
Klinické dysmorfické syndrómy s tumorigenézou
Clinical Dysmorphic Syndromes with Tumorigenesis
Genetic alterations cause predisposition to malignancy by increased cancer risk related to constitutional mutations in growth-regulating or DNA repair genes. Some pediatric malignancies are associated with dysmorphic features in several body areas. Through physical examination, we recognise characteristic signs of genetic dysmorphic disorders, such as somatic overgrowth, undergrowth, macrocephaly, microcephaly and dysmorphic changes of the face, eyes, mouth and lips, heart, gastrointestinal tract, urinary tract, genitalia and skeleton. Recognition of a cancer-associated dysmorphic syndrome allows intensive cancer screening and genetic counseling. Therefore, it is recommended that every child with cancer should be examined by a clinical geneticist. Molecular diagnostics of germinal mutations may very effectively detect families at high risk of malignancy and help provide primary prevention. This work presents clinical syndromes with genetic backround and cancer screening recommendations for 18 syndromes with increased cancer risk.
Key words:
genetic dysmorphic syndromes – RAS-MAPK signaling pathway – malignance in childhood
Molecular diagnostics of Rasopathies was supported by The League against Cancer of the sR.
The authors declare they have no potential conflicts of interest concerning drugs, pruducts, or services used in the study.
The Editorial board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
24. 4. 2012
Accepted:
25. 6. 2012
:
D. Ilenčíková; M. Čižmárová; A. Krajčiová; S. Požgayová; A. Rybárová; L. Kovács
:
II. detská klinika, LF UK a DFNsP Bratislava, Slovenská republika
:
Klin Onkol 2012; 25(Supplementum): 39-48
Genetické zmeny spôsobujú predispozíciu k zvýšenému riziku vzniku nádoru v súvislosti s konštitučnými mutáciami v génoch pre reguláciu rastu alebo opravu DNA. Niektoré detské malignity sú spojené s dysmorfiou vyskytujúcou sa na rôznych častiach tela. Fyzikálnym vyšetrením je možné rozpoznať charakteristické znaky genetických dysmorfických ochorení. Medzi ne patrí: nadmerný či nízky vzrast, makrocefália, mikrocefália a dysmorfické zmeny na tvári, očiach, ústach a perách, srdci, v gastrointestinálnom trakte, na obličkách, genitáliách a skelete. Rozpoznanie dysmorfického syndrómu s malignitou iniciuje pátranie po malignite a genetické poradenstvo pre dieťa a jeho rodinu. Preto sa u každého dieťaťa s malignitou odporúča zvážiť možnosť genetického vyšetrenia. Molekulárna diagnostika zárodočných mutácií môže veľmi účinne odhaliť rodiny s vysokým rizikom malignity a poskytnúť pomoc pri primárnej prevencii. Predložená prehľadová práca predstavuje 18 genetických dysmorfických syndrómov s rizikom malignity, popisuje ich klinické znaky, genetický podklad a odporúčania pre sledovanie pacientov s cieľom skorého zachytenia malignít.
Kľúčové slová:
genetické dysmorfické syndrómy – MAPK-RAS signálna dráha – malignity v detstve
V etiológii kongenitálnych anomálií sú dysregulované signálne dráhy, ktoré môžu byť zapojené aj v procese tumorigenézy. Mnohé štúdie poukazujú na to, že pacienti s kongenitálnymi anomáliami majú oproti kontrolnej skupine detí zvýšené riziko vzniku malignít. Prvá štúdia, ktorá dokladuje 7,5% incidenciu malignít u 1 073 vyšetrených detí s klinickými genetickými syndrómami, bola publikovaná v roku 2005 Merksom et al [1]. Nakoľko sa jedná o skutočne vysoké číslo, predpokladá sa, že klinicky jasne definované syndrómy s malignitou nie sú v pediatrickej praxi dostatočne rozpoznané. Z tohto dôvodu je vhodné, aby každé dieťa s malignitou bolo vyšetrené klinickým genetikom. Klinické syndrómy s dysmorfickými črtami a malignitou môžeme rozdeliť na tie s:
- nadmerným vzrastom,
- nízkym vzrastom,
- rôznorodými dysmorfnými prejavmi a nízkym výskytom malignít,
- mutáciami v Mitogén - aktivovanej proteín kinázovej (MAPK) - RAS signálnej dráhe (RASopatie).
Dysmorfické syndrómy s malignitou a nadmerným vzrastom
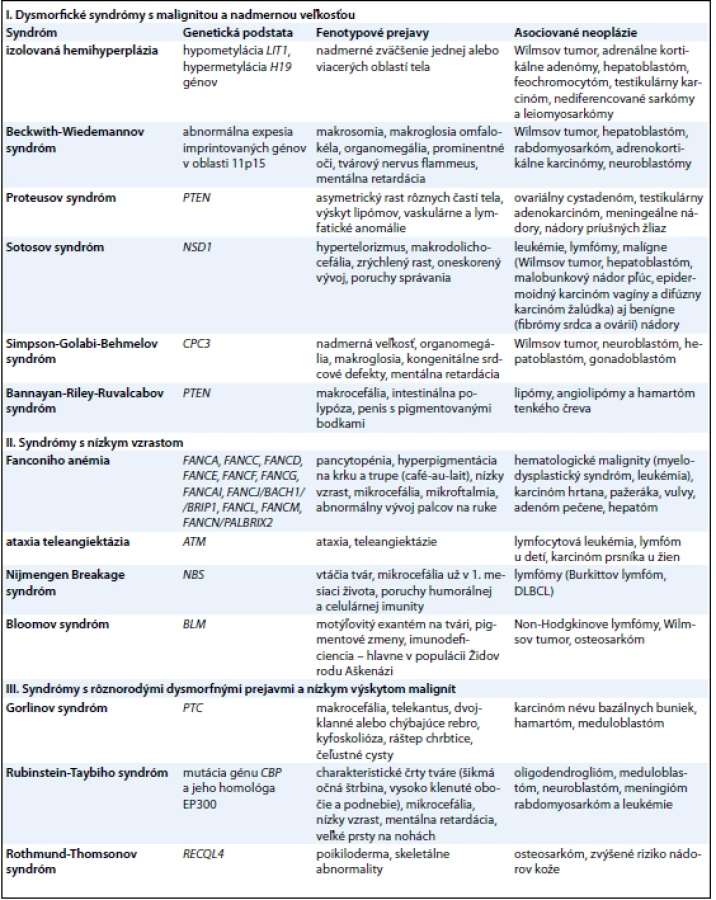
Nadmerná veľkosť je charakterizovaná výškou a váhou so štandardnou odchýlkou 2 – 3 v závislosti od veku a pohlavia. V práci autorov Lapunzina a Cohena et al [2,13] je medzi syndrómami s nadmernou veľkosťou popisovaný častejší výskyt malignít obličiek, pečene, neuroblastómu, osteosarkómu a leukémií. Patria sem predovšetkým: izolovaná hemihyperplázia (IHH), Beckwith - Wiedemannov syndróm (BWS), Proteusov syndróm (PS), Sotosov syndróm (SS), Simpson - Golabi - Behmelov syndróm (SGBS) a Bannayan - Riley - Ruvalcabov syndróm (BRRS) (tab. 1).
Izolovaná hemihyperplázia (IHH)
Predstavuje klinický prejav nadmernej veľkosti jednej alebo viacerých oblastí ľudského tela. Diferencia sa prejavuje medzi veľkosťou kostí a mäkkého tkaniva.
Genetika. Častým genetickým podkladom IHH býva chromozómová aberácia. Vyskytuje sa častejšie v mozaikovej forme a z molekulárnych mechanizmov sa v súvislosti s fenotypom zistila hypometylácia génu LIT1 a hypermetylácia H19 génov (oba sú lokalizované na 11p) [3].
Malignity. U pacientov je zvýšené riziko (6 %) vzniku embryonálnych nádorov, predovšetkým Wilmsovho tumoru [4], adrenálnych kortikálnych adenómov a hepatoblastómu, feochromocytómu, testikulárneho karcinómu, nediferencovaných sarkómov a leiomyosarkómu.
Sledovanie. Pravidelná sonografia a fyzikálne vyšetrenie, sérová hladina AFP a bHCG, vyšetrenie moču (hladina vanilmandľovej kyseliny, katecholamínov, metanefrínu a normetanefrínu v plazme) každé 3 mesiace od dovŕšenia 7. roku života [2].
Beckwith - Wiedemannov syndróm (BWS)
Je charakterizovaný klinickým trias: makrosomiou, makroglosiou a omfalokélou. Pacienti majú typické znaky na tvári, medzi ktoré patria prominentné oči, tvárový nevus flammeus, makroglózia, predné záhyby ušného lalôčika a zadné helikálne jamky. Väčšina pacientov s BWS má pôrodnú výšku viac ako 97. percentil vzhľadom na gestačný vek [5]. Tieto zvýšené rastové parametre sa však v období puberty vyrovnajú. Ďalším znakom tohto syndrómu je organomegália, predovšetkým zväčšenie pečene, pankreasu, sleziny, obličiek a adrenálnych žliaz. Mentálna retardácia alebo opozdený psychomotorický vývoj sa vyskytuje v súvislosti s nálezom duplikácie oblasti 11p15 alebo vplyvom nedetekovaných epizód hypoglykémie, ktoré sú popisované u 30 – 50 % prípadov dojčiat s BWS [6].
Genetika. Prejav ochorenia je viazaný na aberáciu chromozómovej oblasti 11p15. BWS býva vo väčšine prípadov sporadický (85 %), pričom cytogenetické aberácie (translokácie, inverzie a duplikácie) zahŕňajúce oblasť 11p15 sú prítomné v 1 % prípadov. U 10 – 15 % prípadov bol popísaný autozómovo- - dominantný prenos preferenčne z maternálnej línie, preto je odporúčané zrealizovať podrobné a starostlivé genealogické vyšetrenie [7].
Základným genetickým mechanizmom zodpovedným za vznik BWS je abnormálna expresia imprintovaných génov na 11p15. Zmena v dvoch doménach (DMR1 a DMR2), ktoré zahŕňajú imprintované gény a vedú k zmene ich expresie. Doména DMR1 je lokalizovaná na telomerickom konci v 11p15 obsahuje gén IGF2 – exprimovaný paternálne a H19 exprimovaný maternálne. Takmer 25 % pacientov vykazuje stratu imprintingu (LOI) IGF2, čo spôsobí expresiu oboch paternálnych alel. Druhá doména DMR2, ktorá je voči DMR1 uložená centromericky, zahŕňa nasledovné imprintované gény: KCNQ1, LIT1, CDKN1C, PHLDA a SLC22A18. U takmer 50 % pacientov so sporadickým výskytom BWS je identifikovaná hypometylácia DMR2 a LOI LIT1 génu (paternálna uniparentálna dizómia – UPD 11p15 tvorí 20 %, mutácie v géne CDKN1C tvoria 10 %, chromozómové prestavby 1 % a ostatné prípady predstavujú iné epigenetické mechanizmy [8].
Malignity. Pacienti majú zvýšené riziko vývoja embryonálnych tumorov (5 – 10 %), predovšetkým vo veku od 5 – 8 rokov [6,9]. Najčastejšie sú tumory obličiek (Wilmsov tumor), hepatoblastóm, rabdomyosarkóm, adrenokortikálne karcinómy a neuroblastómy [10]. Wilmsov tumor sa najčastejšie vyskytuje v asociácii s UPD 11p15 a H19 hypometyláciou.
Pravidelné sledovanie pacientov s BWS je odporúčané nezávisle od výsledkov molekulárneho vyšetrenia [11]. V čase diagnózy má byť zrealizované vyšetrenie MRI abdomenu a každé 3 mesiace abdominálna sonografia s vyšetrením hladiny sérového AFP [12].
Proteov syndróm (PS)
Je charakterizovaný asymetrickým nadmerným rastom rôznych častí tela [13]. Často sa u pacientov vyvíjajú lipómy, epidermálne névy, vaskulárne a lymfatické anomálie. Na hlave sa môžu nachádzať kraniálne hyperostózy a tvar hlavy je často dolichocefalický. Tvár býva obyčajne dlhá, nízko posadený koreň nosa. Bieseckerom et al [14] boli v roku 2005 stanovené diagnostické kritériá pre PS, a to: hemihyperplázia a lipomatóza.
Genetická podstata nie je doteraz objasnená. Predpokladá sa, že v patogenéze vzniku genetického syndrómu, ako aj vývoja malignít, zohráva úlohu mutácia génu PTEN [15]. Prípady sú však skôr sporadické alebo sa jedná o mozaicizmus [16].
Malignity. V spektre malignít prevažujú ovariálny cystadenóm, testikulárny adenokarcinóm, meningeálne nádory a nádory príušných žliaz. Tieto sa prejavujú často viac početne u jedného pacienta.
Zo sledovaní je odporučené sledovanie MRI mozgu každé dva roky, ročne fyzikálne vyšetrenie a abdominálna sonografia. U detí je riziko predčasného úmrtia v dôsledku vaskulopatie, preto sa odporúča antitrombotická liečba [16].
Sotosov syndróm (SS)
Je autozómovo - dominantné ochorenie, ktoré sa však nadmernou veľkosťou prejavuje len relatívne. Výška a obvod hlavy sa nachádzajú na hornej hranici normy (10 %) normálnych detí. Hoci kostný vek je často akcelerovaný, po dovŕšení 5. roku života sa skôr znižuje. Väčšina pacientov sa prejavuje oneskoreným vývojom a behaviorálnymi prejavmi. Po narodení sú deti hypotonické a makrodolichocefalické. Majú zrýchlený rast, riedke vlasy, prominentné čelo a bradu. Z očných prejavov býva prítomný hypertelorizmus. Na MR mozgu možno zaznamenať miernu dilatáciu cerebrálnych ventrikúl, na EEG nešpecifické zmeny. V niektorých prípadoch sú pozorované kŕče.
Genetika. V roku 2002 bol vyklonovaný gén NSD1 zodpovedný za fenotypový prejav ochorenia. Najčastejším genetickým podkladom je haploinsuficiencia génu, pričom mikrodelécie sa nachádzajú v Kaukazskej populácii menej často (6 – 10 %), avšak u Japoncov boli popísané až v 52 % prípadov [17].
Malignity. Riziko výskytu malignít sa zvyšuje od 5. roku života a pohybuje okolo 3 % [18]. Medzi najčastejšie malignity patria leukémie a lymfómy, pričom embryonálne tumory sa vyskytujú veľmi zriedkavo. Naopak malígne tumory, ktoré boli doteraz popísané, sú veľmi pestré (hepatoblastóm, Wilmsov tumor, malobunkový nádor pľúc, epidermoidný karcinóm vagíny a difúzny karcinóm žalúdka) a patria sem aj benígne tumory (fibrómy srdca a ovárií).
Sledovanie. Každé 4 mesiace sa odporúča fyzikálne vyšetrenie, abdominálna sonografia, vyšetrenie séra a moču.
Simpson - Golabi - Behmelov syndróm (SGBS)
Je charakterizovaný nadmernou veľkosťou diagnostikovanou už prenatálne, hrubými črtami tváre, makroglosiou, organomegáliou, kongenitálnymi srdcovými chybami a mentálnou retardáciou [19].
Genetika. Pilia et al identifikovali v roku 1996 kauzálny gén CPC3 s lokalizáciou na Xq26, preto sa ochorenie dedí ako X viazané ochorenie [20].
Malignity. Riziko vzniku malignít sa pohybuje okolo 10 % a najčastejšie sa jedná o výskyt Wilmsovho tumoru, hepatoblastómu, neuroblastómu, gonadoblastómu [21].
Sledovanie. Odporúča sa ročné fyzikálne vyšetrenie, abdominálna sonografia, analýza moču a biochemický screening pre embryonálne tumory.
Bannayan - Riley - Ruvalcaba syndróm (BRRS)
Je syndróm rozpoznateľný podľa makrocefálie, pri pôrode veľkými rozmermi, intestinálnou polypózou a vývojom pigmentovaných bodiek na penise [22].
Genetika. Dedičnosť je autozómovo - dominantná a vo väčšine prípadov bola zistená mutácia v géne PTEN, podobne ako je to u Cowdenovho syndrómu [23].
Malignity. Práve spektrum malignít odlišuje BRRS od Cowdenovho syndrómu. U BRRS sa vyskytujú predovšetkým benígne tumory: lipómy, angiolipómy a hamartóm tenkého čreva [13], kým u Cowdenovho syndrómu prevažuje výskyt malígnych tumorov (nádory štítnej žľazy, prsníka, ganglioneurómy). Tieto boli u BRRS popísané len mimoriadne zriedkavo [13].
Sledovanie. Ročné fyzikálne vyšetrenie a kolonoskopia po 12. roku života.
Syndrómy s nízkym vzrastom
Nízky vzrast je charakterizovaný výškou a váhou so štandardnou odchýlkou 2 – 3 v závislosti od veku a pohlavia. Do tejto skupiny zaraďujeme predovšetkým syndrómy charakteristické poruchou dozrievania kostnej drene, najčastejšie na podklade chromozómovej instability (tab. 1). Všetky tieto syndrómy sú spojené s vysokým rizikom malignity a sú známe endogénnou precitlivenosťou na rôzne faktory prostredia (žiarenia, chemické látky). Prejav v jadre bunky je charakteristický spontánnou poruchou integrity chromozómov (tvorbou trhlín, zlomov a prestavbou chromozómov).
Fanconiho anémia (FA)
Ochorenie je charakterizované nízkym vzrastom, mikrocefáliou, mikroftalmiou a abnormálnym vývojom palcov na ruke, ktorý sa niekedy vyskytuje s hypopláziou rádia alebo bez nej. Ďalším významným prejavom je generalizovaná hyperpigmentácia na trupe a krku s typickými škvrnami typu „café - au - lait“ (> 50 % prípadov) a niekedy aj s hypopigmentovanými oblasťami. Pri FA je však dominujúcim a vysoko sugestívnym diagnostickým znakom pancytopénia.
Genetika. Ochorenie je autozómovo--recesívne s lokusovou heterogenitou. V kauzálnej súvislosti sa potvrdili mutácie doteraz popísaných génov FANCA, FANCC, FANCD, FANCE, FANCF, FANCG, FANCAI, FANCJ/ BACH1/ BRIP1, FANCL, FANCM a FANCN/ PALBRIX2 [24].
Malignity. Kumulatívna incidencia hematologických malignít je až 40 % [25]. Obyčajne sa manifestuje najprv aplastickou anémiou, s prechodom do myelodysplastického syndrómu a leukémie. Zo solidných tumorov sa zriedkavo môže vyskytnúť adenóm pečene, hepatóm, karcinóm hrtana, pažeráka či karcinóm vulvy [26,27].
Sledovanie. 6-mesačné pravidelné fyzikálne vyšetrenie, kontroly krvného obrazu, kostnej drene a príprava na transplantáciu kostnej drene.
Ataxia teleangiektázia (Louis Barr syndróm) (AT)
Toto ochorenie je popri hlavných prejavoch, ktorými sú ataxia a teleangiektázie, asociované aj s malým počtom jednotlivo sa vyskytujúcich „cafe - au - lait“. Pacienti s AT majú približne sto násobne vyššie riziko vzniku malígneho ochorenia ako bežná populácia.
Genetika. Jediný známy gén asociovaný s touto chorobou je tumorsupresorový gén ATM, v ktorom bolo popísaných viac než 300 mutácií. Produktom génu je proteín ATM zo skupiny fosfatidylinositol 3 - kináz, hrajúci dôležitú úlohu v kontrole nádorového rastu, účasti na kontrole bunkového cyklu a reparačných pochodov [27,28].
Malignity. Viac ako 85 % zo všetkých malignít v detskom veku tvorí lymfocytová leukémia alebo lymfóm. U dospelých s AT sa častejšie vyskytujú rôzne solídne tumory, vrátane karcinómu prsníka u žien [29].
Sledovanie. 6-mesačné pravidelné fyzikálne vyšetrenie, kontroly krvného obrazu, kostnej drene a onkodermatologické vyšetrenie.
Nijmengen Breakage syndróm (NBS)
Typická je mikrocefalia a znaky na tvári (tzv. „vtáčia tvár“ charakteristická ustupujúcim čelom, prominentnou strednou časťou tváre, dlhým nosom, dlhým filtrom a ustupujúcou mandibulou). Ataxia v porovnaní s AT nie je prítomná. Ťažká a progredientná mikrocefalia je nápadná už 1 mesiac po pôrode a prítomná takmer u všetkých detí. Väčšina pacientov má aj väčšie uši a riedke vlasy. Nízky vzrast sa prejavuje predovšetkým po 2. roku života. Je prítomná porucha humorálnej, ako aj celulárnej imunity, preto deti mávajú časté infekty.
Genetika. Podobne ako AT je ochorenie diagnostikované cytogenetiky spontánnymi chromozómovými aberáciami. Jedná sa o autozómovo- - recesívne ochorenie. V homozygótnom stave sú za ochorenie zodpovedné mutácie v géne NBS. Algoritmus genetického vyšetrenia spočíva najprv vo vyšetrení najčastejších, tzv. slovanských mutácií. V prípade neprítomnosti mutácie je potrebné zrealizovať sekvenačné vyšetrenie celej kódujúcej oblasti génu. Ak ani táto analýza pri jasných klinických znakoch nepotvrdí diagnózu, odporúča sa pokračovať vyšetrením funkčnosti proteínu nibrínu. Toto odporúčanie je dané preto, lebo gén NBS (nibrin) je súčasťou proteínového komplexu (MRN) spolu s MRE11 a RAD50, a preto môže byť mutácia lokalizovaná v druhých génoch [27,30].
Malignity. Približne 40 % detí s NBS vyvinie lymfómy (Burkittov lymfóm a DLBCL) alebo leukémiu ešte pred dovŕšením 20. roku života. Počas života môžu pacienti s NBS vyvinúť aj sekundárne malignity – tiež lymfoproliferatívneho charakteru.
Sledovanie. Pre hyperrádiosenzitivitu sa u NBS pacientov odporúča vyhýbať sa ionizujúcemu žiareniu. Aj v liečbe sa uprednostňujú nižšie úvodné dávky cytostatík, s pomalým zvyšovaním dávky a intenzívnejším sledovaním pacientov. Sledovanie trvá mnoho rokov po stanovení diagnózy a pozostáva z 6 - mesačného fyzikálneho vyšetrenia a vyšetrenia krvného obrazu.
Bloomov syndróm (BS)
Incidencia ochorenia je 1 : 10 000 a predominantne postihuje populáciu Židov rodu Aškenázi. Na koži sa po expozícii slnečným žiarením objavujú rôzne pigmentové zmeny a dilatované cievy v podkoží. Na tvári býva typický motýľovitý exantém. Ďalšie príznaky sú v dôsledku imunodeficiencie, mentálny deficit býva zriedkavý.
Genetika. Pri cytogenetickom vyšetrení zistíme vyššiu frekvenciu chromozómových zlomov a prestavieb. Uvedené autozómovo - recesívne ochorenie spôsobujú mutácie v géne BLM, v dôsledku čoho alterujú alebo redukujú BLM proteínovú DNA helikázovú aktivitu ovplyvňujúcim replikačný proces [27,31].
Malignity. Najčastejšie sa vyskytujú Non - Hodgkinove lymfómy, Wilmsov tumor a osteosarkóm.
Sledovanie. 6 - mesačné pravidelné fyzikálne vyšetrenie, kontroly krvného obrazu, kostnej drene v kožnej ambulancii.
Syndrómy s rôznorodými dysmorfnými prejavmi a nízkym výskytom malignít
Existuje množstvo dysmorfických syndrómov asociovaných s tumorigenézou, ktoré sú však v súvislosti s výskytom malignity len zriedkavo publikované a v literatúre ich nájdeme najskôr v rubrike kazuistík. Niektoré z nich však predstavujeme ako príklad klinických syndrómov s dysmorfnými črtami, ktoré patria medzi bežnejšie syndrómy s o niečo vyššou frekvenciou malignít (tab. 1).
Gorlinov syndróm (Syndróm névu bazálnych buniek, NBCCS)
Pacienti s NBCCS majú charakteristické dysmorfické črty tváre, ako je napríklad: makrocefália, frontálne a temporo -parietálne vyčnievajúce čelo a nápadné supraorbitálne brázdy. U 50 % pacientov je prítomná čiastočná makrocefália. Čeľusť je prognatická, koreň nosa je široký a môže sa vyskytnúť aj telekantus alebo dokonca hypertelorizmus. Okrem toho sa u NBCCS vyskytuje množstvo rádiologických znakov ako dvojklanné, fúzované alebo čiastočne chýbajúce rebrá, s výskytom približne u 60 % prípadov. Kyfoskolióza nastáva u 30 – 40 % a rázštep chrbtice (spina bifida occulta) u 60 % prípadov. Menej frekventovanými znakmi sú krátke metakarpy, pre alebo postaxiálna polydaktýlia, syndaktýlia druhého alebo tretieho prstu a Sprengelova deformácia. Až u 90 % pacientov sa do veku 40 rokov vyvinú čeľustné cysty (odontogénne keratocysty).
Genetika. Gorlin a Goltz [32] predpokladali autozómovo - dominantnú dedičnosť s úplnou penetranciou a variabilnou expresivitou. Gén pre túto chorobu bol lokalizovaný na 9q22.3 [33]. Gén PTC (PTCH) bol identifikovaný ako kauzálny gén pre NBCCS [34] a zohráva úlohu v hedgehog signálnej dráhe [35]. Mutovaný sa nachádza aj pri sporadickej forme bazálneho bunkového karcinómu.
Malignity. Viacnásobný karcinóm névu bazálnych buniek sa objavuje po puberte, obzvlášť na tvári a krku, ale tiež aj na trupe a ostatných častiach tela. Taktiež sa môžu objaviť rôzne neoplazmy alebo hamartóm [35]. Asi 5 % detí s NBCCS môže vyvinúť meduloblastóm alebo PNET (neuroektodermálny tumor). Vrchol incidencie PNETu je v 2. roku života [36].
Sledovanie. Každé 3 mesiace kožné vyšetrenie, fyzikálne vyšetrenie a MR mozgu od 1 roku života. Pacienti s NBCCS sú nadmerne citliví na rádioterapiu ionizačným žiarením a u niektorých pacientov sa po krátkej expozícii na ožiarených miestach v bazálnych bunkách vyvinulo nezvyčajne veľké množstvo tumorov [37]. Z tohto dôvodu je potrebné vyhnúť sa ordinovaniu RTG a CT zobrazovacím vyšetreniam.
Rubinstein Taybiov syndróm (RSTS)
Rubinstein Taybiov syndróm (RSTS) je charakterizovaný typickými črtami tváre, mikrocefáliou, širokým a často hranatým palcom a veľkými prstami na nohách, nízkym vzrastom a stredne ťažkou až ťažkou mentálnou retardáciou [38]. Prenatálny rast je často normálny, ale výška, váha a obvod hlavy percentilne klesá v prvých mesiacoch života. V detstve alebo v adolescencii sa môže vyskytovať obezita. IQ skóre je v rozsahu od 25 – 79, priemerné IQ je medzi 36 a 51. Medzi menej časté nálezy patrí koloboma, katarakta, kongenitálne defekty srdca, obličkové abnormality a kryptorchidizmus [39].
Genetika. Ide o autozómovo - dominantné ochorenie. Pacienti s cytogenetickou aberáciou v 16p13.3 [40] pomohli identifikovať CBP gén (CREB viažúci proteín) ako hlavný kauzálny gén pri RSTS [41]. Mikrodelécie CBP génu boli nájdené iba v 10 %, kým mutácia je prítomná u 30 – 50 % pacientov s RSTS [42].
Malignity. V roku 1995 Miller a Rubinstein [43] opísali pacientov s RSTS, ktorí mali zvýšené riziko tvorby tumorov. Medzi viac ako 700 pacientmi, 17 malo malígne tumory a 19 benígne tumory. Dvanásť z týchto tumorov bolo lokalizovaných v nervovom systéme, zahŕňajúc oligodendroglióm, meduloblastóm, neuroblastóm a menigióm. Iné typy tumorov predstavovali rabdomyosarkóm a leukémie.
Sledovanie. Odporúča sa pravidelné klinické sledovanie podľa neurologickej symptomatológie MR mozgu, aby sa včas rozpoznali vyvíjajúce sa malignity, predovšetkým nervového systému.
Rothmund - Thomsonov syndróm (RTS)
Rothmund - Thomsonov syndróm (RST) je charakterizovaný infantilnou poikilodermiou, riedkymi vlasmi, malou postavou, skeletálnymi a dentálnymi abnormalitami, kataraktou a hypogonadizmom. Skeletálne abnormality zahŕňajú dysplázie, absenciu alebo malformácie kostí (ako napríklad absencia radia), osteopeniu a oneskorené formovanie kostí.
Genetika. Ochorenie je autozómovo - recesívne. Kitao et al [44] identifikovali mutácie v géne RECQL4 lokalizovanom na 8q24.3.
Malignita. U RTS bola zaznamenaná zvýšená frekvencia malignít hlavne osteosarkómov [45]. Okrem osteosarkómu je tiež známe zvýšené riziko vzniku karcinómu bazálnych buniek a dlaždicovitých buniek [46].
Sledovanie. Skeletálna rádiografia dlhých kostí a vyšetrenie kože by mali byť povinné do 5. roku pre všetkých pacientov s RTS [47].
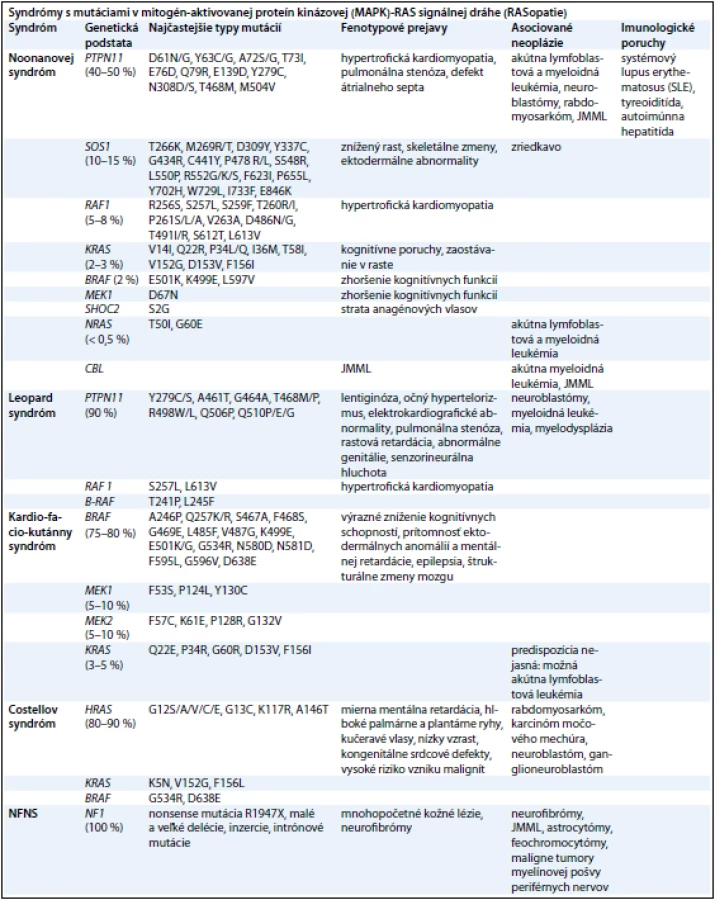
Syndrómy s mutáciami v mitogén - aktivovanej proteín - kinázovej (mapk) - ras signálnej dráhe (rasopatie)
Nedávne genetické štúdie preukázali, že Noonanovej syndróm (NS) a klinicky sa prekrývajúce ochorenia, ako je kardio - facio - kutánny (CFC), Costellov syndróm (CS) a Noonanovej - neurofibromatóza typ 1 (NFNS), sú spôsobené zárodočnými mutáciami v génoch zapojených do Ras - MAPK dráhy, a preto sa súhrnne označujú ako RASopatie (obr. 1). Z hľadiska klinických príznakov ich možno označiť ako neuro facio - kardio - kutánne poruchy [48].
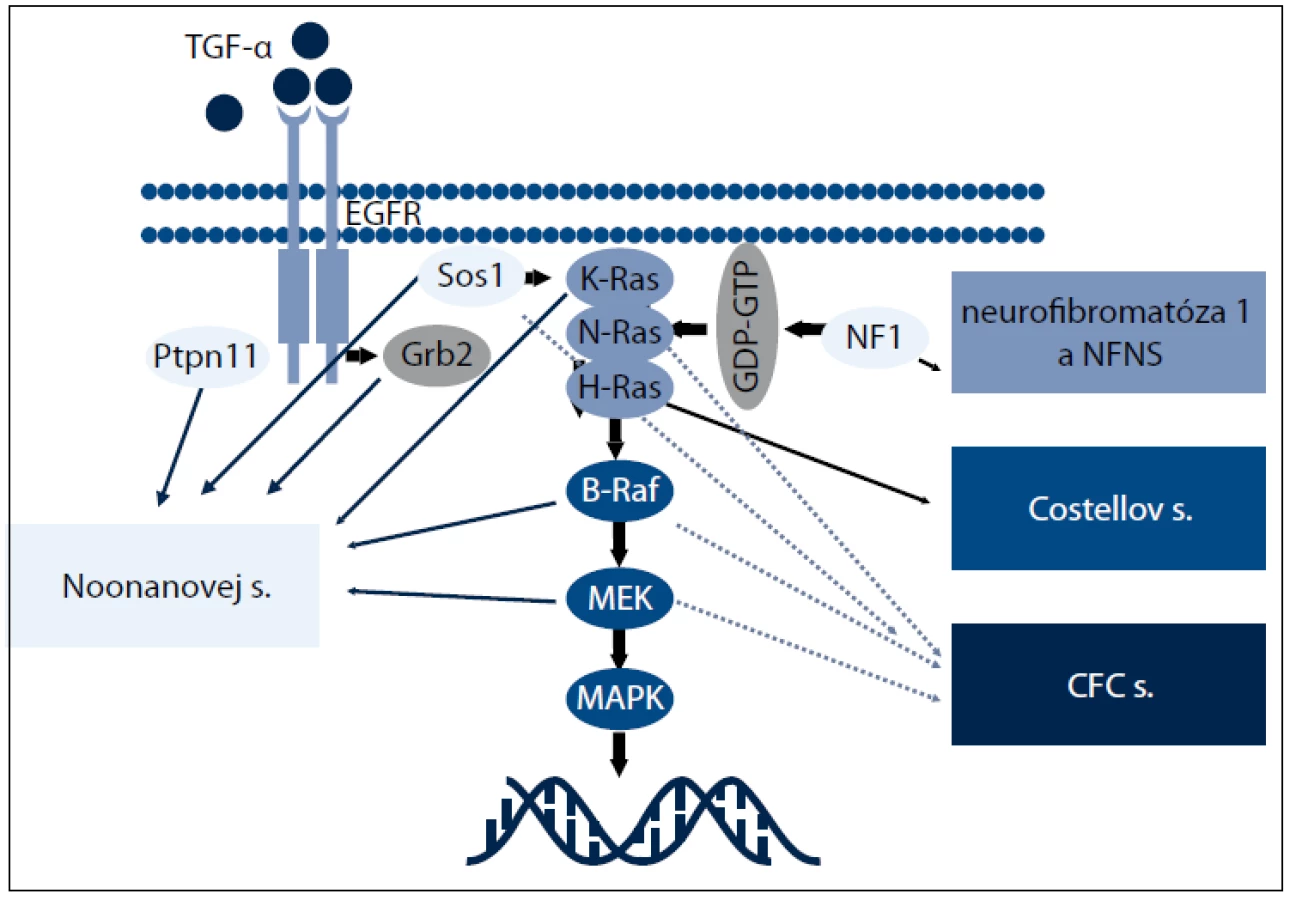
RAS (kódované génmi HRAS, KRAS a NRAS) a mitogénom aktivované proteín - kinázy (MAPK) (kódované génmi RAF, MEK a ERK) sú zapojené do signalizačnej dráhy, ktorá prenáša signály z receptorových proteín - kináz (receptory rastových faktorov) do bunkového jadra. RAS - GTPázy fungujú ako hlavný molekulový prepínač medzi aktívnym GTP - viažucim a inaktívnym GDP - viažucim stavom. Aktivované receptory rastových faktorov získavajú adaptorové proteíny, ktoré aktivujú guanín nukleotid výmenné faktory (guanine nucleotide exchange factors – GEFs), ako SOS1, k presunutiu guaninínových nukleotidov z RAS a dovoľujú ich pasívne naviazanie ku GTP. V GTP - viažucom stave RAS podstupuje konformačné zmeny umožňujúce proteínu viazať a aktivovať efektorové proteíny, ako RAF, čím dochádza k aktivácii MAPK signalizačnej dráhy. Táto interakcia je ukončená hydrolýzou GTP na GDP vnútornou GTPázovou aktivitou RAS, čím dochádza k obnoveniu RAS inaktívnej konformácie. Táto aktivita je zosilnená GTPázu aktivujúcimi proteínmi, akým je neurofibromín, ktorý funguje ako negatívny regulátor RAS signalizácie [49]. Diagnostiku jednotlivých RASopatií sťažujú prekrývajúce sa klinické znaky, široké spektrum fenotypových prejavov [50] (tab. 2). Preto sa molekulárna analýza stala dôležitým kľúčom k potvrdeniu klinickej diagnózy a vytvoreniu vyšetrovacích postupov u jednotlivých pacientov.
Noonanovej syndróm (NS)
Je jedným z najčastejších dedičných monogénových ochorení s incidienciou 1 : 1 000 – 1 : 2 500 živo - narodených detí. Bol prvýkrát opísaný v roku 1963 detskou kadiologičkou Jacqueline Noonanovou [51]. Charakteristickými črtami tohto syndrómu sú predovšetkým: kongenitálne srdcové defekty (pulmonálna stenóza, defekt átrialneho septa a hypertofická kardiomyopatia), nízky vzrast a kranio - faciálne anomálie (hypertelorizmus, nízko posadené a posteriórne orientované uši, ptóza viečok, ametropia, krátky a široký krk, pterýgium coli, nízka vlasová hranica). Často sa objavujú aj deformity hrudníka, hypertenzibilita v kľboch a postihnutie pohlavných orgánov.
Genetika. Hlavným génom zodpovedným za toto ochorenie je PTPN11 (40 – 50 % prípadov), kódujúci SHP2 – tyrozín fosfatázu nereceptorového typu, ktorá funguje ako pozitívny regulátor RAS - MAPK signalizačnej dráhy. V dôsledku mutácie tohto génu dochádza k zmene vlastností proteínového produktu, ktorej fenotypovým prejavom je hypertrofická kardiomyopatia, pulmonálna stenóza a defekt atriálneho septa [52]. Graham et al v roku 2009 zistili, že jednou z možných príčin vzniku NS môže byť aj duplikácia postihujúca PTPN11 lokus, vedúca k zvýšeniu SHP2 a následnej dysregulácii vnútrobunkovej signalizácie [53]. Druhým najčastejšie mutovaným génom pri NS je SOS1 (10 – 15 % prípadov), ktorého proteínovým produktom je RAS - špecifický guanín - nukleotid výmenný faktor. Mutácia v tomto géne sa prejavuje zníženým rastom a skeletálnymi zmenami [54]. Mutácie v géne RAF1 (5 – 8 % prípadov), ktorý kóduje serín - treonínovú kinázu aktivujúcu MEK1 a MEK2, vedú k hypertrofickej kardiomyopatii [55]. V 2 – 3 % prípadov je za diagnózu NS zodpovedná mutácia v KRAS géne, ktorá sa klinicky prejavuje kognitívnymi poruchami a zaostávaním v raste [56]. Za miernejšiu formu NS, ktorej fenotypovým prejavom je mierne zhoršenie kognitívnych schopností, zodpovedajú mutácie v génoch BRAF a MEK1, ktoré sú však typické pre CFC syndróm [57]. Nedávno boli identifikované gény pre zriedkavé formy NS, a to: CBL gén, ktorého mutácia je zodpovedná za juvenilnú myelomonocytovú leukémiu (JMML) [58] a NRAS (menej ako 0,5 % prípadov NS) [59].
Malignity. Za najčastejšie nádory pri NS sa považuje neuroblastóm a akútna lymfoblastová leukémia. Druhoradé sú gliómy (low - grade glioma) a rabdomyosarkóm [60]. Holandská epidemiologická štúdia z roku 2011 uviedla 3,5 - krát vyššie riziko vzniku rakoviny u pacientov s NS a mutáciou v géne PTPN11 v porovnaní s bežnou populáciou [61]. Aktuálne údaje naznačujú, že NS môže byť spojený so širokým spektrom nádorových ochorení, než sa predtým predpokladalo a rovnako aj s miernym zvýšením rizika vzniku a vývoja malignít.
Kardio - facio - kutánny (CFC)
CFC je možné odlíšiť od NS na základe výrazného zníženia kognitívnych schopností, prítomnosti ektodermálnych anomálií, hlavne hyperkeratotických kožných lézií a svetlých kučeravých vlasov, mentálnej retardácie, či menej často sa vyskytujúcej epilepsie a štrukturálnych zmien mozgu.
Genetika. K týmto klinickým prejavom dochádza v dôsledku mutácií v génoch: BRAF (75 – 80 %), MEK1 (5 – 10 %), MEK2 (5 – 10 %) a KRAS (5 %) [62].
Malignity. U tohto syndrómu sa v porovnaní s inými RASopatiami predpokladá znížené riziko vzniku neoplázií [63].
Costellov syndróm (CS)
Fenotypovo sa prejavuje miernou alebo strednou mentálnou retardáciou, hlbokými palmárnymi a plantárnymi ryhami, plnými perami, veľkým jazykom, kučeravými vlasmi, nízkym vzrastom, kongenitálnymi srdcovými defektmi [64].
Genetika. Pre CS sú typické mutácie v HRAS géne.
Malignity. CS je charakteristický predovšetkým vysokým rizikom vzniku malignít (15 – 25 %): rhabdomyosarkómu, karcinómu močového mechúra a neuroblastómu. U CS pacientov sa môže vyvinúť aj neurinóm akustiku a epitelióm [65].
Noonanovej - neurofibromatóza typ 1 (NFNS)
Hoci NF1 a NS predstavujú dva odlišné syndrómy, môžu sa klinicky prekrývať. V takomto prípade ide o NFNS, ktorý bol popísaný v roku 1985 Allansonom et al [66]. Klinickým prejavom tohto syndrómu sú: kraniofaciálne anomálie, nízky vzrast, poruchy učenia [23] a s NF1 - asociované mnohopočetné kožné lézie a neurofibrómy [67].
Genetika. Z molekulárneho hľadiska je príčina prekrývania klinických prejavov oboch syndrómov jasná. Produkty oboch génov (neurofibromín a SHP - 2) sú zapojené do RAS - sprostredkovanej transdukčnej kaskády, kde pôsobia ako antagonisti meniaci bunkovú odpoveď na cytokínové receptory a rastové faktory. Preto sa predpokladá, že je to jedna z hlavných príčin zvýšenej náchylnosti NFNS pacientov k rozvoju hematologických malignít [68]. V roku 2005 DeLuca et al dospeli k záveru, že hlavným génom zodpovedným za vznik NFNS je práve NF1 gén. Toto tvrdenie pramenilo z lokalizácie mutácie u 17 pacientov s NFNS, z ktorých až u 16 bola mutácia prítomná v NF1 géne [34]. Zistilo sa, že v prípade mutácie NF1 génu za vývojové poruchy zodpovedá zárodočná mutácia v jednej alele NF1 [35], kým kožné lézie a tumory sú výsledkom straty funkcie druhej alely NF1 génu v dôsledku somatickej mutácie a klonálnej expanzie buniek zbavených schopnosti inhibície RAS - MAPK dráhy neurofibromínom, ktorý je za normálnych okolností negatívnym regulátorom tejto dráhy [69].
Malignity. Hematologické malignity (JMML, akútna leukémia a lymfómy), nádory mozgu (gliómy, pilocytárne astrocytómy).
Sledovanie spoločné pre všetky Rasopatie
Očné vyšetrenie pri problémoch každé 2 roky, vyšetrenie sluchu, hrudníka, kardiologické, ortopedické a zubné vyšetrenie od 1. roku života a potom každý rok. Pri riziku malígnej hypertermie pri príjme celkovej anestézie. V prípade myopatie vyšetrenie kreatínkinázy. Krvný obraz a zobrazovacie vyšetrenia pri symptómoch upozorňujúcich na malignitu [70].
V súčasnosti sa mnoho štúdií venuje testovaniu látok, ktoré pôsobia ako inhibítory jednotlivých zložiek RAS - MAPK dráhy. Takýmito látkami sú napr. inhibítory RAF a MEK, ako PD - 325901 a sorafenib, zabraňujúce vzniku rakoviny [71]. Zistilo sa však, že nie sú vhodné na dlhodobú liečbu. Ďalšími látkami s inhibičnou aktivitou pôsobiacou na RAS dráhu sú inhibítory HMG - CoA reduktázy či lovastatín, ktorý obnovuje schopnosť učenia sa u myší s mutáciou v NF1 géne [72]. Veľmi vzrušujúcou oblasťou výskumu je v súčasnosti využitie genetických poznatkov v klinickej praxe. Očakávame, že pochopenie molekulovo - genetických mechanizmov vzniku jednotlivých RASopatií a ďalších horeuvedených syndrómov s dysmorfickými črtami prispeje k zlepšeniu diagnostických a terapeutických prístupov u týchto pacientov, predovšetkým malignít.
Molekulová diagnostika Rasopatií je finančne podporená Ligou proti rakovine SR.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Denisa Ilenčíková, PhD.
II. detská klinika
LF UK a DFNsP Bratislava
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: denisa.ilencikova@dfnsp.sk
Obdrženo: 24. 4. 2012
Přijato: 25. 6. 2012
Sources
1. Merks JH, Caron HN, Hennekam RC. High incidence of malformation syndrome in a series of 1,073 children with cancer. Am J Med Genet A 2005; 34A(2): 132 – 143.
2. Lapunzina P. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet 2005; 137C(1): 53 – 71.
3. Martin RA, Grange DK, Zehnbauer B et al. LIT1 and H19 methylation defect in isolated hemihyperplasia. Am J Med Genet A 2005; 134A(2): 129 – 131.
4. Niemitz EL, Feinberg AP, Brandenburg SA et al. Children with idiopathic hemihypertrophy and Beckwith - Wiedemann syndrome have different constitutional epigenotypes associated with Wilms tumor. Am J Hum Genet 2005; 77(5): 887 – 891.
5. Weng EY, Moeschler JB, Graham JM Jr. Logitudinal observations on 15 children with Wiedemann‑Beckwith syndrome. Am J Med Genet 1995; 56(4): 366 – 373.
6. Pettenati MJ, Haines JL, Higgins RR et al. Wiedemann‑Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of literature. Hum Genet 1986; 74(2): 143 – 154.
7. Slavotinek A, Gaunt L, Donnai D. Paternally inherited duplications of 11p15.5 and Beckwith - Wiedemann syndrome. J Med Genet 1997; 34(10): 819 – 826.
8. Weksberk R, Shumann C, Smith AC. Beckwith - Wiedermann syndrome. Am J Med Genet C 2005; 137C(1): 12 – 23.
9. Rump P, Zeegers MP, van Essen AJ. Tumor risk in Beckwith - Wiedemann syndrome: a review and meta‑anlysis. Am J Med Genet A 2005; 136(1): 95 – 104.
10. Bliek J, Glicquel C, Gaston V et al. Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith - Wiedemann syndrome (BWS). J Pediatr 2004; 145(6): 796 – 799.
11. Choyke PL, Siegel MJ, Craft AW et al. Screening for Wilms tumor in children with Beckwith - Wiedemann syndrome or idiopathic hemihypertrophy. Med Pediatr Oncol 1999; 32(3): 196 – 200.
12. Evermann DB, Shuman C, Dzolganovski B et al. Serum alpha - fetoprotein levels in Beckwith - Wiedemann syndrome. J Pediatr 2000; 137(1): 123 – 127.
13. Cohen MM Jr, Neri G, Weksberg R. Overgrowth Syndromes. New York: Oxford University Press 2002 : 75 – 110.
14. Biesecker LG. The multifaceted challenges of Proteus syndrome. JAMA 2001; 285(17): 2240 – 2243.
15. Zhou XP, Marsh DJ, Hampel H et al. Germline and germline mosaic PTEN mutations associated with a Proteus‑like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet 2000; 9(5): 765 – 768.
16. Cohen MM Jr, Gorlin RJ. Noonan‑like/ multiple giant cell lesion syndrome. Am J Hum Genet 1991; 40(2): 159 – 166.
17. Tatton - Brown K, Douglas J, Coleman K et al. Genotype - phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet 2005; 77(2): 193 – 204.
18. Hersh JH, Cole TR, Bloom AS et al. Risk of malignancy in Sotos syndrome. J Pediatr 1992; 120(4 Pt 1): 572 – 574.
19. Golabi M, Rosen L. A new X‑linked mental retardation - overgrowth syndrome. Am J Med Genet 1984; 17(1): 345 – 358.
20. Pilia G, Hughes - Benzie RM, MacKenzie A et al. Mutations in GPC3, a glypican gene, cause the Simpson - Golabi - Behmel overgrowth syndrome. Nat Genet 1996; 12(3): 241 – 247.
21. Li M, Shuman C, Fei Y et al. GPC3 mutation analysis in a spectrum of patients with overgrowth expands the phenotype of Simpson - Golabi - Behmel syndrome. Am J Med Genet 2001; 102(2): 161 – 168.
22. Ruvalcaba RH, Myhre S, Smith DW. Sotos syndrome with intestinal polyposis and pigmentary changes of the genitalia. Clin Genet 1980; 18(6): 413 – 416.
23. Marsh DJ, Dahia PL, Zheng Z et al. Germline mutations in PTEN are present in Bannayan - Zonana syndrome. Nat Genet 1997; 16(4): 33 – 34.
24. Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program 2007; 1 : 29 – 39.
25. Kutler DI, Singh B, Satagopan J et al. A 20‑year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003; 101(4): 1249 – 1256.
26. Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood 2006; 107(11): 4223 – 4233.
27. Krutílková V, Eckschlager T. Přehled syndromů spojených s rizikem nádorů dĕtského vĕku. Klin Onkol 2009; 22 (Suppl): 45 – 49.
28. Ball LG, Xiao W. Molecular basis of ataxia telangiectasia and related diseases. Acta Pharmacol Sin 2005; 26(8): 897 – 907.
29. Seemanová E, Mišovicová N, Schindler D. Louis - Barové syndrom (ataxia teleangiectasia) v konsanguinní rodině. Čes Slov Pediat 2006; 61(11): 666 – 668.
30. Callén E, Samper E, Ramírez MJ et al. Breaks and telomeres and TRF2 - independent end fusions in Fanconi anemia. Hum Mol Genet 2002; 11(4): 439 – 444.
31. Amor - Guéret M. Bloom syndrome, genomic instability and cancer: the SOS‑like hypothesis. Cancer Lett 2006; 236(1): 1 – 12.
32. Gorlin RJ, Goltz RW. Multiple nevoid basal - cell epithelioma, jaw cysts and bifid rib: a syndrome. N Engl J Med 1960; 262 : 908 – 912.
33. Reis A, Küster W, Linss G et al. Localisation of gene for for naevoid basal - cell carcinoma syndrome. Lancet 1992; 339(8793): 617.
34. Hahn H, Gillies S, Negus K et al. Mutation of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85(6): 841 – 851.
35. Stone DM, Phillips H, Noll M et al. The tumour - suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996; 384(6605): 129 – 134.
36. Cowan R, Hoban P, Kelsey A et al. The gene for the naevoid basal cell carcinoma syndrome acts as tumour - suppressor gene in medulloblastoma. Br J Cancer 1997; 76(2): 141 – 145.
37. Featherstone T, Taylor AM, Harnden DG. Studies on the radiosensitivity of cells from patients with basal cell naevus syndrome. Am J Hum Genet 1983; 35(1): 58 – 66.
38. Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities A possible mental retardation syndrome. Am J Dis Child 1963; 105 : 588 – 608.
39. Hennekam RC. Rubistein‑Taybi syndrome. Eur J Hum Genet 2006; 14(9): 981 – 985.
40. Lacombe D, Saure R, Taine L et al. Confirmation of assigment of a locus for Rubistein‑Taybi syndrome gene to 16p13.3. Am J Med Genet 1992; 44(1): 126 – 128.
41. Petrij F, Giles RH, Dauwerse HG et al. Rubistein‑Taybi syndrome caused by mutations in the transcriptional co‑activator CBP. Nature 1995; 376(6538): 348 – 351.
42. Bartsch O, Schmidt S, Richter M et al. DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubistein‑Taybi syndrome (RSTS) and in another patient with incomplete RSTS. Hum Genet 2005; 117(5): 485 – 493.
43. Miller RW, Rubinstein JH. Tumors in Rubistein‑Taybi syndrome. Am J Med Genet 1995; 56(1): 112 – 115.
44. Kitao S, Shimamoto A, Goto M et al. Mutations in RECQL4 cause a subset of cases of Rothmund - Thomson syndrome. Nat Genet 1999; 22(1): 82 – 84.
45. Cumin I, Cohen JY, David A et al. Rothmund - Thomson syndrome complicated by osteosacroma. Med Pediatr Oncol 1996; 26(6): 414 – 416.
46. Piquero‑Casals J, Okubo AY, Nico MM. Rothmund - Thomson syndrome in three siblings and development of cutaneous squamous cell carcinoma. Pediatr Dermatol 2002; 19(4): 312 – 316.
47. Wang LL, Levy ML, Lewis RA et al. Clinical manifestations in cohort of 41 Rothmund - Thomson syndrome patients. Am J Med Genet 2001; 102(1): 11 – 17.
48. Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol 2010; 1(1): 2 – 26.
49. Wittinghofer A. Signal transduction via Ras. Biol Chem 1998; 379(8 – 9): 933 – 937.
50. Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 2011; 25(1): 161 – 179.
51. Noonan JA, Ehmke DA. Associated non cardiac malformations in children with congenital hearth disease. J Pediatr 1963; 63 : 468 – 470.
52. Tartaglia M, Kalidas K, Shaw A et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype - phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002; 70(6): 1555 – 1563.
53. Graham JM Jr, Kramer N, Bejjani BA et al. Genomic duplication of PTPN11 is an uncommon cause of Noonan syndrome. Am J Med Genet A 2009; 149A(10): 2122 – 2128.
54. Lepri F, De Luca A, Stella L et al. SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype - phenotype correlations. Hum Mutat 2011; 32(7): 760 – 762.
55. Pandit B, Sarkozy A, Pennacchio LA et al. Gain‑of - function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 2007; 39(8): 1007 – 1012.
56. Schubbert S, Zenker M, Rowe SL et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet 2006; 38(3): 331 – 336.
57. Koudova M, Seemanova E, Zenker M. Novel BRAF mutation in a patient with LEOPARD syndrome and normal intelligence. Eur J Med Genet 2009; 52(5): 337 – 340.
58. Martinelli S, De Luca A, Stellacci E et al. Heterozygous germline mutations in the CBL tumor - suppressor gene cause a Noonan syndrome‑like phenotype. Am J Hum Genet 2010; 87(2): 250 – 257.
59. Cirstea IC, Kutsche K, Dvorsky R et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 2010; 42(1): 27 – 29.
60. Kratz CP, Rapisuwon S, Reed H et al. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet 2011; 157(2): 83 – 89.
61. Jongmans MC, van der Burgt I, Hoogerbrugge PM et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet 2011; 19(8): 870 – 874.
62. Niihori T, Aoki Y, Narumi Y et al. Germline KRAS and BRAF mutations in cardio - facio - cutaneous syndrome. Nat Genet 2006; 38(3): 294 – 296.
63. van Den Berg H, Hennekam RC. Acute lymphobastic leukaemia in a patient with cardiofaciocutaneous syndrome. J Med Genet 1999; 36(10): 799 – 800.
64. Lin AE, Alexander ME, Colan SD et al. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: a Ras/ MAPK pathway syndrome. Am J Med Genet A 2011; 155A(3): 486 – 507.
65. Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet 2005; 137C(1): 72 – 77.
66. Allanson JE, Hall JG, van Allen MI. Noona phenotype associated with neurofibromatosis. Am J Med Genet 1985; 21(3): 457 – 462.
67. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda Md, USA, July 13 – 15, 1987. Neurofibromatosis 1988; 1(3): 172 – 178.
68. Opitz JM, Weaver DD. The neurofibromatosis - Noonan syndrome. Am J Med Genet 1985; 21(3): 477 – 490.
69. Hüffmeier U, Zenker M, Hoyer J et al. A variable combination of features of Noonan syndrome and neurofibromatosis type I are caused by mutations in the NF1 gene. Am J Med Genet A 2006; 140(24): 2749 – 2756.
70. Romano JA, Pierpont ME, Roberts AN et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 2010; 126(4): 746 – 759.
71. Maertens O, Brems H, Vandesompele J et al. Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum Mutat 2006; 27(10): 1030 – 1040.
72. Li W, Cui Y, Kushner SA et al. The HMG - CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 2005; 15(21): 1961 – 1967.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2012 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Birt-Hogg-Dubé Syndrome
- The Clinical Importance of a Genetic Analysis of Moderate-Risk Cancer Susceptibility Genes in Breast and Other Cancer Patients from the Czech Republic
- Hereditary Diffuse Gastric Cancer
- Clinical Dysmorphic Syndromes with Tumorigenesis