regulated Kinase) náležejících do signální dráhy MAPK (Mitogen-Activated Protein Kinase), která je u melanomů často deregulována, a tyto proteiny jsou klienty Hsp90. Dále byla stanovena hladina cyklinu D1 a Hsp70. Významné rozdíly v hladinách proteinů Raf a ERK před a po podání 17AAG nebyly pozorovány, zato hladina cyklinu D1 byla u většiny nádorů snížena a hladina Hsp70 zvýšena. Vzhledem k výsledkům obou studií je pravděpodobné, že trvání biologické aktivity inhibitoru 17AAG je krátkodobé a u jednotlivých klientních proteinů se liší. Tyto dočasné změny hladin onkogenních proteinů zřejmě nejsou pro zastavení růstu nádoru dostatečné [54] a v dávkování 17AAG je nutný vývoj nových postupů umožňujících jeho pozvolné uvolňování.
Klinické studie kombinující 17AAG a cytotoxickou chemoterapii, jako např. paklitaxel [55] nebo irinotekan [56], zaznamenaly lepší klinické účinky, přesto u žádného z pacientů nedošlo k úplné ani částečné odpovědi podle kritérií RECIST (Response Evaluation Criteria in Solid Tumors). Klinické hodnocení této kombinované terapie nadále pokračuje a je možné, že s novými způsoby podání 17AAG se její účinky zvýší. Studie testující 17AAG a gemcitabin prokázala protinádorovou aktivitu a tato kombinace je testována ve druhé fázi klinického hodnocení. Naproti tomu kombinace 17AAG a cisplatiny vykázala závažnou toxicitu [57]. Spojení 17AAG a cílené protinádorové terapie je ještě účinnější, neboť 17AAG může potencovat účinky inhibice specifického molekulárního cíle. Byla testována kombinace 17AAG a bortezomibu, která přinesla výraznou léčebnou odpověď. Zdánlivě protichůdné využití inhibitorů molekulárního chaperonu a proteazomu přináší efekt kumulace toxických agregátů proteinů vzniklých inhibicí Hsp90. Hsp70 indukovaný 17AAG dále prokazoval neuroprotektivní funkci, a snížil tak negativní účinky monoterapie bortezomibem [58]. Nadějná se zdá být i terapie kombinující 17AAG a trastuzumab u Her-2 pozitivních nádorů prsu rezistentních k trastuzumabu. Her-2 je klientním proteinem Hsp90 a k jeho inhibici je velmi citlivý. Úloha trastuzumabu při této odpovědi je zatím neznámá [59]. Jedním z faktorů omezujících terapeutické využití 17AAG je výskyt primárních i získaných rezistencí, které jsou způsobeny zvýšenou expresí P-glykoproteinu (P-gp) [60] nebo ztrátou či mutací genu NQO1 (NAD(P)H chinon dehydrogenáza 1), který je nezbytný pro metabolizaci 17AAG na hydrochinon [61] s výrazně větší afinitou k Hsp90 [62]. Inhibicí Hsp90 se také indukuje stresová odpověď v buňce vyvolaná uvolněním a aktivací transkripčního faktoru HSF-1 (Heat Shock Factor 1). Tento faktor se váže na responzivní elementy HSE (Heat Shock Elements) nacházející se v promotorech mnoha genů kódujících stresové proteiny, jako jsou Hsp90, Hsp70 a Hsp27 [63]. Indukce stresové odpovědi může také vyvolat rezistenci k léčbě. U buněčných linií bylo zjištěno, že umlčení exprese Hsp70 nebo Hsp27 pomocí RNA interference má pozitivní vliv na zvýšení citlivosti a potlačení rezistence k 17AAG. Kombinovaná inhibice všech těchto proteinů by proto mohla významně zvyšovat účinnost terapie [64].
Analog 17DMAG (alvespimycin) je rozpustný ve vodě a umožňuje jak intravenózní, tak i orální podání. Nedávno byly publikovány výsledky tří klinických studií. Při jedné byl 17DMAG podáván intravenózně denně po dobu pěti (16 mg/m2) nebo tří dnů (25 mg/m2) ve třítýdenních intervalech pacientům se solidními nádory. Tyto dávky byly dobře tolerovány, nebyla však pozorována významná odpověď, pouze u 7 % pacientů došlo ke stabilizaci onemocnění [65]. Další dvě studie pro následující klinické hodnocení doporučily dávkování 21 nebo 24 mg/m2 intravenózně dvakrát týdně [66,67]. Toto dávkovací schéma bylo klinicky úspěšnější, u 29 % pacientů s pokročilými malignitami došlo ke stabilizaci onemocnění po dobu 2–22 měsíců s mediánem 4 měsíce [66] a u 3 ze 17 pacientů s leukemií došlo k remisi [67].
IPI-504 (retaspimycin) je také rozpustný ve vodě a je podáván intravenózně. Klinické hodnocení druhé fáze bylo prováděno na pacientech s nemalobuněčným karcinomem plic. IPI-504 byl aplikován infuzí ve dnech 1, 4, 8 a 11 v cyklu 21 dní. Podávaná dávka 400 mg/m2 byla snížena na 225 mg/m2 z důvodu hepatotoxicity. Objektivní odpověď na léčbu byla zaznamenána u 7 % pacientů [68].
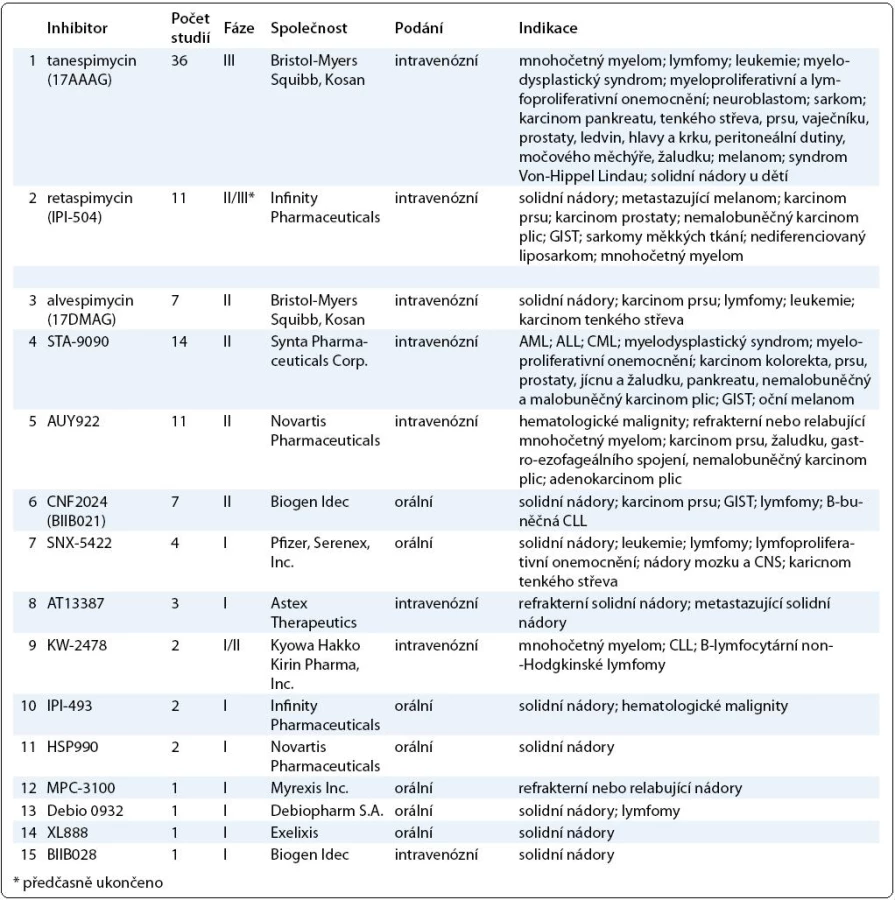
Výsledky klinických studií dalších inhibitorů Hsp90 zatím nebyly publikovány, v tab. 1 je uveden přehled studií inhibitorů Hsp90 registrovaných v databázi ClinicalTrials.gov k únoru 2011.
Závěr
Hsp90 je díky svému univerzálnímu výskytu v nádorových buňkách považován za důležitý cíl protinádorové léčby. Inhibicí Hsp90 dochází k narušení mnoha onkogenních signálních drah současně, což umožňuje využití inhibitorů Hsp90 v léčbě různých typů nádorových onemocnění. Klinické testování nejdéle známých N-koncových inhibitorů 17AAG a 17DMAG však prozatím nepřináší očekávané výsledky, neboť kromě nízké odpovědi nádorů se projevily i závažné negativní vedlejší účinky terapie. Negativní výsledky mohou být způsobeny farmakodynamickými a farmakokinetickými vlastnostmi testovaných látek, které však mohou být zlepšeny novými modifikacemi či syntézami jejich analogů [44]. Aplikace inhibitorů rovněž indukuje expresi stresových proteinů, např. Hsp70, Hsp27 a ko-chaperonu HOP, které mají anti-apoptotické vlastnosti, a tím mohou interferovat s degradací klientních proteinů [63]. Východiskem by mohla být kombinovaná terapie zaměřená na více součástí chaperonového systému a nalezení markerů, které by pomohly předvídat odpověď buňky na terapii. Předběžné analýzy nově objevených C-koncových inhibitorů, jako je novobiocin a jeho deriváty, ukazují, že se jedná o nadějná léčiva. Hsp90 proto stále zůstává významným cílem protinádorové léčby.
Práce byla podpořena granty IGA MZ ČR NS/9812-4 a RECAMO CZ.1.05/2.1.00/03.010.1
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
RNDr. Bořivoj Vojtěšek, DrSc.
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: vojtesek@mou.cz
Obdrženo: 18. 4. 2011
Přijato: 25. 5. 2011
Sources
1. Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 2009; 16(6): 574–581.
2. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 2010; 11(7): 515–528.
3. Horwich AL, Apetri AC, Fenton WA. The GroEL/GroES cis cavity as a passive anti-aggregation device. FEBS Lett 2009; 583(16): 2654–2662.
4. Wegele H, Müller L, Buchner J. Hsp70 and Hsp90 – a relay team for protein folding. Rev Physiol Biochem Pharmacol 2004; 151 : 1–44.
5. Nakamoto H, Vigh L. The small heat shock proteins and their clients. Cell Mol Life Sci 2007; 64(3): 294–306.
6. Laskey RA, Honda BM, Mills AD et al. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 1978; 275(5679): 416–420.
7. Besche HC, Haas W, Gygi SP et al. Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 2009; 48(11): 2538–2549.
8. Brugge JS, Erikson E, Erikson RL. The specific interaction of the Rous sarcoma virus transforming protein, pp60src, with two cellular proteins. Cell 1981; 25(2): 363–372.
9. Sreedhar AS, Kalmár E, Csermely P et al. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett 2004; 562(1–3): 11–15.
10. Tsutsumi S, Neckers L. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci 2007; 98(10): 1536–1539.
11. Eustace BK, Sakurai T, Stewart JK et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol 2004; 6(6): 507–514.
12. Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 2000; 25(1): 24–28.
13. Hainzl O, Lapina MC, Buchner J et al. The charged linker region is an important regulator of Hsp90 function. J Biol Chem 2009; 284(34): 22559–22567.
14. Hawle P, Siepmann M, Harst A et al. The middle domain of Hsp90 acts as a discriminator between different types of client proteins. Mol Cell Biol 2006; 26(22): 8385–5395.
15. Minami Y, Kimura Y, Kawasaki H et al. The carboxy-terminal region of mammalian HSP90 is required for its dimerization and function in vivo. Mol Cell Biol 1994; 14(2): 1459–1464.
16. Young JC, Obermann WM, Hartl FU. Specific binding of tetratricopeptide repeat proteins to the C-terminal 12-kDa domain of hsp90. J Biol Chem 1998; 273(29): 18007–18010.
17. Prodromou C, Panaretou B, Chohan S et al. The ATPase cycle of Hsp90 drives a molecular ‚clamp‘ via transient dimerization of the N-terminal domains. EMBO J 2000; 19(16): 4383–4392.
18. Panaretou B, Siligardi G, Meyer P et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol Cell 2002; 10(6): 1307–1318.
19. Richter K, Walter S, Buchner J. The Co-chaperone Sba1 connects the ATPase reaction of Hsp90 to the progression of the chaperone cycle. J Mol Biol 2004; 342(5): 1403–1413.
20. Lee P, Shabbir A, Cardozo C et al. Sti1 and Cdc37 can stabilize Hsp90 in chaperone complexes with a protein kinase. Mol Biol Cell 2004; 15(4): 1785–1792.
21. Roe SM, Ali MM, Meyer P et al. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004; 116(1): 87–98.
22. Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem 2008; 283(27): 18473–18477.
23. Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med 2004; 10(2): 47–51.
24. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100(1): 57–70.
25. Kitano H. Cancer robustness: tumour tactics. Nature 2003; 426(6963): 125.
26. Kobayashi S, Boggon TJ, Dayaram T et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005; 352(8): 786–792.
27. Pashtan I, Tsutsumi S, Wang S et al. Targeting Hsp90 prevents escape of breast cancer cells from tyrosine kinase inhibition. Cell Cycle 2008; 7(18): 2936–2941.
28. Sangster TA, Lindquist S, Queitsch C. Under cover: causes, effects and implications of Hsp90-mediated genetic capacitance. Bioessays 2004; 26(4): 348–362.
29. Gatenby RA, Maini PK. Mathematical oncology: cancer summed up. Nature 2003; 421(6921): 321.
30. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005; 5(10): 761–772.
31. Whitesell L, Mimnaugh EG, De Costa B et al. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A 1994; 91(18): 8324–8328.
32. Kamal A, Thao L, Sensintaffar J et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003; 425(6956): 407–410.
33. Whitesell L, Bagatell R, Falsey R. The stress response: implications for the clinical development of hsp90 inhibitors. Curr Cancer Drug Targets 2003; 3(5): 349–358.
34. Blagg BS, Kerr TD. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med Res Rev 2006; 26(3): 310–338.
35. DeBoer C, Meulman PA, Wnuk RJ et al. Geldanamycin, a new antibiotic. J Antibiot (Tokyo) 1970; 23(9): 442–447.
36. Supko JG, Hickman RL, Grever MR et al. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol 1995; 36(4): 305–315.
37. Porter JR, Ge J, Lee J et al. Ansamycin inhibitors of Hsp90: nature’s prototype for anti-chaperone therapy. Curr Top Med Chem 2009; 9(15): 1386–1418.
38. Roe SM, Prodromou C, O’Brien R et al. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 1999; 42(2): 260–266.
39. Soga S, Shiotsu Y, Akinaga S et al. Development of radicicol analogues. Curr Cancer Drug Targets 2003; 3(5): 359–369.
40. Chiosis G, Timaul MN, Lucas B et al. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol 2001; 8(3): 289–299.
41. Taldone T, Chiosis G. Purine-scaffold Hsp90 inhibitors. Curr Top Med Chem 2009; 9(15): 1436–1446.
42. Cheung KM, Matthews TP, James K et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg Med Chem Lett 2005; 15(14): 3338–3343.
43. Marcu MG, Chadli A, Bouhouche I et al. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem 2000; 275(47): 37181–37186.
44. Holzbeierlein JM, Windsperger A, Vielhauer G. Hsp90: a drug target? Curr Oncol Rep 2010; 12(2): 95–101.
45. Smith JR, Clarke PA, de Billy E et al. Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene 2009; 28(2): 157–169.
46. Westerheide SD, Bosman JD, Mbadugha BN et al. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem 2004; 279(53): 56053–56060.
47. Zhang T, Li Y, Yu Y et al. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem 2009; 284(51): 35381–35389.
48. Kim YS, Alarcon SV, Lee S et al. Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem 2009; 9(15): 1479–1492.
49. Burris HA 3rd, Berman D, Murthy B et al. Tanespimycin pharmacokinetics: a randomized dose-escalation crossover phase 1 study of two formulations. Cancer Chemother Pharmacol 2011; 67(5): 1045–1054.
50. Chandran T, Katragadda U, Teng Q et al. Design and evaluation of micellar nanocarriers for 17-allyamino-17-demethoxygeldanamycin (17-AAG). Int J Pharm 2010; 392(1–2): 170–177.
51. Xiong MP, Yáñez JA, Kwon GS et al. A cremophor-free formulation for tanespimycin (17-AAG) using PEO-b-PDLLA micelles: characterization and pharmacokinetics in rats. J Pharm Sci 2009; 98(4): 1577–1586.
52. Grem JL, Morrison G, Guo XD et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol 2005; 23(9): 1885–1893.
53. Banerji U, O’Donnell A, Scurr M et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 2005; 23(18): 4152–4161.
54. Solit DB, Osman I, Polsky D et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res 2008; 14(24): 8302–8307.
55. Ramalingam SS, Egorin MJ, Ramanathan RK et al. A phase I study of 17-allylamino-17-demethoxygeldanamycin combined with paclitaxel in patients with advanced solid malignancies. Clin Cancer Res 2008; 14(11): 3456–3461.
56. Tse AN, Klimstra DS, Gonen M et al. A phase 1 dose-escalation study of irinotecan in combination with 17-allylamino-17-demethoxygeldanamycin in patients with solid tumors. Clin Cancer Res 2008; 14(20): 6704–6711.
57. Hubbard J, Erlichman C, Toft DO et al. Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Invest New Drugs 2011; 29(3): 473–480.
58. Richardson PG, Badros AZ, Jagannath S et al. Tanespimycin with bortezomib: activity in relapsed/refractory patients with multiple myeloma. Br J Haematol 2010; 150(4): 428–437.
59. Modi S, Stopeck AT, Gordon MS et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol 2007; 25(34): 5410–5417.
60. Tsuruo T, Naito M, Tomida A et al. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci 2003; 94(1): 15–21.
61. Kelland LR, Sharp SY, Rogers PM et al. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst 1999; 91(22): 1940–1949.
62. Gaspar N, Sharp SY, Pacey S et al. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res 2009; 69(5): 1966–1975.
63. Kim HR, Kang HS, Kim HD. Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life 1999; 48(4): 429–433.
64. McCollum AK, TenEyck CJ, Stensgard B et al. P-Glycoprotein-mediated resistance to Hsp90-directed therapy is eclipsed by the heat shock response. Cancer Res 2008; 68(18): 7419–7427.
65. Ramanathan RK, Egorin MJ, Erlichman C et al. Phase I pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. J Clin Oncol 2010; 28(9): 1520–1526.
66. Kummar S, Gutierrez ME, Gardner ER et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur J Cancer 2010; 46(2): 340–347.
67. Lancet JE, Gojo I, Burton M et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010; 24(4): 699–705.
68. Sequist LV, Gettinger S, Senzer NN et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol 2010; 28(33): 4953–4960.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2011 Issue 5
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Parciální regrese ložisek Erdheimovy-Chesterovy nemoci v CNS po léčbě 2-chlorodeoxyadenosinem a jejich kompletní vymizení při léčbě lenalidomidem
- Neuroendoskopická biopsie tumoru mozku
- Sekundární angiosarkomy po konzervativní léčbě nádorů prsu
- Co pacientky potřebují vědět před operací mamárního karcinomu