
Brazilský příběh mutace p53 R337H
:
J. Šmardová 1,2; J. Koptíková 3
:
Ústav patologie, LF MU a FN Brno
1; Ústav experimentální biologie, PřF MU, Brno
2; Institut biostatistiky a analýz, LF a PřF MU, Brno
3
:
Klin Onkol 2014; 27(4): 247-254
:
Review
Nádorový supresor p53 patří k „evergreenům“ molekulární onkologie. Byl objeven v roce 1979 a od té doby je intenzivně zkoumán. Protein p53 má „pouhých“ 393 aminokyselinových zbytků a funkce téměř každého jednoho z nich je pečlivě prozkoumaná a známá. Somatické mutace se vyskytují v téměř všech kodonech p53, jsou extrémně časté a vyskytují se téměř ve všech typech nádorů. Vrozené mutace p53 jsou vzácné, ale výrazně penetrantní a jsou typicky spojeny s rozvojem širokého spektra nádorů. V roce 2001 však v oblasti výzkumu p53 došlo k nečekanému objevu: v jižní Brazílii byla objevena alela R337H, která byla zcela netypicky spojena s jediným typem nádoru – dětským adrenokortikálním karcinomem – a vyznačovala se nízkou penetrancí. Nastala doslova honba za dalšími informacemi o fungování a důsledcích mutace R337H. Ta během několika málo let přinesla nejenom spoustu poznatků o vlastnostech této konkrétní varianty p53, ale i o obecnějších principech fungování mutovaných variant p53. Také se ukázalo, že všechny alely R337H, které jsou v jižní Brazílii masivně rozšířené, pocházejí z jediného zdroje, tj. mají společného předka.
Klíčová slova:
Li ‑ Fraumeniho syndrom – adrenokortikální karcinomy – gen p53 – R337H
Úvod
Nádorový supresor p53 patří k „evergreenům“ molekulární onkologie. Byl objeven v roce 1979 [1,2] a v roce 2003 se stal molekulou roku. p53 je transkripční faktor, který prostřednictvím svých cílových genů zprostředkovává adekvátní reakci buňky na nejrůznější typy stresu. Při poškození DNA, hladovění, nedostatku kyslíku, kritickém zkrácení telomer či vlivem nefyziologicky vysoké exprese některých onkogenů je p53 v buňce stabilizován a aktivuje transkripci svých cílových genů. Může zastavit buněčný cyklus, indukovat mechanizmy opravující DNA, vyvolat senescenci nebo apoptózu. Proto je p53 klíčovým nádorovým supresorem, který velmi komplexně chrání buňku před neoplastickou transformací. Svědčí o tom i vysoká frekvence, s jakou se u lidských nádorů vyskytují somatické mutace p53. Lze je detekovat asi u 40 % všech nádorů a vyskytují se téměř u všech nádorových typů.
Vrozené mutace nádorového supresoru p53 jsou spojené s Li ‑ Fraumeniho syndromem (LFS). Tento vzácný autozomálně dominantní syndrom predisponuje k vývoji širokého spektra nádorů v mladém věku a u 60 – 80 % postižených je způsoben mutací p53 v zárodečné linii. Specifické kumulativní riziko vývoje nádorů je u nositelů vrozené mutace p53 18 % pro ženy ve věku 20 let, 49 % pro ženy ve věku 30 let, 77 % pro ženy ve věku 40 let a 93 % pro ženy ve věku 50 let. Pro muže ve stejných věkových kategoriích riziko dosahuje 10 %, 21 %, 33 % a 68 % [3,4]. Mezi typické a nejčastější nádory v rámci LFS patří nádory prsu (27,8 %), sarkomy měkkých tkání (13,8 %), nádory mozku (12,9 %), adrenokortikální karcinomy (10,9 %), osteosarkomy (8,7 %), leukemie (3,8 %) a další (graf 1). Vedle „klasického“ LFS byl definován také Li ‑ Fraumeniho‑like syndrom (LFLS), který má ve srovnání s LFS méně striktní kritéria, a to jak z hlediska spektra nádorů, tak z hlediska věku pacientů, při kterém se nádory objevují. Také podíl detekovaných vrozených mutací p53 je u rodin s LFLS nižší než u rodin s LFS [4].
![Zastoupení jednotlivých typů nádorů mezi pacienty postiženými LFS.
Převzato a upraveno dle databáze http://www-p53.iarc.fr, verze R17 z listopadu 2013 [5].](https://pl-master.mdcdn.cz/media/image/5a494b381a12fc48ada06ef4300b944e.jpg?version=1537794135)
Poslední verze R17 databáze mutací p53 IARC (International Agency for Research on Cancer) z listopadu 2013 (http:/ / www ‑ p53.iarc.fr) [5,6] zahrnuje 1 360 vrozených a 27 721 somatických mutací p53 a umožňuje srovnání spektra a distribuce zárodečných (graf 2) a somatických (graf 3) mutací genu p53. Podobně jako u somatických, také u vrozených mutací převládají bodové mutace zaměňující v mutovaném proteinu p53 jednu aminokyselinu za jinou (73,2 vs 73,5 %). Nejčastěji se zárodečné i somatické mutace vyskytují v té části genu p53, která kóduje centrální DNA vazebnou doménu, a také většina tzv. „hot ‑ spot“ kodonů, tedy kodonů nejčastěji postižených mutací, je společná pro somatické a zárodečné mutace. Jsou to kodony 175, 245, 248, 273 a 282. U somatických mutací je to navíc kodon 249, zatímco u zárodečných mutací je vůbec nejčastěji mutovaným – vedle kodonu 248 – kodon 337, který ovlivňuje oligomerizační doménu proteinu p53.
![Spektrum zárodečných mutací genu p53.
Převzato a upraveno dle databáze http://www-p53.iarc.fr, verze R17 z listopadu 2013 [5].](https://pl-master.mdcdn.cz/media/image/c4a40cfb2492b4a3ad45b05175861441.jpg?version=1537796514)
![Spektrum somatických mutací genu p53.
Převzato a upraveno dle databáze http://www-p53.iarc.fr, verze R17 z listopadu 2013 [5].](https://pl-master.mdcdn.cz/media/image/b580e920b243a2f9c32ce3f15adddfbd.jpg?version=1537796105)
Vrozené mutace p53 – existuje vztah mezi genotypem a fenotypem LFS?
Jednou z prvních prací, která poukázala na možnou souvislost mezi typem zárodečné mutace v genu p53 a fenotypem LFS – tj. spektrem nádorů a věkem pacientů, ve kterém se nádory objevují ,byla práce Varleye et al (1998). Autoři zde popsali málo obvyklou tichou mutaci v kodonu 125, která vedla k aberantnímu sestřihu exonu 4 a expresi tří abnormálních transkriptů. Spektrum nádorů u postižené rodiny bylo neobvyklé –žádný sarkom, žádný nádor v dětském věku a pro LFS neobvyklý výskyt adrenokortikálního karcinomu v dospělém věku 45 let [7]. Autoři tehdy z literárních údajů dohledali další rodinu se stejnou mutací a povšimli si zarážející podoby fenotypu –ačkoliv v této rodině byly nalezeny nádory v dětském věku (mozku a adrenokortikální), nevyskytl se žádný sarkom a vyskytl se opět adrenokortikální nádor u dospělé ženy staré 33 let [8].
S ohledem na „brazilský příběh“ mutace R337H je velice zajímavá i další práce stejného týmu. Ten v ní publikoval výsledky analýzy genu p53 u 14 nepříbuzných dětských pacientů s adrenokortikálním karcinomem. U 11 z nich (78,6 %) překvapivě detekovali zárodečnou mutaci v genu p53. Ještě překvapivější bylo zjištění, že spektrum mutací p53 bylo velmi úzké, dvě mutace, P152L a R158H, byly nalezeny u devíti dětí. V rodinách nemocných dětí se další nádory mezi nositeli stejných zárodečných mutací vyskytovaly poměrně vzácně, což naznačovalo, že se jedná o mutace s relativně nízkou penetrancí [9].
Další příspěvek k otázce vztahu typu zárodečné mutace p53 a fenotypu LFS přinesli Olivier et al (2003). Analyzovali data z databáze IARC týkající se 265 rodin a 226 mutací p53. Zjistili, že nádory mozku se častěji vyskytují v rodinách, které mají mutaci způsobující záměnu aminokyseliny v DNA vazebné oblasti p53 zajišťující kontakt s malým žlábkem DNA, zatímco adrenokortikální nádory významně častěji souvisejí se změnami aminokyselin umístěných na protilehlé straně proteinu p53 mimo přímý kontakt s DNA. Mutace p53, které způsobují úplnou ztrátu proteinu a funkce 53, jsou významně spojeny s časným výskytem nádorů mozku [10].
Jiný přístup k řešení stejně položené otázky zvolili Monti et al (2007). Tito autoři rozdělili mutace p53 podle míry zachování transkripčně aktivačních vlastností výsledného proteinu p53. Zjistili, že míra transaktivační aktivity proteinu p53 ovlivňuje míru rizika, tedy penetranci, spektrum nádorů i věk, ve kterém se vyvíjejí [11].
Adrenokortikální karcinomy
Adrenokortikální karcinomy (adrenal cortical carcinoma – ACC) jsou maligní nádory, které vycházejí z buněk kůry nadledvin. Vyskytují se v každém věku s převahou v dětství a poté v dospělosti ve 4. – 5. dekádě. Jsou často (až v 50 % případů) hormonálně aktivní. Jejich incidence v ČR dosahuje přibližně 1 – 2 případů na 1 mil. obyvatel a rok [12], což odpovídá celosvětové incidenci ACC u dospělých 1,7 – 2,0 na 1 mil. obyvatel a rok [13].
Dětské ACC se typicky objevují do pátého roku života, přičemž častější jsou u dívek. Pacienti s malými operabilními nádory mají celkové přežití (overall survival – OS) asi 80 %, pacienti s neoperabilním nádorem mají šanci na přežití jen malou, pacienti s velkými operabilními nádory mají střední výhled. Asi 2/3 pacientů mají operabilní nádor. Dětské ACC bývají velmi často spojeny s mutacemi nádorového supresoru p53 [14,15], zatímco u dospělých s ACC je výskyt mutací p53 méně častý [16 – 18].
Dětské adrenokortikální karcinomyjsou velmi vzácné, incidence se v USA uvádí asi 0,3 případu na 1 mil. dětí do 20 let [19]. V nádorovém registru dětí v Manchesteru bylo mezi lety 1954 – 1986 zachyceno 12 dětských případů ACC, incidence byla stanovena na 0,38 případů na 1 mil. dětí do 15 let [20]. Odhad incidence ACC ve Francii je 0,2 případu na mil. dětí [21]. Naproti tomu se odhadovalo, že v jižní Brazílii (ve státech Sao Paulo, Paraná a Minas Gerais; obr. 1) je výskyt dětských ACC podstatně vyšší, dosahuje 3,4 – 4,2 případu na 1 mil. dětí. Přesnější stanovení incidence bylo provedeno v roce 2006, a to pro stát Paraná na základě úmrtí na ACC hlášených ve státní nemocnici v Curitibě v letech 1998 – 2003. Mortalita byla určena na 1,6 případu na 1 mil. dětí do věku 15 let a na základě podílu přežívajících (0,542) byla incidence odhadnuta na 3,5 případu na 1 mil. dětí do 15 let [22], tedy převyšující incidenci v jiných oblastech světa více než 10krát! Populace v jižní Brazílii je velmi heterogenní se silným evropským (portugalským, italským, španělským, německým) zastoupením, spolu s brazilskými indiány a africkými vlivy. Nemoc se vyskytuje u všech etnik. Příčina tak častého výskytu dětských případů ACC byla dlouho nejasná a ani obecnější rodinná genetická predispozice k vývoji nádorů nebyla dlouho zaznamenána.
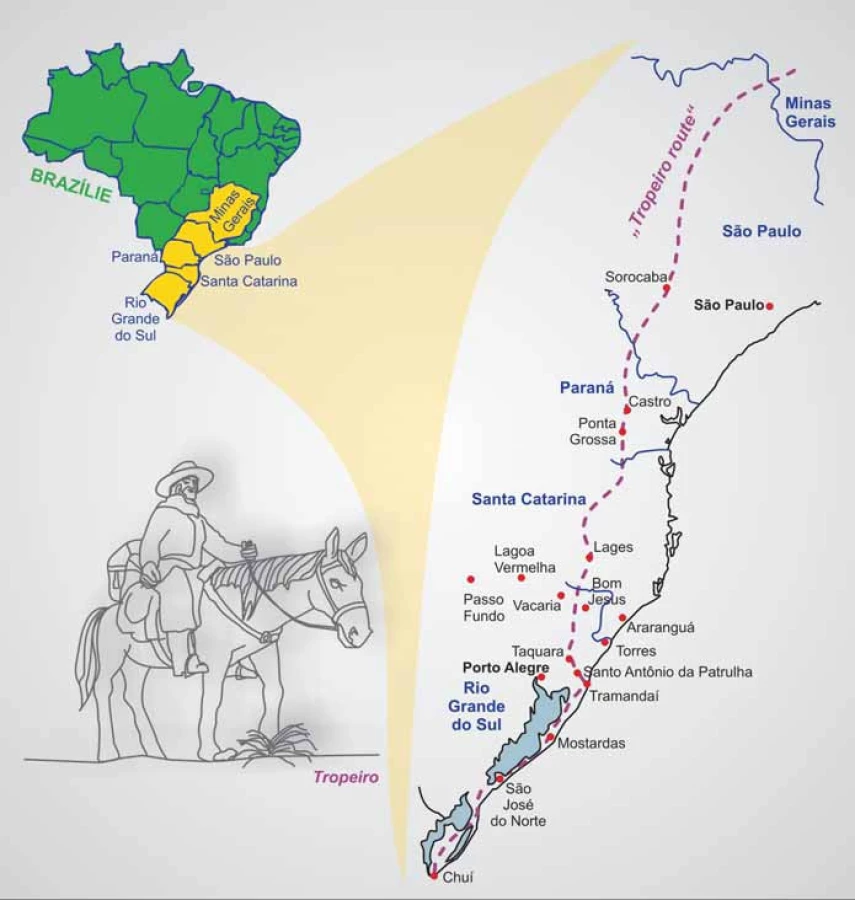
Brazilský příběh mutace R337H
Začátek příběhu – 2001
Příběh mutace R337H začal v roce 2001. Kolektiv autorů [23] tehdy shromáždil dostupné informace o 92 dětech s ACC léčených v nemocnici v Curitibě mezi roky 1966 a 1999. Na základě získaných dat autoři oslovili 55 rodin a vyzvali je, aby se zúčastnily genetického testování. Následně bylo vyšetřeno 36 probandů s ACC, z toho 29 dívek a 7 chlapců, a celkem 186 členů rodin včetně 21 rodičů a 29 sourozenců. Výsledky analýzy byly doslova šokující. Stejnou zárodečnou mutaci p53: R337H mělo 35 ze 36 probandů (97 %!). Tato mutace byla nalezena také u rodičů, sourozenců a dalších příbuzných, ovšem bez výskytu jiných nádorů v rodinné anamnéze. Nádor byl diagnostikován pouze u jediného příbuzného prvního stupně. Pouze jedna rodina (z 25) splňovala kritéria LFS, zbylých 24 rodin nemělo známky zvýšené predispozice k nádorům. Násobný výskyt ACC byl zaznamenán ve čtyřech rodinách. Zdálo se, že celkově je potenciál mutace R337H vyvolávat nádory nízký, má nízkou penetranci a je výrazně orgánově specifický, což je jev v oblasti p53 zcela výjimečný. Velké rozmanitosti a „nespecifičnosti“ mutací p53 se vymyká snad jen somatická mutace v kodonu R249S spojená s hepatocelulárním karcinomem a expozicí aflatoxinům a hepatitidě B. V tomto případě se ale jedná o mutaci somatickou a navíc spojenou s konkrétním kancerogenem. Mutace R337H byla u dětí s ACC zděděná a autoři nezaznamenali v postižené oblasti výskyt žádného známého mutagenu.
Funkční a strukturní analýza mutace R337H
Funkční formou proteinu p53 je tetramer, tj. dimer dimerů. Jak již bylo uvedeno výše, nejčastěji se zárodečné i somatické mutace p53 projevují záměnami aminokyselin v centrální DNA vazebné doméně. Je ale známo i několik mutací, které narušují strukturu oligomerizační domény (vymezenou kodony 310 – 360), znemožňují vytvoření tetrameru a tak narušují funkci proteinu p53. Mutace R337H spadá do oligomerizační domény. Funkční analýzou tohoto mutanta se zabývali již autoři původní práce z roku 2001. Zjistili, že v transkripčně aktivačních testech dosahuje mutant R337H téměř stejné aktivity jako standardní varianta p53, zachovává si schopnost indukovat apoptózu a také schopnost suprimovat růst kolonií osteosarkomových buněk SAOS ‑ 2 [23]. Vysokou transkripčně aktivační schopnost mutanta R337H potvrdili i další autoři [24].
Podrobná strukturní analýza ukázala, že tetramerizační doména s histidinem v pozici 337 má strukturu velice podobnou standardní variantě, ale je méně stabilní, především je velice citlivá k pH. Mutant R337H ztrácí aktivitu již při fyziologických hodnotách pH nad 7,7 [25]. Také se zjistilo, že protein p53 s histidinem v pozici 337 má oproti standardnímu proteinu výrazně vyšší tendenci tvořit fibrily podobné amyloidu [26]. Autoři těchto poznatků spekulovali, že možná právě extrémní citlivost mutanta R337H k pH vysvětluje jeho výrazné spojení s ACC. Nadledvinky procházejí během prenatálního i postnatálního vývoje rozsáhlou remodelací, která vyžaduje bezchybnou indukci apoptózy. Během apoptózy v buňkách dochází k navýšení pH až na hodnoty kolem 7,9, což je hodnota, která mutanta R337H destabilizuje. Ve vyvíjejících se nadledvinkách se tak může vytvářet mikroprostředí, které narušuje nádorově supresorovou funkci p53 a může přispívat k vývoji nádorů [13,25].
Specifický funkční dopad mutace R337H byl později nalezen také ve vztahu k rozvoji variability v počtu kopií. Variabilita v počtu kopií (copy number variability – CNV) představuje jednu z forem polymorfizmu savčího genomu. CNV jsou nerovnováhy – ztráty nebo zisky – DNA, které mění diploidní stav postiženého lokusu. Obvykle mají rozsah od 50 pb do asi 1 Mpb. Odhaduje se, že výskytem CNV může být postiženo asi 15 % lidského genomu a že v průměru každý člověk má ve svém genomu asi 12 CNV [27]. Navýšení počtu CNV v genomu (v zárodečné i somatické linii) může být odrazem zvýšené genetické nestability a může úzce souviset s vývojem nádorů [28,29]. Nositelé vrozené mutace p53 mají zhruba 3násobně zvýšený počet CNV oproti zdravým kontrolám, což může zvyšovat genetickou nestabilitu a tím riziko vývoje nádorů a také vysvětlovat anticipaci mutací p53 [30]. Ačkoliv v pozdější studii nebylo prokázáno zvýšení celkového počtu CNV u nositelů mutace p53 oproti kontrolám bez mutace, bylo detekováno významné (více než 5násobné) navýšení tzv. vzácných CNV. Při porovnání spektra CNV u zdravých kontrol a nositelů různých typů mutací p53 se ukázalo, že spektrum CNV nositelů mutace R337H se výrazně liší od spektra nositelů „hot ‑ spot“ mutací v DNA vazebné doméně a podobá se mnohem více kontrolním vzorkům. To by odpovídalo celkově mírnějšímu dopadu mutace R337H [31].
Dalším příspěvkem k potenciálně specifickému účinku mutace R337H byla práce Maceda et al (2012). Ti prokázali v krvi nositelů mutace R337H vyšší hodnoty indikátorů oxidativního stresu v porovnání s kontrolami bez mutace p53. To by naznačovalo, že právě zvýšený oxidativní stres by mohl být příčinou onkogenních vlastností této varianty p53. Na druhou stranu není zatím jasné, zda je to specifický účinek právě této varianty nebo zda podobné důsledky mají i další mutace p53 [32].
Penetrance alely R337H poprvé
Již původní práce o mutaci R337H silně naznačovala, že se pravděpodobně jedná o alelu p53 s nízkou penetrancí [23]. Ve stejné době byla publikována práce jiných autorů, kteří ve stejné oblasti Brazílie (Sao Paulo) prováděli podobnou analýzu. Vyšetřili 55 pacientů, 37 dospělých a 18 dětí s benigním nebo maligním ACC bez další rodinné historie nádorů, 21 asymptomatických blízkých příbuzných a 60 zdravých nepříbuzných kontrol. Mutaci R337H nalezli u 19 pacientů, z toho u 14 dětí (14/ 18 – 77,7 %) a 5 dospělých (5/ 37 – 13,5 %). Mutaci zachytili i mezi zdravými příbuznými prvního stupně, nedetekovali ji mezi kontrolami [16]. Podobně uspořádaná studie ze stejné oblasti tyto výsledky podpořila [33].
Detailní analýza penetrance alely R337 byla provedena na rozsáhlejším souboru. Byly vyšetřeny rodiny 30 probandů s ACC, nositelů mutace R377H. Celkem bylo vyšetřeno 927 osob, z toho 695 členů rodin probandů. Žádná z vyšetřených rodin nesplnila kritéria LFS, sedm rodin bylo zařazeno mezi LFLS. Z 695 jedinců z rodin probandů bylo 240 (34,5 %) nositelů mutace R337H. Tato mutace však nebyla nalezena u žádné kontrolní osoby. Mezi rodiči a prarodiči bylo 50 % nositelů mutace, nebyla tedy detekována jediná mutace R337H de novo. Mezi příbuznými 1. a 2. stupně bylo nalezeno 31 dalších nádorů. Celková penetrance tak byla odhadnuta na 9,9 %. Jinými slovy, každý 10. nositel mutace R337H onemocní nádorem, a to s největší pravděpodobností právě dětským adrenokortikálním karcinomem, protože mezi nositeli mutace R377H s nádorem představují nemocní s ACC 57 %, zatímco u klasického LFS (jehož penetrance dosahuje celoživotně asi 80 %) ACC představují výrazně menší – 10,9% podíl všech nádorů (graf 1) [34].
Je mutace R337H spojena s LFS a LFLS? Penetrance alely R337H podruhé
K dosti odlišným výsledkům než autoři předchozí studie dospěli Achatz et al (2007), kteří zvolili odlišnou strategii. Studovali 45 brazilských (Sao Paulo, Porto Alegre, Rio de Janiero) nepříbuzných jedinců z rodin, které splňovaly kritéria LFS nebo LFL. Ve 13 případech (28,9 %) nalezli zárodečnou mutaci p53, z toho šest pacientů (46,1 %) mělo mutaci R337H. Rodiny s mutací R337H měly široké spektrum nádorů, včetně karcinomů prsu (30,4 %), mozku (10,7 %), sarkomů (10,7 %) a ACC (8,9 %). Mezi 57 jedinci bez nádorové historie nebyla nalezena ani jedna mutace p53. Závěr autorů byl jasný a vůči práci Figueireda et al (2006) významně se vymezující – mutace R337H predisponuje k vývoji širokého spektra nádorů a neliší se tak od jiných mutací p53 [35].
Závěry studie byly ovšem velmi rychle napadeny a zpochybněny. Autoři práce Ribeiro et al (2007) byli přesvědčeni, že žádná ze studovaných rodin [35] nesplňovala kritéria LFS, ale pouze LFLS. Dále upozornili, že studie má jiný design a jinak definuje problém. V původní studii [34] by podle stejných kritérií bylo zahrnuto pouze 7 probandů a jejich rodin ze 30, ostatních 23 dětí/ rodin nejevilo žádné znaky zvýšené dispozice k nádorům kromě ACC. Navíc poukázali na další odlišnosti ve spektru nádorů nositelů mutace R337H a nositelů jiných mutací p53 než jen odlišná frekvence ACC. V této souvislosti byla na místě otázka, zda změněná dispozice k nádorům není dána kombinací R337H a nějakého dalšího kooperujícího genetického faktoru [36].
Obraz o penetranci alely R337H, o jejím vztahu ke spektru nádorů, ke kterým predisponuje, a také o frekvenci jejího výskytu v jižní Brazílii rozšířily další tři studie. V té první byla zárodečná mutace R337H hledána mezi 123 ženami s nádorem prsu a 223 adekvátními zdravými kontrolami. Mutace byla nalezena u tří žen s nádorem (2,4 %), ale u žádné kontroly. Anamnéza dvou z těchto tří žen odpovídala kritériím LFLS. Závěr autorů? Alela R337H může významně navýšit riziko vývoje nádoru prsu, tedy nejenom ACC, i když pravděpodobně v souvislosti s nějakými dalšími genetickými vlivy [37]. Podobně uspořádaná studie v Rio de Janeiru našla mezi 390 pacientkami s nádorem prsu dvě nositelky mutace R337H (0,5 %), zatímco mutace se nenašla ani u jedné z 324 zdravých žen. Obě nositelky mutace byly s nádorem prsu diagnostikovány v mladém věku (pod 40 let) a uváděly další případy nádoru prsu mezi příbuznými [38]. Ve třetí studii bylo analyzováno 750 zdravých žen – dobrovolnic ve věku 40 – 69 let – z Porto Alegre. Mutace R337H byla nalezena u dvou žen (0,3 %), které byly příbuzné 4. stupně a udávaly historii výskytu různých nádorů v rodině. Rodiny nesplňovaly kritéria LFS ani LFLS. Dalším testováním v rodinách byli nalezeni tři další nositelé mutace R337H, z toho u jedné ženy se vyvinul karcinom prsu ve 36 letech. Byla tak opakovaně prokázána nízká penetrance alely R337H. Frekvence výskytu této alely v jižní Brazílii byla stanovena na 0,0013, tedy asi 10 – 20krát vyšší než je výskyt ostatních mutací p53 souvisejících s vývojem LFS nebo LFLS [39]. Některé zdroje uvádějí frekvenci výskytu dokonce ještě vyšší [40].
Do diskuze o penetranci alely R337H a spektru nádorů, s nimiž je spojena, pak přispěli i autoři, kteří se primárně ve své práci zabývali jiným problémem (viz níže). Analýzou 12 rodin s vrozenou mutací R337H došli k závěru, že spektrum nádorů, které se vyvíjejí v souvislosti s touto mutací, je široké a neomezuje se pouze na ACC a že celoživotní míra penetrance alely R337H je srovnatelná s jinými mutacemi p53. Upozornili ale na specifickou dynamiku penetrance. Ta podle nich dosahuje asi 15 % ve věku 10 – 15 let, pak nastává plató a teprve po 30. roku života nastává strmé navýšení výskytu nádorů. Tito autoři tvrdí, že celoživotní riziko vývoje nádoru se tedy zřejmě neliší od ostatních mutací p53 spojených s LFS a LFLS, ale nádory mají tendenci se objevovat až v relativně vyšším věku [41].
Mají nositelé mutace společného předka?
Diskuzi o možném efektu zakladatele, tedy o možnosti jednoho společného zdroje alely R337H, započali již autoři původní práce Ribeiro et al (2001). Do analýzy zahrnuli 17 nepříbuzných nositelů vrozené mutace R337H a 10 jejich příbuzných. Vyšetřili čtyři polymorfní (intergenové a intragenové) markery a došli k závěru, že efekt zakladatele je nepravděpodobný, že alespoň některé vyšetřené alely vznikly nezávisle [23].
Naproti tomu další práce přinesla zcela opačný výsledek. Ve své práci Pinto et al (2004) vyšetřili dva vysoce polymorfní intragenové markery u 22 pacientů – nepříbuzných nositelů mutace R337H, a 60 kontrol. A došli k závěru, že je velmi pravděpodobné, že existoval jeden společný předek všech nositelů mutace R337H [42]. Jejich výsledek pak významně podpořila práce Garritana et al (2010). Tito autoři provedli velmi detailní haplotypovou analýzu 12 nepříbuzných rodin (celkem 48 nositelů alely R337H) s využitím 29 polymorfních markerů. Jedenáct vyšetřovaných rodin pocházelo z jižní Brazílie, jedna rodina s portugalskými kořeny byla zachycena ve Francii [24]. Závěr práce byl zcela jednoznačný. Všichni nositelé mutace R337H mají stejný haplotyp (A3), což jednoznačně potvrdilo efekt zakladatele. Na základě svých výsledků navíc autoři došli k závěru, že společný předek velmi pravděpodobně pochází z Evropy, nejpravděpodobněji je Portugalec [41].
Mnoho měst a vesnic jižní Brazílie vznikalo v 18. a 19. století podél osy známé jako „tropeiro route“. Tropeiros byli obchodníci, převážně portugalského původu, kteří dlouhodobě cestovali mezi Sao Paulem a Porto Alegre a významně se podíleli na osidlování této oblasti (obr. 1). Je tedy možné, že právě tyto specifické historické a demografické okolnosti přispěly k tak rozsáhlému rozšíření alely R337H. K dlouhodobé perzistenci alely R337H v populaci pak mohla významně přispět také její nízká penetrance, případně specifická dynamika penetrance [41].
Efekt zakladatele je také silně podpořen opakovaným zjištěním, že vrozená mutace R337H se zatím nikdy neobjevila de novo, vždy je detekována u jednoho z rodičů probanda [16,33,34]. V jedné studii byl dokonce zachycen případ probanda – homozygota , který měl v obou alelách genu p53 mutaci R337H, oba jeho rodiče byli heterozygoti [16].
Za hranicí brazilského příběhu R337H
Brazilský příběh R337H je příběhem o vztahu mutace R337H a dětských ACC odehrávající se v jižní Brazílii. Na tento sevřený, úzce vymezený a překvapující příběh ale navazují další otázky, které ho přesahují.
Je ACC jediný typ nádoru pevněji spjat s mutací R337H?
Jednou z nich je otázka, zda je ACC jediný typ nádoru, k němuž R337H predisponuje, nebo zda existují další typy nádorů pevněji spjatých s touto konkrétní mutací. Kromě již zmíněného vztahu k vývoji karcinomu prsu [37] byl nalezen další typ nádoru, který je dokonce výrazně – srovnatelně s ACC – spojen s vrozenou mutací R337H. Je to dětský karcinom choroidálního plexu, který se také vyskytuje v jižní Brazílii se zvýšenou incidencí a který je ve více než 60 % případů spojen právě s touto mutací [43,44]. A dále samozřejmě platí i výše zmíněné nálezy Achatzové et al (2007) svědčící o tom, že alela R337H je u mnoha rodin (a pravděpodobně v souvislosti s nějakými dalšími genetickými okolnostmi) spojena s rozvojem širokého spektra nádorů typického pro LFS a LFLS [35].
Je mutace R337H jedinou alelou p53 výrazněji predisponující k ACC?
Hledání odpovědi na otázky, zda je mutace nádorového supresoru p53 R337H jediná, která tak výrazně predisponuje k ACC, a pokud existují další mutace, co mají společného, je složitější. V úvodu je zmíněna práce Varleye et al (1999), ve které byly detekovány vrozené mutace P152L a R158H opakovaně se vyskytující u dětských ACC. Obě mutace jsou teplotně závislé, a tedy jen částečně inaktivující [45] a mají nízkou penetranci [9]. Později byly publikovány další případy vrozených mutací p53, které měly nízkou penetranci, byly spojeny s dětskými případy ACC, ale nikoliv s LFS či LFLS. Byly to mutace R175L [46] nebo sestřihová mutace vedoucí k expresi proteinu p53 s pozměněným C-koncem [47]. Tyto práce zapadaly do představy, že možná právě jen částečná ztráta funkce a nízká penetrance mutací p53 je to, co takové mutace spojuje přednostně s predispozicí k vývoji CCA v dětství [48]. Na základě hluboké znalosti do značné míry extrémního příběhu mutace R337H a dalších literárních údajů navrhl Zambetti model tzv. gradientového efektu vrozených mutací p53. Podle tohoto modelu lze mutace p53 odstupňovat podle míry zachované funkce, přičemž míra zbytkové aktivity p53 koreluje s fenotypem, tj. s mírou susceptibility k nádorům. Výsledný dopad typu vrozené mutace p53 je ale výrazně modifikován dalšími genetickými vlivy, především polymorfizmem MDM2 (SNP309) a několika intragenovými polymorfizmy samotného genu p53 (obr. 2) [48 – 51].
![Gradientový efekt dopadu mutací p53.
Někteří mutanti p53 jsou zcela inaktivní (R175H), zatímco jiní si ponechávají částečnou aktivitu, případně se jejich aktivita blíží aktivitě standardní varianty p53 (R337H). Stupeň aktivity p53 koresponduje s mírou susceptibility k nádorům. Výsledný dopad mutace p53 je modifi kován dalšími genetickými vlivy, především polymorfi zmy genu MDM2 a p53. Převzato a upraveno dle [48].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ef954588cdf52e1077a37fbc7b0a9993.jpg)
Recentně byl ustaven mezinárodní registr a tkáňová banka dětských případů ACC. V nádorové tkáni 48 případů, které jsou v bance uloženy, bylo detekováno 23 nezávislých vrozených mutací p53. Většina mutací se vyskytovala v typických „hot ‑ spot“ kodonech a pouze tři mutace se vyskytly opakovaně: R175H, R273C a R337H. Celkově se tedy zdá, že mutované alely p53 s nízkou penetrancí výrazně predisponují k vývoji dětských ACC, ale zároveň se dětské případy ACC často vyskytují v rodinách s LFS či LFLS, které mají v zárodečné linii variantu p53 s vysokou penetrancí [52].
Závěry I
Odhalení úzkého spojení mezi mutací R337H a vývojem dětských adrenokortikálních karcinomů bylo na poli p53 zcela mimořádným objevem. Ač je tento nádorový supresor u lidských nádorů tak často mutován a ačkoliv známe tolik různých mutací tohoto genu, tak specifický vztah mezi jedním typem mutace a konkrétním nádorem, jako je vztah R337H a dětských ACC, je velmi neobvyklý. Hlavní závěry tohoto příběhu lze formulovat v několika bodech:
- Incidence dětských ACC je v jižní Brazílii více než 10krát vyšší než v jiných oblastech světa.
- Výskyt dětských ACC v jižní Brazílii je extrémně často spojen s vrozenou mutací nádorového supresoru p53 R337H.
- Frekvence výskytu alely R337H v jižní Brazílii je 0,0013, tedy asi 10 – 20krát vyšší, než je výskyt ostatních mutací p53 souvisejících s vývojem LFS nebo LFLS.
- Varianta proteinu p53 nesoucí v pozici 337 histidin namísto argininu (R337H) si zachovává vysokou transkripčně aktivační schopnost, ale je extrémně senzitivní k pH.
- Penetrance alely R337H je nízká.
- Všichni nositelé alely R337H mají společného předka, vrozená mutace R337H se neobjevuje de novo.
Závěry II
Závěry uvedené v předchozím odstavci jsou výsledkem mnohaletého úsilí několika skvělých vědeckých týmů. Jsou samy o sobě strhující. A přesto lze formulovat ještě závěry další, přerůstající hranice jedné konkrétní badatelské oblasti a přinášející hluboké poučení.
Při pečlivém sběru a studiu prací, které byly postupně publikovány, je čtenář na jedné straně vtažen do dramatického příběhu odhalování souvislostí, na straně druhé chvílemi i zmaten a znejistěn, jak se věci vlastně mají a kde je pravda. Aby posléze zjistil, že odhalování pravdy, nahlížení toho „jak to vlastně doopravdy je“, platnost všech získaných odpovědí je velmi ovlivněna už samotnou formulací otázky, designem studie a přesností interpretace dat. To je jistě poučení uplatnitelné v každé oblasti lidské činnosti.
Neméně obecnou platnost má i další zkušenost související s tímto konkrétním příběhem. V roce 2007 se v Lyonu konal mezinárodní workshop o mutantech p53 a LFS, a v jeho rámci přednáška dvou brazilských autorek, Marie ‑ Isabely Achatz a Patricie Ashton ‑ Prolla, které tehdy hovořily o některých aspektech brazilského příběhu, mimo jiné o efektu zakladatele. Nejenom trochu romantizující úvahy o tom, zda společným předkem všech současných nositelů mutace p53 byl úspěšný a okouzlující tropeiro, či chrabrý, silný a neméně okouzlující (nebo násilnický?) konkvistador, ale především příležitost dozvědět se podrobnosti přímo z epicentra dění tehdy rozpoutala jiskřivou a vášnivou diskuzi, která nebrala konce. Přednáška byla naštěstí poslední ve své sekci, která byla závěrečnou sekcí jednacího dne, a tak nekončící příval dotazů a ochota obou žen na ně odpovídat vedly k tomu, že ten den končila konference s dvouhodinovým zpožděním! A zážitek – stimulující, motivující, okouzlující – to byl nepochybně pro všechny účastníky. A i ten patří do historie a ke vkladům brazilského příběhu mutace p53 R337H.
Práce byla podpořena grantem IGA MZ ČR č. NT/13784-4/2012.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prof. RNDr. Jana Šmardová, CSc.
Ústav patologie
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: janasmarda@seznam.cz
Obdrženo: 28. 4. 2014
Přijato: 19. 5. 2014
Sources
1. Lane DP, Crawford LV. T antigen is bound to host protein SV40 - transformed cells. Nature 1979; 278(5701): 261 – 263.
2. Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40 transformed cell and uninfected embryonal carcinoma cells. Cell 1979; 17(1): 43 – 52.
3. Robles AI, Harris CC. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol 2010; 2(3): a001016. doi: 10.1101/ cshperspect.a001016.
4. Malkin D. Li ‑ Fraumeni syndrome. Genes Cancer 2011; 2(4): 475 – 484. doi: 10.1177/ 1947601911413466.
5. Petitjean A, Mathe E, Kato S et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007; 28(6): 622 – 629.
6. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2010; 2(1): a001008. doi: 10.1101/ cshperspect.a001008.
7. Varley JM, Chapman P, McGown G et al. Genetic and functional studies of germaline TP53 splicing mutation in a Li ‑ Fraumeni‑like family. Oncogene 1998; 16(25): 3291 – 3298.
8. Warneford SG, Witton LJ, Townsend ML et al. Germ‑line splicing mutation of the p53 gene in a cancer ‑ prone family. Cell Growt Differ 1992; 3(11): 839 – 846.
9. Varley JM, McGown G, Thorncroft M et al. Are there low ‑ penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet 1999; 65(4): 995 – 1006.
10. Olivier M, Goldgar DE, Sodha N et al. Li ‑ Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 2003; 63(20): 6643 – 6650.
11. Monti P, Ciribilli Y, Jordan J et al. Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clin Cancer Res 2007; 13(13): 3789 – 3795.
12. Broul M, Schraml J, Sticova E et al. Adrenokortikální karcinom. Urol Pro Praxi 2010; 11(4): 200 – 203.
13. Custodio G, Komechen H, Figueiredo RO et al. Molecular epidemiology of adrenocortical tumors in southern Brazil. Mol Cell Endocrinol 2012; 351(1): 44 – 51. doi: 10.1016/ j.mce.2011.10.019.
14. Ribeiro RC, Figueiredo B. Childhood sdrenocortical tumours. Eur J Cancer 2004; 40(8): 1117 – 1126.
15. Rodriguez ‑ Galindo C, Figueiredo BC, Zambetti GP et al. Biology, clinical characteristics, and managment of adrenocortical tumors in children. Pediatr Blood Cancer 2005; 45(3): 265 – 273.
16. Latronico AC, Pinto EM, Domenice S et al. An inherited mutation outsider the highly conserved DNA‑binding domain of the p53 tumor suppressor protein in chlidren and adults with sporadic adrenocortical tumors. J Clin Endocrin Met 2001; 86(10): 4970 – 4973.
17. Waldmann J, Patsalis N, Fendrich V et al. Clinical imapct of TP53 alterations in adrenocortical carcinomas. Langenbecks Arch Surg 2012; 397(2): 209 – 216. doi: 10.1007/ s00423 ‑ 011 ‑ 0868 ‑ 6.
18. Hermann LJ, Heinze B, Fassnacht M et al. TP53 germline mutations in adult patiens with adrenocortical carcinoma. J Clin Endocrinol Metab 2012; 97(3): E476 – E485. doi: 10.1210/ jc.2011 ‑ 1982.
19. Bernstein L, Gurney JG. Carcinomal and other malignant epithelial neoplasm. In: Ries LA, Smith MA, Gurney JG et al (eds). Cancer and survival among children and adolescents: United States SEER program 1975 – 1995. Bethesda, MD: NIH 1999 : 139 – 147.
20. Birch JM, Blair V. Increase in childhood carcinomas in North ‑ West England. Lancet 1988; 1(8589): 833.
21. Desandes E, Clavel J, Berger C et al. Cancer incidence among children in France, 1990 – 1999. Pediatr Blood Cancer 2004; 43(7): 749 – 757.
22. Pianovski MA, Maluf EM, de Carvalho DS et al. Mortality rate of adrenocortical tumors in children under 15 years of age in Curitiba, Brazil. Pediatr Blood Cancer 2006; 47(1): 56 – 60.
23. Ribeiro RC, Sandrini F, Figueiredo B et al. An inhereted p53 mutation that contributes in a tissue ‑ specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci USA 2001; 98(16): 9330 – 9335.
24. Chompret A, Brugieres L, Ronsin M et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000; 82(12): 1932 – 1937.
25. Di Giammarino EL, Lee AS, Cadwell C et al. A novel mechanism of tumorigenesis involving pH ‑ dependent destabilization of a mutant p53 tetramer. Nat Struct Biol 2002; 9(1): 12 – 16.
26. Lee AS, Galea C, DiGiammarino EL et al. Reverzible anuloid formation by the p53 tetramerization domain and a cancer‑associated mutant. J Mol Biol 2003; 327(3): 699 – 709.
27. Li W, Olivier M. Current analysis platforms and methods for detecting copy number variation. Physiol Genomics 2013; 45(1): 1 – 16. doi: 10.1152/ physiolgenomics.00082.2012.
28. Shlien A, Malkin D. Copy number variations and cancer. Genome Medicine 2009; 1(6): 62. doi: 10.1186/ gm62.
29. Kuiper RP, Lichtenberg MJ, Hoogerbrugge N et al. Germline copy number variation and cancer risk. Curr Opin Genet Dev 2010; 20(3): 282 – 289. doi: 10.1016/ j.gde.2010.03.005.
30. Shlien A, Tabori U, Marshall CR et al. Excessive genomic DNA copy number variation in the Li ‑ Fraumeni cancer predisposition syndrome. Proc Natl Acad Sci USA 2008; 105(32): 11264 – 11269. doi: 10.1073/ pnas.0802970105.
31. Silva AG, Achatz MI, Krepischi AC et al. Number of rare germline CNVs and TP53 mutation types. Orphanet J Rare Dis 2012; 7 : 101. doi: 10.1186/ 1750 ‑ 1172 ‑ 7 ‑ 101.
32. Macedo GS, Lisbôa da Motta L, Giacomazzi J et al. Increased oxidative damage in carriers of the germline TP53 p.R337H mutation. PLOS One 2012; 7(10): e47010. doi: 10.1371/ journal.pone.0047010.
33. Sandrini F, Villani DP, Tucci S et al. Inheritance of R337H p53 gene mutation in children with sporadic adrenocortical tumor. Horm Metab Res 2005; 37(4): 231 – 235.
34. Figueiredo BC, Sandrini R, Zambetti GP et al. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J Med Genet 2006; 43(1): 91 – 96.
35. Achatz MI, Olivier M, Le Calvez F et al. The TP53 mutation, R337H, is associated with Li ‑ Fraumeni and Li ‑ Fraumeni‑like syndromes in Brazilian families. Cancer Lett 2007; 245(1 – 2): 96 – 102.
36. Ribeiro RC, Rodriguez ‑ Galindo C, Figueiredo BC et al. Germline TP53 R337H mutation is not sufficient to establish Li ‑ Fraumeni or Li ‑ Fraumeni‑like syndrome. Cancer Lett 2007; 247(2): 353 – 355.
37. Assumpção JG, Seidinger AL, Mastellaro MJ et al. Association of the germline TP53 R337H mutation with brest cancer in southern Brazil. BMC Cancer 2008; 8 : 357. doi: 10.1186/ 1471 ‑ 2407 ‑ 8 ‑ 357.
38. Gomes MC, Kotsopoulos J, Leao de Alemida G et al.The R337H mutation in TP53 and breast cancer in Brazil. Hered Cancer Clin Pract 2012; 10(1): 3 – 6. doi: 10.1186/ 1897 ‑ 4287 ‑ 10 ‑ 3.
39. Palmeto EI, Schuler ‑ Faccini L, Caleffi M et al. Detection of R337H, a germline TP53 mutation predispozing to multiple cancers, in asymptomatic women participat-ing in a brest cancer screening program in Southern Brazil. Cancer Lett 2008; 261(1): 21 – 25. doi: 10.1016/ j.canlet.2007.10.044.
40. Achatz MI, Hainaut P, Ashton ‑ Prolla P. Highly prevalent TP53 mutation predisposing to many cancers in the Brazilian population: a case for newborn screening? Lancet Oncol 2009; 10(9): 920 – 925. doi: 10.1016/ S1470 ‑ 2045(09)70089 ‑ 0.
41. Garritano S, Gemignani F, Palmeto EI et al. Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: evidence for a flunder effect. Hum Mutat 2010; 31(2): 143 – 150. doi: 10.1002/ humu.21151.
42. Pinto EM, Billerbeck AE, Villares MC et al. Flunder effect for the highly převaleny R337H mutation of the tumor suppressor p53 in Brazilian patiens with adrenocortical tumors. Arg Bras Endocrinol Metabol 2004; 48(5): 647 – 650.
43. Seidinger AL, Mastellaro MJ, Fortes FP et al. Association of the highly převaleny TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in Southeast Brazil. Cancer 2011; 117(10): 2228 – 2235. doi: 10.1002/ cncr.25826
44. Custodio G, Taques GR, Figueiredo BC et al. Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in Southern Brazil. PLoS One 2011; 6(3): e18015. doi: 10.1371/ journal.pone.0018015.
45. Shiraishi K, Kato S, Han SY et al. Isolation of temperature‑sensitive p53 mutations from a comprehensive missense mutation library. J Biol Chem 2004; 279(1): 348 – 355.
46. West AN, Ribeiro RC, Jenkins J et al. Identification of a novel germ line variant hotspot mutant p53 - R175L in pediatric adrenal cortical carcinoma. Cancer Res 2006; 66(10): 5056 – 5062.
47. Pinto EM, Ribeiro RC, Kletter GB et al. Inherited germline TP53 mutation encodes a protein with an aberrant C‑terminal motif in a case of pediatric adrenocortical tumor. Fam Cancer 2011; 10(1): 141 – 146. doi: 10.1007/ s10689 ‑ 010 ‑ 9392 ‑ z.
48. Zambetti GP. The p53 mutation „gradient effect“ and its clinical implications. J Cell Physiol 2007; 213(2): 370 – 373.
49. Bond GL, Hu W, Levine A. A single nucleotide polymorphism in the MDM2 gene: from a molecular and cellular explanation to clinical effect. Cancer Res 2005; 65(13): 5481 – 5484.
50. Bougeard G, Baert ‑ Desurmont S, Tournier I et al. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li ‑ Fraumeni syndrome. J Med Genet 2006; 43(6): 531 – 533.
51. Grochola LF, Zeron ‑ Medine J, Mériaux S et al. Single‑nucleotide polymorhisms in the p53 signaling pathway. Cold Spring Harb Perspect Biol 2010; 2(5): a001032. doi: 10.1101/ cshperspect.a001032.
52. Pinto EM, Ribeiro RC, Figueiredo BC et al. TP53‑associated pediatric malignancies. Genes Cancer 2011; 2(4): 485 – 490. doi: 10.1177/1947601911409745.
Labels
Paediatric clinical oncology Paediatric radiology Surgery Clinical oncology RadiotherapyArticle was published in
Clinical Oncology

2014 Issue 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Brazilian Story of the R337H p53 Mutation
- Acupuncture in the Treatment of Symptoms of Oncological Diseases in the Western World
- Paraneoplastic Vasculitis in a Patient with Cervical Cancer
- Screening of Malnutrition Risk Versus Indicators of Nutritional Status and Systemic Inflammatory Response in Newly Diagnosed Lung Cancer Patients