
Molekulárně cytogenetická analýza chromozomových aberací v buňkách nízkostupňových gliomů a její přínos pro klasifikaci nádoru
Molecular Cytogenetic Analysis of Chromosomal Aberrations in Cells of Low Grade Gliomas and Its Contribution for Tumour Classification
Background:
Low-grade gliomas represent a heterogeneous group of primary brain malignancies. The current diagnostics of these tumors rely strongly on histological classification. With the development of molecular cytogenetic methods several genetic markers were described, conributing to a better distinction of glial subtypes. The aim of this study was to assess the frequency of acquired chromosomal aberrations in low ‑ grade gliomas and to search for new genomic changes associated with higher risk of tumor progression.
Patients and Methods:
We analysed biopsy specimens from 41 patients with histological diagnosis of low-grade glioma using interphase fluorescence in situ hybridization (I ‑ FISH) and single nucleotide polymorphism (SNP) array techniques (19 females and 22 males, medium age 42 years).
Results:
Besides notorious and most frequent finding of combined deletion of 1p/ 19q (81.25% patients) several other recurrent aberrations were described in patients with oligodendrogliomas: deletions of p and q arms of chromosome 4 (25% patients), deletions of the short arms of chromosome 9 (18.75% patients), deletions of the long arms of chromosome 13 and monosomy of chromosome 18 (18.75% patients). In biopsy specimens from patients with astrocytomas, we often observed deletion of 1p (24% patients), amplification of the long arms of chromosome 7 (16% patients), deletion of the long arm of chromosome 13 (20% patients), segmental uniparental disomy (UPD) of the short arms of chromosome 17 (60% patients) and deletion of the long arms of chromosome 19 (28% patients). In one patient we detected a shuttered chromosome 10 resulting from chromothripsis.
Conclusion:
Using a combination of I ‑ FISH and SNP array, we detected not only known chromosomal changes but also new or less frequent recurrent aberrations. Their role in cancer ‑ cell progression and their impact on low ‑ grade gliomas classification remains to be elucidated in a larger cohort of patients.
Key words:
oligodendroglioma – astrocytoma – SNP array – interphase FISH – glioma
This work was supported by grants of Internal Grant Agency of the Czech Ministry of Health No. NT/13212-4, PRVOUK-P27/LF1/1 a RVO-VFN64165.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
5. 11. 2013
Accepted:
29. 1. 2014
Authors:
H. Lhotská 1; Z. Zemanová 1; F. Kramář 2; L. Lizcová 1; K. Svobodová 1; Š. Ransdorfová 3; D. Bystřická 1; Z. Krejčík 3; P. Hrabal 2; A. Dohnalová 4; M. Kaiser 5; K. Michalová 1
Authors‘ workplace:
Centrum nádorové cytogenetiky, Ústav lékařské biochemie a laboratorní diagnostiky
1. LF UK a VFN v Praze
1; Oddělení neurochirurgie, 1. LF UK a ÚVN Praha
2; Oddělení cytogenetiky, Ústav hematologie a krevní transfuze, Praha
3; Fyziologický ústav, 1. LF UK v Praze
4; Neurochirurgie, Krajská nemocnice Liberec
5
Published in:
Klin Onkol 2014; 27(3): 183-191
Category:
Original Articles
Overview
Východiska:
Nízkostupňové gliomy představují heterogenní skupinu primárních mozkových nádorů. Jejich současná diagnostika je založena hlavně na histologické klasifikaci. S rozvojem molekulární cytogenetiky však bylo objeveno několik markerů umožňujících lépe definovat daný gliomový subtyp. Cílem studie bylo sledovat získané chromozomové aberace v buňkách nízkostupňových gliomů molekulárně cytogenetickými metodami a hledat nové genomové změny, které by mohly souviset s progresí nádoru.
Soubor pacientů a metody:
Technikami interfázní fluorescenční in situ hybridizace (I ‑ FISH) a single nucleotide polymorphism (SNP) array jsme vyšetřili vzorky od 41 pacientů s histologicky potvrzenými nízkostupňovými gliomy (19 žen a 22 mužů, medián věku 42 let).
Výsledky:
Kromě nejčastější známé aberace, tj. kombinované delece krátkých ramen chromozomu 1 a dlouhých ramen chromozomu 19 (u 81,25 % pacientů), jsme u pacientů s oligodendrogliomy detekovali další rekurentní aberace – delece krátkých a/ nebo dlouhých ramen chromozomu 4 (25 % nemocných), delece krátkých ramen chromozomu 9 (18,75 % pacientů), delece dlouhých ramen chromozomu 13 a monozomii chromozomu 18 (18,75 % pacientů). U pacientů s astrocytomy jsme často pozorovali deleci krátkých ramen chromozomu 1 (24 % nemocných), amplifikaci dlouhých ramen chromozomu 7 (16 % nemocných), deleci dlouhých ramen chromozomu 13 (20 % nemocných), segmentální uniparentální disomii (UPD) na krátkých ramenech chromozomu 17 (60 % pacientů) a deleci dlouhých ramen chromozomu 19 (28 % nemocných). U jednoho pacienta jsme pozorovali tzv. chromothripsis chromozomu 10.
Závěr:
V pilotní studii jsme kombinací metod I ‑ FISH a SNP array detekovali nejen známé chromozomové změny, které jsou typické pro jednotlivé subtypy nádorů, ale také nové nebo méně časté rekurentní aberace. Jejich úlohu v progresi nádorových buněk, stejně jako jejich význam z hlediska klasifikace nízkostupňových gliomů však bude nezbytné ověřit v dalších studiích na větších souborech nemocných.
Klíčová slova:
oligodendrogliom – astrocytom – SNP array – interfázní FISH – gliom
Východiska
Mezi nejčastější primární nádory centrální nervové soustavy (CNS) patří mozkové gliomy. Jedná se o heterogenní skupinu maligních nádorů skládající se z různých histologických subtypů. V současné době jsou gliomy děleny na základě klasifikace Světové zdravotnické organizace (Word Health Organization – WHO) z roku 2007 [1]. Nejčastější jsou astrocytomy, oligodendrogliomy a smíšené oligoastrocytomy. Jednotlivé subtypy lze pak rozdělit do čtyř stupňů podle stupně malignity, a to na tzv. nízkostupňové gliomy (grade I a II, low ‑ grade) a gliomy vyšších stupňů (grade III a IV, high‑grade). Histologický subtyp a stupeň gliomu jsou spojeny s maligním potenciálem, odpovědí na léčbu a přežitím pacienta [2].
Astrocytomy jsou v rámci primárních mozkových nádorů nejčastěji se vyskytujícím typem gliomu s roční incidencí 4,8 případu na 100 000 obyvatel. Pravděpodobnost 5letého přežití nemocných s tímto subtypem mozkového nádoru činí pouze 15 %. Incidence nízkostupňových astrocytomů je 1,2 případu/ 100 000 obyvatel, zde přežije prvních pět let od diagnózy až 43 % pacientů [3]. V případě nízkostupňových oligodendrogliomů je roční výskyt 0,3 případu a tito pacienti mají 65% pravděpodobnost 5letého přežití [3].
Léčba mozkových gliomů je problematická, protože jejich difuzní charakter neumožňuje úplnou chirurgickou resekci. Výjimku z tohoto pravidla tvoří pilocytický astrocytom (pilocytic astrocytoma, WHO grade I) vyskytující se převážně u pediatrických pacientů. Radikální resekce znamená v tomto případě vyléčení nemocného. Z důvodu neúplné, z biologického hlediska nemožné resekce pak dochází u ostatních gliomů k relapsu onemocnění a v případě gliomů nižších stupňů (grade II) také k malignímu zvratu [4]. Určitou představu o charakteru nádoru dává vyšetření magnetickou rezonancí s kontrastní látkou a magnetická rezonanční spektroskopie, které umožňují s vysokou senzitivitou určit, zda se jedná o nízko ‑ či vysokostupňový gliom [5]. Nicméně současná klasifikace gliomů je stále založena na histopatologických rysech nádoru, jimiž je např. jaderný pleomorfizmus, zvýšená buněčnost a mitotická aktivita, proliferace endoteliálních buněk a nekróza. Takovéto rozlišení jednotlivých subtypů je však často subjektivní, a to i za použití specifických imunohistochemických markerů. Z tohoto důvodu byly gliomy podrobeny molekulárně genetické a cytogenetické analýze, která umožnila definovat v genomu několik oblastí asociovaných s určitým subtypem gliálního nádoru. Velmi významným nálezem u difuzních astrocytomů je např. specifická mutace IDH1 R132H, resp. IDH2 R172M. V současné době je tato mutace považována za první krok v tumorigenezi většiny difuzních astrocytomů (diffuse astrocytoma) [6]. K nejčastěji popisovaným aberacím patří amplifikace dlouhých ramen chromozomu 7 vyskytující se u 55 % difuzních astrocytomů (WHO grade II) [7,8], dále amplifikace krátkých ramen chromozomu 7, spojená s amplifikací genu EGFR, která je charakteristickým nálezem u gliomů vyšších stupňů (anaplastický astrocytom, WHO grade III a glioblastom, WHO grade IV). Kombinovaná delece v oblasti 1p a 19q je pak spojena s oligodendrogliálním subtypem nádorů a bývá nalézána až v 90 % nízkostupňových oligodendrogliomů (WHO grade II) a v 70 % anaplastických oligodendrogliomů (WHO grade III)[1,9,10]. Tato aberace je obvykle důsledkem nebalancované translokace mezi chromozomy 1 a 19, při které vzniká derivovaný chromozom der(1;19)(q10;p10) [11,12]. Podle některých autorů znamená přítomnost kombinované delece 1p/ 19q pro pacienta dobrou prognózu zejména díky pomalejšímu růstu nádoru a lepší odpovědi na radio ‑ a chemoterapii [13 – 15]. Další často se vyskytující aberací jsou mutace nebo delece genu TP53 (lokalizovaného v oblasti 17p13.1) a s tím související segmentální uniparentální dizomie (uniparental disomy – UPD) na krátkých ramenech chromozomu 17 [2]. Pro gliomy vyšších stupňů je typická přítomnost monozomie chromozomu 10, delece RB1 genu (v oblasti 13q14), delece genu PTEN (v oblasti 10q23.3), delece genu CDKN2A (p16, v oblasti 9p21) atd. [16 – 20].
Navzdory velkému množství studií, které se zabývají specifickými markery a chromozomovými aberacemi typickými pro toto nádorové onemocnění, charakteristické změny umožňující definovat jednotlivé subtypy nízkostupňových gliomů již při úvodní diagnóze doposud zůstávají nepopsané. V naší pilotní studii jsme provedli detailní analýzu buněk mozkových nádorů získaných od 41 pacientů s histologicky potvrzenými nízkostupňovými gliomy metodami molekulární cytogenetiky, které umožňují nejen cílenou detekci známých chromozomových aberací, ale i celogenomovou analýzu nádorových buněk. Naším hlavním cílem bylo získat co nejširší přehled o získaných chromozomových změnách vyskytujících se u nízkostupňových astrocytomů a oligodendrogliomů a případně hledat nové aberace, které by mohly být spojeny se zvýšeným rizikem transformace do nádorů vyšších stupňů.
Soubor pacientů a metody
Vyšetřili jsme vzorky nádorové tkáně získané od 41 nemocných s histologicky prokázanými nízkostupňovými gliomy (WHO grade II), kteří byli v letech 2005 – 2013 operováni na Neurochirurgické klinice 1. LF UK a ÚVN v Praze a na Neurochirurgickém oddělení KN v Liberci. Všem pacientům byl předložen formulář pro informovaný souhlas a do projektu byli zařazeni jen ti nemocní, kteří s navrženým léčebným postupem a odběrem vzorků pro výzkumné účely souhlasili.
Odběr a zpracování vzorků
Nádorová tkáň byla odebírána během neurochirurgických operací, a její odběr tak pro pacienta nepředstavoval nadbytečnou zátěž. Čerstvě odebraná tkáň byla přenesena do odběrového média (PBS, heparin), ve kterém byla homogenizována a rozdělena na dvě části. První část byla zpracována standardním cytogenetickým postupem (hypotonie, fixace) a z těchto fixovaných buněčných suspenzí byly připraveny mikroskopické preparáty pro I ‑ FISH. Druhá část homogenizátu byla centrifugována (15 000 rpm, 4 °C) a z buněčné pelety byla prostřednictvím QIAamp DNA Blood Mini kitu (Qiagen Inc., Germantown, MD) izolována genomická DNA (gDNA). Hodnota koncentrace a čistoty gDNA byla určena pomocí NanoQuant Infinite M200 spektrofotometru. Nespotřebované fixované buněčné suspenze i izolovaná gDNA byly zamraženy a archivovány pro případné další analýzy.
Interfázní fluorescenční in situ hybridizace (I ‑ FISH)
K detekci nejčastěji se vyskytujících chromozomových aberací jsme použili metodu dvoubarevné I ‑ FISH s panelem přímo značených lokus specifických (LSI) a/ nebo centromerických (CEP) sond Vysis (Abbott Molecular, Des Plaines, IL, USA). Sonda pro sledovanou oblast byla vždy použita v kombinaci s odlišně značenou kontrolní sondou. Hybridizace byla provedena dle návodu doporučeného výrobcem. Přehled DNA sond použitých u jednotlivých nádorových subtypů je uveden v tab. 1. Buňky byly vizualizovány pomocí DAPI (4,6 - diamid ‑ 2 - fenylindol). U každého vzorku bylo dvěma nezávislými pozorovateli zhodnoceno celkem 200 interfázních jader (na preparát a hybridizační směs). Obraz z fluorescenčního mikroskopu AXIO Imager Z1 (Zeiss) byl zpracován specializovaným softwarem pro I ‑ FISH (ISIS, MetaSystemTM). Hodnota „cut‑off“ byla stanovena na 5 % pro delece či monozomie a na 2,5 % pro amplifikace či nadpočetné chromozomy [16].

Single nucleotide polymorphism (SNP) array
K průkazu změn v genomu nádorových buněk jsme využili SNP array Human CytoSNP ‑ 12 BeadChip (Illumina, San Diego, CA). Na čip bylo hybridizováno 200 ng gDNA dle návodu popsaného výrobcem. Výsledný produkt byl naskenován pomocí BeadStation 500 stanice (Illumina) a výsledky byly analyzovány prostřednictvím programu Illumina KaryoStudio (verze 1.4.3.0). Tento program používá k detekci nebalancovaných změn cnv Partition algoritmus (verze 3.0.7.0.), který je založený na poměru očekávané intenzity sond oproti naměřené intenzitě (Log R ratio) a dále pak na předpokládané frekvenci B alely (B allele frequency).
Statistika
Použitím Mantel ‑ Cox testu jsme statisticky porovnali celkové přežití (overall survival – OS) mezi různými skupinami pacientů. Celkové přežití bylo měřeno od data diagnózy do úmrtí či poslední kontroly pacienta.
Výsledky
Klinická data
Vyšetřili jsme 41 nemocných s histologicky prokázanými nízkostupňovými gliomy, z toho bylo 16 pacientů s oligodendrogliomem a 25 nemocných s difuzním astrocytomem. Celkem se jednalo o 19 žen a 22 mužů. Pacienti s difuzními astrocytomy byli zařazeni do skupiny nízkostupňových astrocytárních gliomů (low-grade astrocytoma – LGA). Nemocní s oligodendrogliálním typem nádorů pak tvořili druhou analyzovanou skupinu (low-grade oligodendroglioma – LGO). Medián věku pacientů s LGA byl v době diagnózy 34 let a u nemocných s LGO 50 let. Medián doby sledování byl v celém souboru 74 měsíců (rozmezí 3 – 160 měsíců). Průměrné OS bylo pro nemocné s astrocytomy 76 měsíců (rozmezí 8 – 154 měsíců) a pro nemocné s oligodendrogliomy 86 měsíců (rozmezí 13 – 167 měsíců). V průběhu studie došlo k progresi nebo recidivě nádoru u 19 pacientů (z toho bylo 10 pacientů s astrocytomy a 9 pacientů s oligodendrogliomy). Patnáct nemocných v průběhu studie zemřelo (10 s astrocytomy, 5 s oligodendrogliomy).
Výsledky I ‑ FISH a SNP array analýz
Celkem u 37 nemocných korespondovaly výsledky I ‑ FISH a SNP array analýz s histologickým a klinickým nálezem a potvrdily tak původní diagnózu. U čtyř nemocných s histologicky definovaným LGA (TKA4, TKA11, TKA13, TKA19) byla detekována kombinovaná delece 1p/ 19q, která se typicky vyskytuje hlavně u oligodendrogliomů (tab. 2). Výsledek I ‑ FISH a SNP array analýz se shodoval v 96,7 % případu. U čtyř pacientů byla z důvodu nedostatečného množství materiálu provedena buď jenom I ‑ FISH (TKA22, TKA24), nebo jen SNP (TKO16, TKO17) array analýza (tab. 2, 3). Ve většině vzorků byly detekovány tři až čtyři chromozomy s aberacemi (rozmezí 2 – 9), přičemž u LGA jsme průměrně nalézali 4,2 aberace na vzorek a u LGO 3,3 aberace na vzorek. Jednalo se o delece, amplifikace či uniparentální disomie (UPD). U šesti pacientů (tři s LGA, tři s LGO) nebyla detekována žádná chromozomová aberace. V obou vyšetřovaných souborech převažoval výskyt delecí/ monozomií nad amplifikacemi/ trizomiemi (graf 1, 2).


Oligodendrogliomy
Nejčastějším nálezem u nemocných s oligodendrogliálním typem nádoru byla kombinovaná delece celých ramen 1p/ 19q, která se vyskytovala u 81,25 % analyzovaných vzorků. Ve většině případů byl však tento nález provázen přítomností dalších aberací, z nichž se rekurentně vyskytovaly delece na chromozomu 4 (p i q ramena) u 25 % pacientů, delece 9p u 18,75 % nemocných, delece 13q a monozomie chromozomu 18rovněž u 18,75 % pacientů. Čtyři z devíti pacientů s kombinovanou delecí 1p/ 19q a další aberací detekovanou v době diagnózy během studie zemřeli. U dalšího pacienta z této skupiny došlo k progresi. U tří nemocných s delecí 1p/ 19q nebyly nalezeny žádné další chromozomové změny. V celém souboru pak nebyly pozorovány žádné aberace v chromozomových oblastech 1q, 3q, 19p a na chromozomech 2, 5, 16, 20 a 21. Výsledky LGO vzorků byly shrnuty v grafu 1 a tab. 3.
Astrocytomy
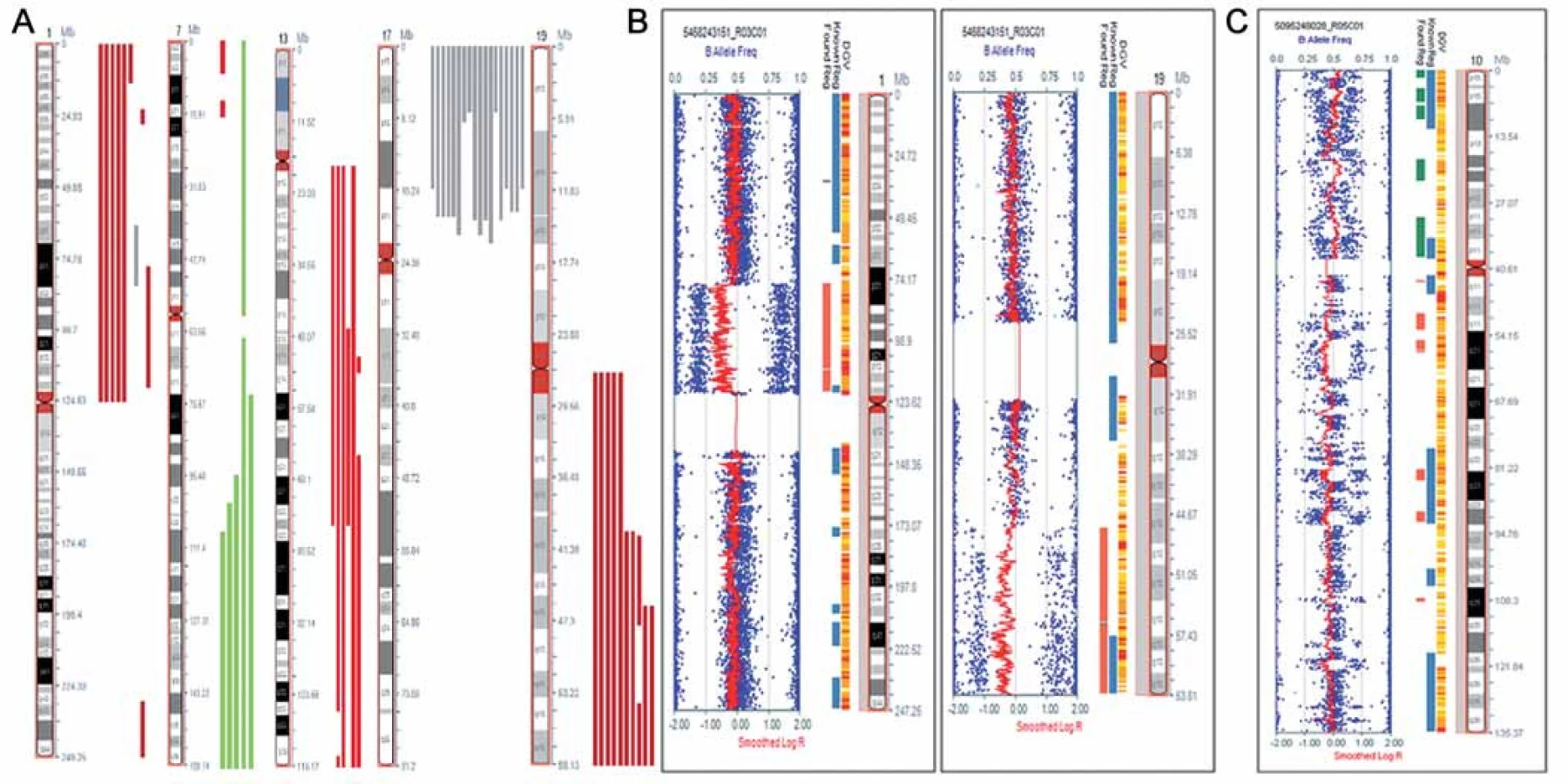
Nálezy ve vzorcích astrocytálních nádorů byly více heterogenní, než tomu bylo u oligodendrogliomů. Mezi často se opakující aberace patřily delece zasahující krátká ramena chromozomu 1nalezené ve 24 % vzorků, amplifikace dlouhých ramen chromozomu 7 (oblast q31.1 - qter) přítomná v 16 % vzorků, delece na chromozomu 13 (oblast q21.3 – q22.3) detekovaná u 20 % případů a delece dlouhých ramen chromozomu 19 prokázaná ve 28 % (obr. 1A, graf 1,tab. 2). Vůbec nejčastějším nálezem v této skupině byla segmentovaná UPD na krátkých ramenech chromozomu 17 (oblast p13.1 – p13.3), kterou jsme detekovali v 60 % případů. U těchto nemocných jsme pozorovali trend k horšímu OS než u ostatních nemocných, i když pravděpodobně vzhledem k nízkému počtu vyšetřených pacientů nebyl zatím rozdíl statisticky významný (p = 0,844). Sedm ze 14 nemocných s detekovanou UPD na chromozomu 17 během studie zemřelo. Ve čtyřech případech LGA byla nalezena kombinovaná delece 1p/ 19q. V jednom ze vzorků (TKA4) jsme pozorovali atypickou kombinovanou deleci, která nezasahovala celá ramena chromozomu 1a 19, ale jen části chromozomu 1 (oblast p12 – p31.1) a chromozomu 19 (oblast q13.2 – q13.43), a tedy vznikla jiným mechanizmem, než je nebalancovaná aberace der(1;19)(q10;p10) (obr. 1B). V dalším vzorku (TKA7) jsme pozorovali fragmentovaný chromozom 10s velkým počtem delecí a amplifikací (obr. 1C). V této skupině postihovaly nebalancované změny všechny chromozomy, žádné aberace nebyly detekovány pouze na krátkých ramenech chromozomu 4 (4p) a na krátkých ramenech chromozomu 16 (16p) (graf 2).
Diskuze
Nízkostupňové gliomy patří k nejčastějším typům mozkových nádorů. Jejich difuzní charakter a komplikovaná histologická klasifikace je řadí mezi obtížně diagnostikovatelné typy malignit. Z terapeutického hlediska je pro pacienta zásadní již při úvodní diagnóze jednoznačně určit, zda se jedná o oligodendrogliom, astrocytom či oligoastrocytom. Histologická klasifikace však může být vzhledem k vysoké heterogenitě těchto nádorů nejednoznačná. Z tohoto důvodu jsou cytogenetické a molekulární metody dalším vhodným nezávislým diagnostickým přístupem. Jejich výsledky nejen doplňují histologické nálezy, ale v některých případech mohou i přispět k upřesnění původní diagnózy [21,22].
V naší studii výsledky molekulární cytogenetické analýzy potvrdily histologický nález u 92,6 % nemocných. U čtyř pacientů s histologicky diagnostikovaným astrocytomem jsme prokázali přítomnost kombinované delece 1p/ 19q, tj. aberace typické pro oligodendrogliomy, a upřesnili tak původní diagnózu na smíšený oligoastrocytom. Celkem u 10 nemocných s LGO jsme kromě specifické delece 1p/ 19q detekovali i další genomové změny, které by mohly mít vliv na prognózu onemocnění. U čtyř těchto nemocných již došlo k progresi nádoru nebo k exitu. Nepříznivý vliv dalších chromozomových aberací v buňkách s delecí 1p/ 19q na průběh onemocnění jsme rovněž pozorovali v naší dřívější studii u nemocných s anaplastickými oligodendrogliomy a oligoastrocytomy (WHO grade III) [16]. V obou vyšetřovaných skupinách jsme detekovali aberace charakteristické pro daný subtyp nízkostupňového difuzního nádoru v četnostech shodujících se s výskytem popsaným v literatuře [7,8,23,24]. Jednotlivé soubory se však mezi sebou lišily v zahrnutí konkrétních chromozomů v chromozomových aberacích. U oligodendrogliomů nebyly detekovány žádné aberace u chromozomů 2, 5, 16, 20 a 21 a v oblastech 1p, 3q a 19p, zatímco u astrocytomů byly takovéto chromozomové oblasti jen dvě – 4p a 16p. Z toho je možné usuzovat, že v případě nemocných s difuzními astrocytomy může docházet k nahodilému vzniku aberací u různých chromozomů, a bude tedy složitější definovat nějaký společný molekulárně cytogenetický ukazatel. Nelze vyloučit, že roli v této nahodilosti změn má právě specifická mutace IDH1 a IDH2.
Podle dosud publikovaných nálezů i podle našich vlastních pozorování je společným rysem astrocytomů absence chromozomových aberací v oblasti 4p[7,8,23,24]. Naopak u oligodendrogliomů, jak popsali Idbaih et al a Rossi et al, dochází k rekurentní ztrátě krátkých ramen chromozomu 4 [25,26]. V našem souboru LGO jsme nalezli celkem čtyři nemocné s delecí 4p (25 %) (graf 2, tab. 3). Tato oblast zahrnuje geny regulující buněčný cyklus (např. CTBP1), tumor supresory (SLIT2, FGFR3, APBB2), geny regulující buněčnou diferenciaci (např. PHOX2B) a geny regulující apoptózu (např. UCHL1). Ztráta exprese těchto genů vlivem delece v oblasti 4p by mohla vést ke zvýšené proliferaci nádorové buňky či tumorogenezi obecně a tím přispět ke vzniku rezistence k léčbě. Skutečný prognostický význam této delece však bude nutné ověřit v dalších studiích na dostatečně velkých souborech pacientů.
Za jeden z možných mechanizmů zodpovědných za iniciaci a progresi nádoru je v současné době považována segmentální uniparentální disomie [27]. UPD může zapříčinit duplikaci heterozygotní somatické či vrozené mutace vedoucí k inaktivaci tumor supresorového genu, nebo naopak ke zvýšené proliferaci díky přítomnosti dvou kopií mutovaného onkogenu. Jedním z takových genů je i TP53 lokalizovaný v oblasti 17p13.1. Molekulární abnormality zahrnující dráhu genu TP53 jsou obvykle jednou z raných událostí v patogenezi difuzních astrocytomů [1]. Populační studie dokázaly, že u difuzních astrocytomů je incidence vzniku mutace TP53 přibližně 53 %, u oligoastrocytomů 44 % a u oligodendrogliomů 13 % [28]. V našem souboru LGA jsme v 60 % případů detekovali segmentální UPD na krátkých ramenech chromozomu 17. Vzhledem k tomu, že se v této oblasti vyskytuje TP53, je možné, že pozorovaná segmentální UPD je asociovaná s mutací v tomto genu. V další studii proto budeme pokračovat v analýze mutačního stavu TP53 u těchto nemocných. Přestože se zatím jedná pouze o malou skupinu a výsledky nejsou statisticky významné, statistická analýza celkového přežití ukázala trend v horším přežívání u pacientů s potvrzenou UPD 17p ve srovnání s nemocnými bez této aberace. Aby bylo možné tento trend statisticky potvrdit, bude jistě nezbytné vyšetřit větší soubory pacientů s LGA a případně doplnit i mutační analýzu TP53 genu.
Dalším nedávno popsaným mechanizmem zodpovědným za vznik chromozomových aberací je jev označovaný jako „chromothripsis“. Tento jev je charakteristický tím, že v jediný kritický moment dochází k rozpadu jednoho či více chromozomů v nádorové buňce na desítky až stovky malých částí. Některé z těchto částí jsou pak v průběhu dalšího buněčného dělení ztraceny nebo naopak amplifikovány, zatímco zbylé jsou reparačními mechanizmy opět náhodně spojeny dohromady, čímž vznikají vysoce přestavěné chromozomy. Chromothripsis byla poprvé popsána u pacientky s chronickou lymfocytární leukemií (chronic lymphocytic leukemia – CLL) a podle dosud publikovaných literárních údajů je přítomna zhruba u 2 – 3 % všech typů maligních onemocnění [29,30]. Zdá se však, že její skutečná incidence bude ve skutečnosti mnohem vyšší, než se původně předpokládalo, a to zejména u agresivnějších typů nádorů. Klinický význam chromothripsis u různých typů nádorových onemocnění zatím není zcela jasný vzhledem k heterogenitě a dosud relativně malému počtu publikovaných případů [31]. V našem souboru jsme takto rozpadlý chromozom 10, pravděpodobně v důsledku chromothripsis, pozorovali u jednoho pacienta s difuzním astrocytomem. Klinický stav tohoto pacienta je v současné době dobrý a budeme jej dále sledovat.
Závěr
Molekulárně cytogenetickými metodami jsme u 16 pacientů s LGO a 25 pacientů s LGA potvrdili přítomnost známých chromozomových aberací charakteristických pro dané subtypy nízkostupňových gliomů. Kromě toho jsme u 10 nemocných s LGO detekovali další rekurentní změny, nejčastěji delece 4p, 9p či 13q. Jaký je přesný význam těchto genomových změn a zda může přítomnost dalších aberací v nádorových buňkách s prognosticky významnou delecí 1p/ 19q ovlivnit prognózu nemocných, je stále předmětem diskuze. V naší studii jsme u pacientů s těmito nálezy pozorovali zvýšené riziko progrese nádoru, i když rozdíl zatím nebyl statisticky významný. U čtyř nemocných s difuzními astrocytomy jsme díky molekulárně cytogenetickým metodám mohli odlišit nádor s oligodendrogliální buněčnou složkou od čistě astrocytálního typu gliomu a upřesnit tak původní diagnózu. Kromě toho jsme u nemocných s LGA detekovali řadu dalších zajímavých chromozomových aberací, jako byla např. chromothripsis chromozomu 10nebo kombinovaná delece 1p/ 19q s atypickými zlomovými místy. Významným nálezem byla zejména segmentální UPD 17p přítomná u více než poloviny pacientů s LGA, u kterých jsme rovněž pozorovali trend k horšímu OS. Ačkoliv mnoho otázek týkajících se klasifikace a progrese nízkostupňových gliomů zůstává stále nezodpovězeno, kombinací nových přístupů vzniká další užitečný analytický nástroj umožňující upřesnit diagnózu a v některých případech i prognózu onemocnění. Naše pilotní výsledky tak mohou sloužit jako východisko pro další molekulárně cytogenetické studie na větších souborech nemocných.
Práce byla podpořena granty IGA MZ ČR č. NT/13212-4, PRVOUK-P27/LF1/1 a RVO-VFN64165.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
Mgr. Halka Lhotská
Centrum nádorové cytogenetiky
Ústav lékařské biochemie a laboratorní diagnostiky
1. LF UK a VFN
U Nemocnice 2
128 08 Praha 2
e-mail: halka.buryova@vfn.cz
Obdrženo: 5. 11. 2013
Přijato: 29. 1. 2014
Sources
1. Louis DN, Ohgaki H, Wiestler OD et al. The 2007 WHO classification of tumours of the centralnervous system. Acta Neuropathologica 2007; 114(2): 97 – 109.
2. Huttner A. Overview of primary brain tumors pathologic classification, epidemiology, molecular biology, and prognostic markers. Hematol Oncol Clin North Am 2012; 26(4): 715 – 732. doi: 10.1016/ j.hoc.2012.05.004.
3. Crocetti E, Trama A, Stiller C et al. Epidemiology of glial and non‑glial brain tumours in Europe. Eur J Cancer 2012; 48(10): 1532 – 1542. doi: 10.1016/ j.ejca.2011.12.013.
4. Godard S, Getz G, Delorenzi M et al. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res 2003; 63(20): 6613 – 6625.
5. Bulik M, Jancalek R, Vanicek J et al. Potential of MR spectroscopy for assessement of glioma grading. Clin Neurol Neurosurg 2013; 115(2): 146 – 153. doi: 10.1016/ j.clineuro.2012.11.002.
6. Labussiére M, Idbaih A, Wang XW et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010; 74(23): 1886 – 1890. doi: 10.1212/ WNL.0b013e3181e1cf3a.
7. Idbaih A, Marie Y, Lucchesi C et al. BAC array CGH distinguishes mutually exclusive alterations that define clinicogenetic subtypes of gliomas. Int J Cancer 2008; 122(8): 1778 – 1786.
8. Roerig P, Nessling M, Radlwimmer B et al. Molecular classification of human gliomas using matrix‑based comparative genomic hybridization. Int J Cancer 2005; 117(1): 95 – 103.
9. Jeuken JW, von Deimling A, Wesseling P. Molecular pathogenesis of oligodendroglial tumors. J Neurooncol 2004; 70(2): 161 – 181.
10. Cairncross G, Jenkins R. Gliomas with 1p/ 19q codeletion: a.k.a. oligodendroglioma. Cancer J 2008; 14(6): 352 – 357. doi: 10.1097/ PPO.0b013e31818d8178.
11. Jenkins RB, Blair H, Ballman KV et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 2006; 66(20): 9852 – 9861.
12. Griffin CA, Burger P, Morsberger L et al. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 2006; 65(10): 988 – 994.
13. Cairncross G, Berkey B, Shaw E et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup radiation therapy oncology group trial 9402. J Clin Oncol 2006; 24(18): 2707 – 2714.
14. Cairncross G, Wang MH, Shaw E et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long‑term results of RTOG 9402. J Clin Oncol 2013; 31(3): 337 – 343. doi: 10.1200/ JCO.2012.43.2674.
15. Jurga LM, Malý M. Úskalia kombinovanej rádiochemoterapie a bioterapie malígnych gliómov. Klin Onkol 2006; 19(6): 317 – 320.
16. Zemanová Z, Kramar F, Babicka L et al. Molecular cytogenetic stratification of recurrent oligodendrogliomas: utility of interphase fluorescence in situ hybridization (I ‑ FISH). Folia Biol 2006; 52(3): 71 – 78.
17. Wiltshire RN, Rasheed BK, Friedman HS et al. Comparative genetic patterns of glioblastoma multiforme: potential diagnostic tool for tumor classification. Neuro Oncol 2000; 2(3): 164 – 173.
18. Holland H, Ahnert P, Koschny R et al. Detection of novel genomic aberrations in anaplastic astrocytomas by GTG ‑ banding, SKY, locus ‑ specific FISH, and high density SNP ‑ array. Pathol Res Pract 2012; 208(6): 325 – 330. doi: 10.1016/ j.prp.2012.03.010.
19. Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet 2012; 205(12): 613 – 621. doi: 10.1016/ j.cancergen.2012.10.009.
20. Vránová V, Necesalová E, Kuglík P et al. Screening of genomic imbalances in glioblastoma multiforme using high‑resolution comparative genomic hybridization. Oncol Rep 2007; 17(2): 457 – 464.
21. Cowell JK, Lo KC, Luce J et al. Interpreting aCGH ‑ defined karyotypic changes in gliomas using copy number status, loss of heterozygosity and allelic ratios. Exp Mol Pathol 2010; 88(1): 82 – 89. doi: 10.1016/ j.yexmp.2009.09.014.
22. Belaud ‑ Rotureau MA, Meunier N, Eimer S et al. Automatized assessment of 1p36 – 19q13 status in gliomas by interphase FISH assay on touch imprints of frozen tumours. Acta Neuropathol 2006; 111(3): 255 – 263.
23. Wiltshire RN, Herndon JE, Lloyd A et al. Comparative genomic hybridization analysis of astrocytomas – prognostic and diagnostic implications. J Mol Diagn 2004; 6(3): 166 – 179.
24. Li YB, Wang DP, Wang L et al. Distinct genomic aberrations between low ‑ grade and high‑grade gliomas of chinese patients. PLoS One 2013; 8(2): e57168. doi: 10.1371/ journal.pone.0057168.
25. Idbaih A, Crinière E, Ligon KL et al. Array‑based genomics in glioma research. Brain Pathol 2010; 20(1): 28 – 38. doi: 10.1111/ j.1750 - 3639.2009.00274.x.
26. Rossi MR, Gaile D, Laduca J et al. Identification of consistent novel submegabase deletions in low ‑ grade oligodendrogliomas using array‑based comparative genomic hybridization. Genes Chromosomes Cancer 2005; 44(1): 85 – 96.
27. Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med 2009; 15(3): 120 – 128. doi: 10.1016/ j.molmed.2009.01.005
28. Okamoto Y, Di Patre PL, Burkhard C et al. Population‑based study on incidence, survival rates, and genetic alterations of low ‑ grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol 2004; 108(1): 49 – 56.
29. Stephens PJ, Greenman CD, Fu B et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144(1): 27 – 40. doi: 10.1016/ j.cell.2010.11.055.
30. Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell 2012; 148(1 – 2): 29 – 32. doi: 10.1016/ j.cell.2012.01.006.
31. Gaiser T, Gaiser MR, Hirsch D et al. Chromothripsis and focal copy number alterations determine poor outcome in malignant melanoma. J Invest Dermatol 2013; 133: S56 – S56. doi:10.1038/ jid.2013.96.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2014 Issue 3
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Velmi pozdní následky radioterapie – limitující faktor současných radioterapeutických technik
- Invertovaný papilóm a jeho zriedkavé formy
- Nádorová onemocnění ve starším věku
- Role bevacizumabu ve druhé linii léčby glioblastomu – zmařené naděje?