
Extraoseální Ewingův sarkom, primární postižení děložního čípku – kazuistika
Extraoseus Ewing‘s Sarcoma, Primary Affection of Uterine Cervix – Case Report
Background:
Ewing‘s sarcoma is usually diagnosed in adolescents and young adults, peak of incidence is around 15 years of age. Primary localization is mostly in the skeleton of long bones and chest wall. Primary extraosseous involvement rarely occurs, incidence increases with age.
Case:
We present a case report of a 57‑year ‑ old patient with locally advanced tumors of the cervix, clinical stage IIB. Due to histological and molecular genetic examination revealing EWS‑ ERG fusion gene, Ewing‘s sarcoma was diagnosed. CT revealed pathological pelvic lymphadenopathy and multiple pulmonary bilateral methastases, scintigraphy did not prove any affection of skeleton. The patient underwent a two‑stage intensive chemotherapy regimens VIDE (vincristine, ifosfamide, doxorubicin, etoposide) and VAI (vincristine, actinomycin D, ifosfamide). During the second phase, concomitant radiotherapy of pelvis was aplied. According to PET/ CT, complete remission was achieved. Whole ‑ lung irradiation was applied in consolidation of the result.
Conclusion:
Primary Ewing‘s sarcoma of the cervix is an extremely rare disease. To our knowledge, only 12 cases was presented until this time. The average age at time of diagnosis was 35 years. Unlike the previous reports, we initially diagnosed distant metastases. The treatment was led according to the protocol Ewing 2008 designed for primary skeletal Ewing‘s sarcoma. Currently, 18 months after the therapy, the patient is without signs of disease. However, long‑term follow-up is necessary.
Key words:
Ewing‘s sarcoma – uterine cervix – cytogenetics – EWS-ERG – chemotherapy – radiotherapy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
3. 6. 2015
Accepted:
25. 7. 2015
:
O. Bílek 1; M. Holánek 1; M. Zvaríková 1; P. Fabian 2; B. Robešová 3; M. Procházková 4; D. Adámková Krákorová 1
:
Klinika komplexní onkologické péče, Masarykův onkologický ústav, Brno
1; Oddělení klinické a experimentální patologie, Masarykův onkologický ústav, Brno
2; Centrum molekulární bio logie a genové terapie, Interní hematologická a onkologická klinika LF MU a FN Brno
3; Oddělení radiologie, Masarykův onkologický ústav, Brno
4
:
Klin Onkol 2015; 28(4): 284-287
:
Case Report
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amko2015284
Východiska:
Ewingův sarkom bývá nejčastěji diagnostikován u adolescentů a mladých dospělých s vrcholem incidence kolem 15. roku věku. Vychází nejčastěji ze skeletu dlouhých kostí a hrudní stěny. Vzácné je primární extraoseální postižení. Tvoří 11 – 24 % z celkové incidence, výskyt stoupá s věkem.
Popis případu:
Předkládáme kazuistiku 57leté pacientky s lokálně pokročilým tumorem děložního čípku klinického stadia IIB. Na základě histologického a molekulárně‑genetického vyšetření s nálezem přestavby EWS‑ ERG byl diagnostikován Ewingův sarkom. CT trupu dále odhalilo patologickou pánevní lymfadenopatii a vícečetná plicní ložiska oboustranně, scintigrafie skeletu neprokázala kostní postižení. Pacientka absolvovala dvoufázovou intenzivní chemoterapii režimy VIDE (vinkristin, ifosfamid, doxorubicin, etoposid) a VAI (vinkristin, aktinomycin D, ifosfamid). V rámci druhé fáze proběhla konkomitantní radioterapie pánve. Bylo dosaženo kompletní remise onemocnění podle PET/ CT. V rámci konsolidace byla léčba doplněna velkoobjemovým ozářením plic.
Závěr:
Primární Ewingův sarkom děložního čípku je extrémně vzácné onemocnění. Dosud bylo zaznamenáno pouze 12 případů s průměrným věkem v době diagnózy 35 let, na rozdíl od naší kazuistiky bez iniciálního průkazu vzdálených metastáz. Léčba byla vedena podle protokolu Ewing 2008 navrženého pro primárně kostní formu Ewingova sarkomu. Aktuálně je pacientka 18 měsíců po ukončení léčby bez známek onemocnění, dlouhodobé sledování je nezbytné.
Klíčová slova:
Ewingův sarkom – děložní čípek – cytogenetika – EWS-ERG – chemoterapie – radioterapie
Úvod
Nádory rodiny Ewingova sarkomu (ES) mají předpokládaný původ v mezenchymální buňce [1]. Bývají diagnostikovány zejména u adolescentů a mladých dospělých, incidence dosahuje vrcholu kolem 15. roku věku, poté rychle klesá, nad věkovou hranicí 40 let se vyskytuje zřídka [2]. Vychází nejčastěji ze skeletu, zejména z dlouhých kostí a hrudní stěny. Metastazuje hematogenně, nejčastěji do plic, kostí a kostní dřeně. Vzácně bývá diagnostikován primární extraoseální Ewingův sarkom (EES), tvoří 11 – 24 % z celkové incidence ES, výskyt stoupá s věkem [2]. V literatuře se nejčastěji popisuje postižení ledvin, Zöllner et al evidují 152 případů [3], dále např. plic [4], žaludku [5], jater [6], urologických [7] či gynekologických orgánů.
Popis případu
Pacientka (57 let) vyšetřovaná pro bolesti v hypogastriu a gynekologické krvácení byla v září 2012 odeslána na naše pracoviště s histologickým nálezem nízce diferencovaného spinocelulárního karcinomu děložního čípku, místy neuroendokrinního až malobuněčného vzhledu. Klinicky byl popsán exofytický, bohatě vaskularizovaný tumor hrdla děložního infiltrující parametria, stadium IIB. Z nádorových markerů jsme zjistili elevaci CA ‑ 125, HE4 a NSE, ostatní laboratorní nálezy byly bez pozoruhodností, s normální hodnotou laktátdehydrogenázy a alkalické fosfatázy. Opakované histologické vyšetření prokázalo high‑grade kulatobuněčný sarkom s vysokou mitotickou aktivitou, imunohistochemicky byl pozitivní vimentin, CD99 a EMA, naopak negativní cytokeratin, desmin, protein S100, Melan A, HMB ‑ 45, chromogranin, NSE a CD56 (obr. 1, 2). Na základě molekulárně‑genetického vyšetření s nálezem přestavby EWS‑ ERG, t(21;22) byl dvěma nezávislými patology potvrzen Ewingův sarkom. CT trupu odhalilo zvětšenou dělohu na podkladě tumoru, patologickou pánevní lymfadenopatii do velikosti 30 mm (obr. 3) a vícečetná plicní ložiska do 13 mm oboustranně. Scintigrafie skeletu neprokázala kostní postižení.
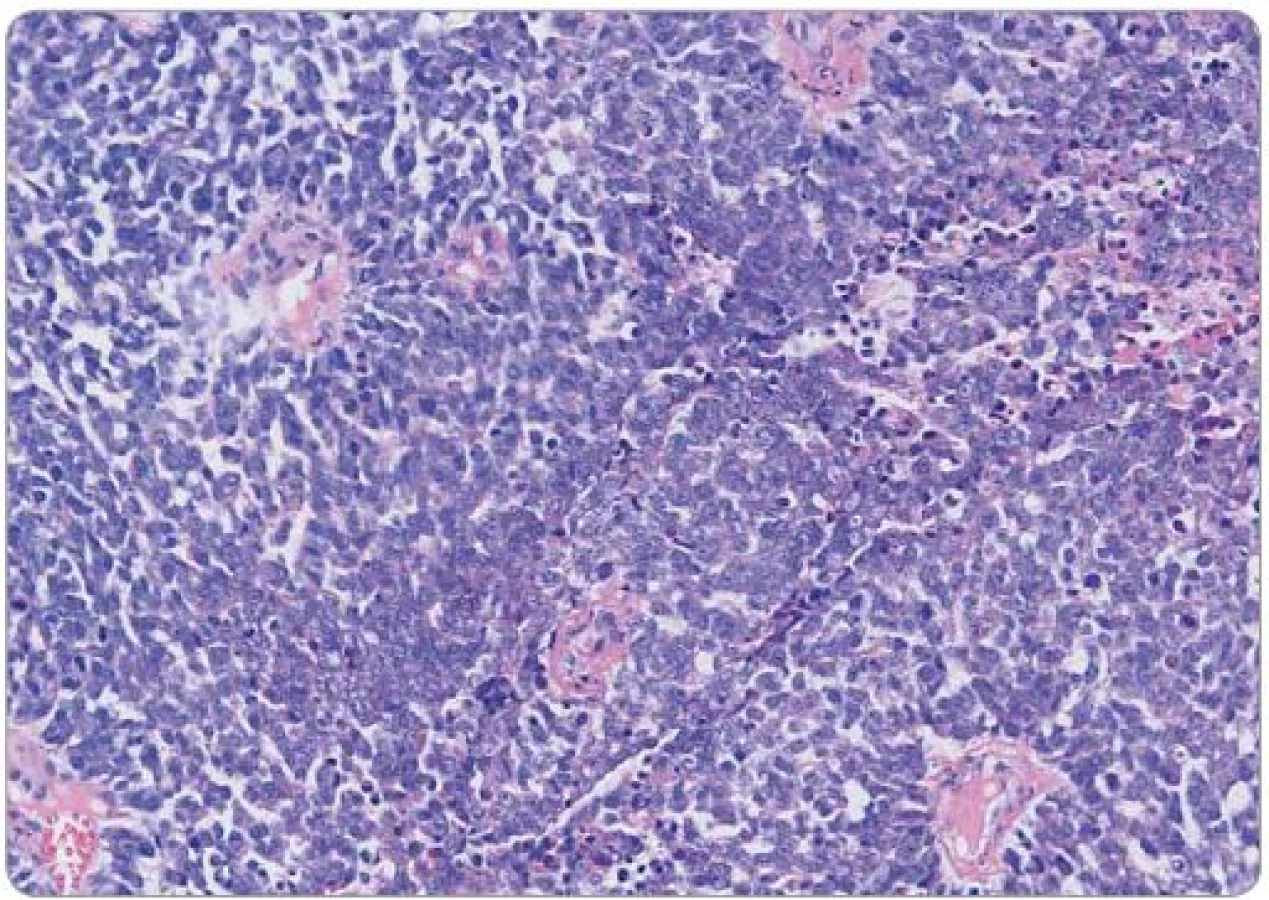
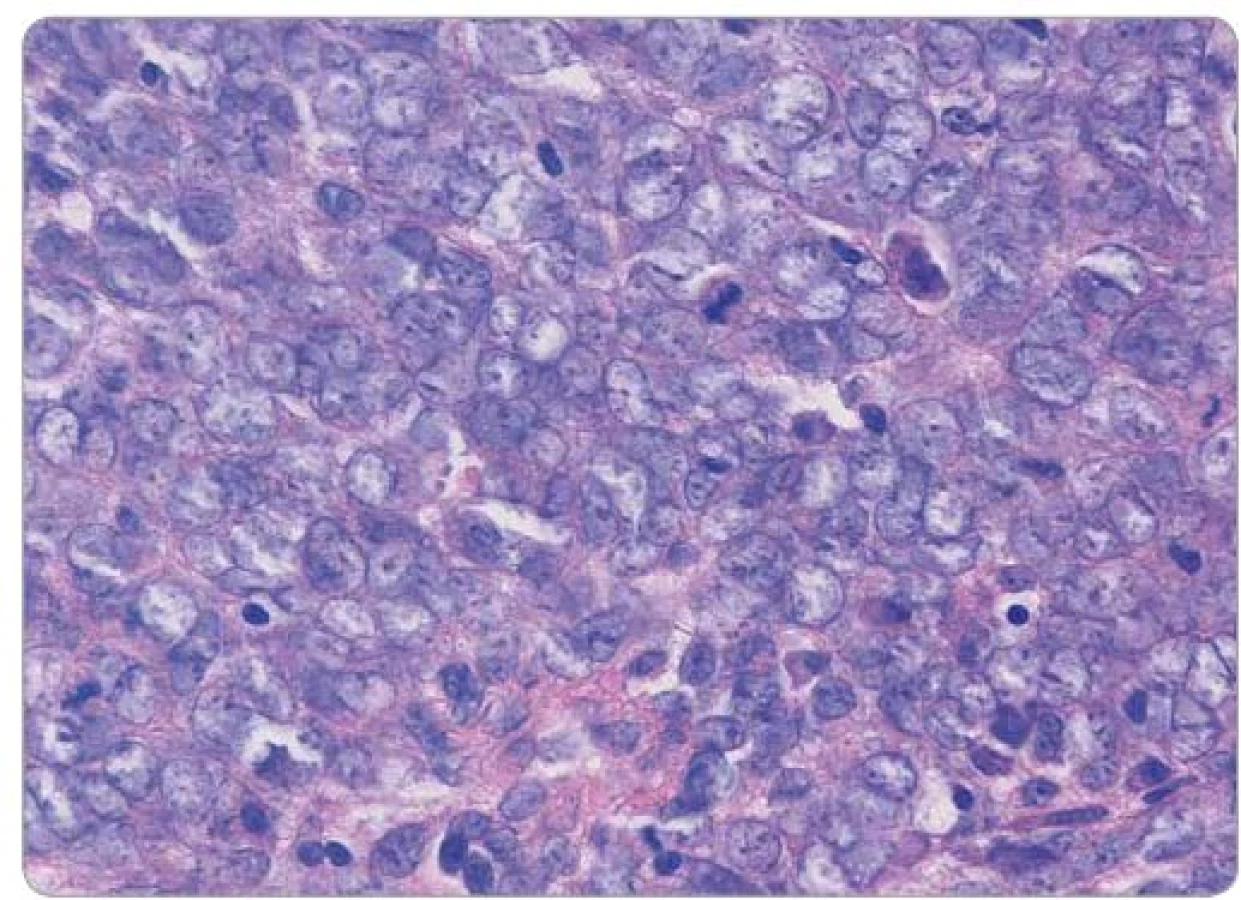

Na základě závěru multioborové komise byla se souhlasem pacientky zahájena systémová chemoterapie podle protokolu Ewing 2008, první fáze v režimu VIDE 6× (vinkristin 1,50 mg/ m2 den 1, ifos-famid 3 000,00 mg/ m2 den 1 – 3, etoposid 150,00 mg/ m2 den 1 – 3, doxorubicin 20,00 mg/ m2, mesna 3 000,00 mg/ m2). Vzhledem k protrahované hematologické toxicitě G3–G4, slizniční toxicitě a celkově špatné toleranci ze strany pacientky bylo nutné od třetího cyklu chemoterapie redukovat dávky cytostatik až o 25 %. Po šesti cyklech chemoterapie bylo dosaženo výrazné regrese onemocnění podle CT, vyšetření PET nezaznamenalo metabolickou aktivitu. Operační resekce primárního tumoru nebyla indikována vzhledem k přetrvávajícímu postižení parametrií. Následovala konsolidační chemoterapie v režimu VAI 8× (vinkristin 1,50 mg/ m2 den 1, aktinomycin D 0,75 mg/ m2 den 1 – 2, ifosfamid 3 000,00 mg/ m2 den 1 – 2, mesna 3 000,00 mg/ m2 den 1 – 2). Druhý a třetí cyklus chemoterapie byl podán v konkomitanci s radioterapií, standardně bez aktinomycinu, aplikována byla zevní radioterapie na oblast pánve v dávce 45 Gy a brachyterapie v celkové dávce 30 Gy. Jako konsolidace klinické remise potvrzené na základě PET/ CT (obr. 3) bylo na samotný závěr provedeno velkoobjemové ozáření plic v dávce 15 Gy. Aktuálně je pacientka 18 měsíců po ukončení léčby bez známek onemocnění, riziko relapsu je však stále považováno za vysoké.
Diskuze
Ewingův sarkom je histologicky agresivní, nízce diferencovaný nádor tvořený malými kulatými buňkami. Tento histologický obraz může sdílet např. s intraabdominálním desmoplastickým tumorem, rhabdomyosarkomem, lymfoblastickým lymfomem, s dalšími nádory dětského věku jako např. neuroblastomem, dále s malobuněčným karcinomem či melanomem [8]. Imunohistochemický fenotyp může zahrnovat expresi EMA, NSE, CD99, Bcl ‑ 2, CD117, p53, S100, charakteristická je přítomnost vimentinu [9]. V případě naší pacientky dominovala difuzní membránová pozitivita CD99. Jedná se o transmembránový protein typu mucinu s extracelulární doménou obsahující 100 aminokyselin, transmembránová doména zahrnuje 25 a cytoplazmatická 35 aminokyselin. Zastává úlohu adhezní molekuly při transendoteliální migraci leukocytů, dále zprostředkovává TCR/ CD3 dependentní aktivaci T lymfocytů a reguluje transport MHC třídy I z Golgiho komplexu na membránu [11]. Jeho downregulace je významná u Hodgkinových a Reed ‑ Sternbergových buněk [12]. Přítomnost tohoto proteinu je pro ES typická, nikoli však dostatečně specifická k samostatnému diagnostickému využití. Ukázalo se, že má zásadní význam pro onkogenní fenotyp ES blokádou diferenciace prostřednictvím ovlivnění intracelulárních signálních drah, zejména RAS/ MAPK zvýšením fosforylace kinázy ERK 1,2. Tyto údaje naznačují nové možnosti terapeutického přístupu s cílem modulace diferenciace nádorových buněk [10]. Prognostický vliv exprese jednotlivých imunohistochemických znaků nebyl prokázán [9].
ES je spojen se specifickými translokacemi vznikajícími fúzí genu EWS s geny rodiny ETS (E26 transformation ‑ specific), v 85 % s genem FLI1 [13]. V případě naší pacientky byla diagnostikována přestavba EWS‑ ERG, t(21;22)(q22;q12) vyskytující se v 10 % případů ES, jiné aberace jsou vzácné [14]. Vzhledem k málo specifickému histologickému obrazu ES je molekulárně‑biologické vyšetření z hlediska diferenciální diagnostiky zcela zásadní. Standardně se provádí z nativní nádorové tkáně a kostní dřeně, v tomto případě se podařilo přestavbu EWS‑ ERG jednoznačně prokázat z formalínem fixované nádorové tkáně zalité do parafínu metodou RT‑PCR a přímým sekvenováním. Byl zvažován i možný prognostický význam typu chromozomální aberace, podle studie dle De Alavy et al se jevila přítomnost translokace EWS‑ FLI1 u pacientů s lokalizovaným onemocněním v korelaci se signifikantním prodloužením celkového přežití (overall survival – OS) [15]. Ve světle pozdějších prací reflektujících efekt intenzivních terapeutických postupů se však tento faktor ukázal být méně významný [16,17].
Z prognostických faktorů ES dominuje především přítomnost či nepřítomnost vzdálených metastáz a jejich lokalizace. Plicní postižení je spojeno s delším OS ve srovnání s metastatickým postižením jiných orgánů. V případě operace je zásadní radikalita výkonu a histopatologické procento nekróz po indukční chemoterapii. Prognóza se zhoršuje s věkem pacienta, dále je uváděn vliv objemu primárního tumoru (> 200 cm3). Méně významná je hladina laktátdehydrogenázy a alkalické fosfatázy před zahájením léčby [14]. Prognostické faktory EES jsou podobné, rovněž je nejvýznamnější rozsah onemocnění, déle přežívají pacienti se subkutánní lokalizací tumoru [18,19].
Léčba EES se odvíjí od primární lokalizace tumoru, podle současných doporučení by měla vycházet z léčebných protokolů oseálního ES. V 80. a 90. letech 20. století se využívaly postupy protokolů RMS ‑ 88 a RMS ‑ 96 navržené pro léčbu rabdomyosarkomu [20,21]. Chemoterapii z těchto režimů použili v léčbě ES děložního čípku Tsao et al [31], Farzaneh et al [35] a Li et al [36]. V jiných dříve popsaných případech byly použity různé varianty kombinované chemoterapie, zpravidla v adjuvantním podání, protože žádná z dříve popsaných pacientek neměla v době stanovení diagnózy vzdálené metastázy [26 – 36]. Naši pacientku jsme léčili chemoterapií a radioterapií podle protokolu Euro EWING 99, resp. aktualizovanou verzí Ewing 2008. Sestává z indukční kombinované chemoterapie VIDE (vinkristin, ifosfamid, doxorubicin, etoposid) následované lokální léčbou s preferencí chirurgické, je‑li možná. Na základě stratifikace pacientů podle rizikových faktorů pokračuje konsolidační chemoterapií v režimu VAI nebo VAC (vinkristin, aktinomycin D, ifosfamid nebo cyklofosfamid) [22], v indikovaných případech doplněné radioterapií. U vysoce rizikových pacientů s negativními prognostickými faktory je možná vysokodávková chemoterapie s následnou transplantací periferních hemopoetických kmenových buněk (PBSC).
Závěr
EES gynekologických orgánů je extrémně vzácný. Bylo popsáno primární postižení ovarií [23], těla děložního [24] či vulvy [25]. V anglicky psané literatuře bylo v letech 1987 – 2015 zaznamenáno 12 případů primárního postižení děložní čípku [26 – 36] s průměrným věkem pacientek v době diagnózy 35 let. V našem případě bylo onemocnění zjištěno ve věku 57 let, což se odrazilo na horší toleranci intenzivní chemoterapie. Léčba byla založena na multidisciplinární spolupráci v rámci protokolu Ewing 2008. Pacientka je 18 měsíců po ukončení léčby bez známek onemocnění, dlouhodobé sledování je nezbytné.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Obdrženo: 3. 6. 2015
Přijato: 25. 7. 2015
MUDr. Ondřej Bílek
Klinika komplexní onkologické péče
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: bilek@mou.cz
Sources
1. Lin PP, Wang Y, Lozano G. Mesenchymal stem cells and the origin of Ewing’s sarcoma. Sarcoma 2011 : 276463. doi: 10.1155/ 2011/ 276463.
2. Bleyer WA, Barr RD. Cancer in adolescents and young adults, bone cancer. New York: Springer ‑ Verlag 2007 : 203 – 215.
3. Zöllner, S, Dirksen U, Jürgens H et al. Renal Ewing tumors. Ann Oncol 2013; 24(9): 2455 – 2461. doi: 10.1093/ annonc/ mdt215.
4. Hwang, SK, Kim DK, Park SI. Primary Ewing‘s sarcoma of the lung. Korean J Thorac Cardiovasc Surg 2014; 47(1): 47 – 50.
5. Kim HS, Kim S, Min YD et al. Ewing‘s sarcoma of the stomach; rare case of Ewing‘s sarcoma and suggestion of new treatment strategy. J Gastric Cancer 2012; 12(4): 258 – 261. doi: 10.5230/ jgc.2012.12.4.258.
6. Ozaki Y, Miura Y, Koganemaru S et al. Ewing sarcoma of the liver with multilocular cystic mass formation: a case report. BMC Cancer 2015; 15(1): 16. doi: 10.1186/ s12885 ‑ 015 ‑ 1017 ‑ 3.
7. Sharma P, Bakshi H, Chheda Y et al. Primary Ewing’ssarcoma of penis – a rare case report. Indian J Surg Oncol 2011; 2(4): 332 – 333. doi: 10.1007/ s13193 ‑ 011 ‑ 0112 ‑ 4.
8. Hayes ‑ Jordan A, Anderson PM. The diagnosis and management of desmoplastic small round cell tumor: a review. Curr Opin Oncol 2011; 23(4): 385 – 389. doi: 10.1097/ CCO.0b013e3283477aab.
9. Kavalar R, Pohar Marinšek Z, Jereb B et al. Prognostic value of immunohistochemistry in the Ewing’s sarcoma family of tumors. Med Sci Monit 2009; 15(8): CR442 – CR452.
10. Rocchi A, Manara MC, Sciandra M et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J Clin Invest 2010; 120(3): 668 – 680. doi: 10.1172/ JCI36667.
11. Kim SH, Choi EY, Shin YK et al. Generation of cells with Hodgkin’s and Reed ‑ Sternberg phenotype through downregulation of CD99 (Mic2). Blood 1998; 92(11): 4287 – 4295.
12. Hahn JH, Kim MK, Choi EY et al. CD99 (MIC2) regulates the LFA ‑ 1/ ICAM‑1 - mediated adhesion of lymphocytes, and its gene encodes both positive and negative regulators of cellular adhesion. J Immunol 1997; 159(5): 2250 – 2258.
13. Procházka P, Vícha A, Kodet R et al. Nádory ze skupiny Ewingova sarkomu – molekulární biologie a genetika. Klin Onkol 2007; 20(2): 205 – 208.
14. Bajčiová V, Štěrba J, Tomášek J et al. Nádory adolescentů a mladých dospělých. Praha: Grada 2011 : 108 – 114.
15. De Alava, E, Kawai A, Healey JH et al. EWS ‑ FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing‘s sarcoma. J Clin Oncol 1998; 16(4): 1248 – 1255.
16. Le Deley MC, Delattre O, Schaefer KL et al. Impact of EWS ‑ ETS Psion type on disease progression in Ewing’s sarcoma/ peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro‑E.W.I.N.G. 99 trial. J Clin Oncol 2010; 28(12): 1982 – 1988. doi: 10.1200/ JCO.2009.23.3585.
17. Van Doorninck JA, Ji L, Schaub B et al. Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol 2010; 28 : 1989 – 1994. doi: 10.1200/ JCO.2009.24.5845.
18. Orr WS, Denbo JW, Billups C et al. Analysis of prognostic factors in extraosseous Ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital experience. Ann Oncol 2012; 19(12): 3816 – 3822. doi: 10.1245/ s10434 ‑ 012 ‑ 2458 ‑ 4.
19. Ladenstein R, Pötschger U, Le Deley MC et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro‑EWING 99 trial. J Clin Oncol 2010; 28(20): 3284 – 3291. doi: 10.1200/ JCO.2009.22.9864.
20. Thacker MM, Temple HT, Scully SP. Current treatment for Ewing‘s sarcoma. Expert Rev Anticancer Ther 2005; 5(2): 319 – 331.
21. Spiller M, Bisogno G, Ferrari A et al. Prognostic factors in localized extraosseous Ewing family tumors. Pediatr Blood Cancer 2006; 46(10): A ‑ PD.024, 434.
22. Le Deley MC, Paulussen M, Lewis I et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard ‑ risk ewing sarcoma: results of the randomized noninferiority Euro‑EWING99-R1 trial. J Clin Oncol 2014; 32(23): 2440 – 2448. doi: 10.1200/ JCO. 2013.54.4833.
23. Hou MM, Xi MR, Yang KX. A rare case of extraosseous Ewing sarcoma primarily arising in the ovary. Chin Med J2013; 126(23): 4597.
24. Park JY, Lee S, Kang HJ et al. Primary Ewing‘s sarcoma – primitive neuroectodermal tumor of the uterus: a case report and literature review. Gynecol Oncol 2007; 106(2): 427 – 432.
25. Farley J, O‘Boyle JD, Heaton J et al. Extraosseous Ewing sarcoma of the vagina. Obstet Gynecol 2000; 96(5 Pt 2): 832 – 834.
26. Russin VL, Valente PT, Hanjani P. Psammoma bodies in neuroendocrine carcinoma of the uterine cervix. Acta Cytol 1987; 31(6): 791 – 795.
27. Sato S, Yajima A, Kimura N et al. Peripheral neuroepithelioma (peripheral primitive neuroectodermal tumor) of the uterine cervix. Tohoku J Exp Med 1996; 180(2): 187 – 195.
28. Horn LC, Fischer U, Bilek K. Primitive neuroectodermal tumor of the cervix uteri. A case report. Gen Diagn Pathol 1997; 142(3 – 4): 227 – 230.
29. Cenacchi G, Pasquinelli G, Montanaro L et al. Primary endocervical extraosseous Ewing’s sarcoma/ PNET. Int J Gynecol Pathol 1998; 17(1): 83 – 88.
30. Pauwels P, Ambros P, Hattinger C et al. Peripheral primitive neuroectodermal tumour of the cervix. Virchows Arch 2000; 436(1): 68 – 73.
31. Tsao AS, Roth LM, Sandler A et al. Cervical primitive neuroectodermal tumor. Gynecol Oncol 2001; 83(1): 138 – 142.
32. Malpica A, Moran CA. Primitive neuroectodermal tumor of the cervix: a clinicopathologic and immunohistochemical study of two cases. Ann Diagn Pathol 2002; 6(5): 281 – 287.
33. Goda JS, Nirah B, Mayur K. Primitive neuroectodermal tumour of the cervix: a rare entity. Internet J Radiol 2007; 6 : 3.
34. Snijders ‑ Keilholz A, Ewing P, Seynaeve C et al. Primitive neuroectodermal tumor of the cervix uteri: a case report – changing concepts in therapy. Gynecol Oncol 2005; 98(3): 516 – 519.
35. Farzaneh F, Rezvani H, Boroujeni PT et al. Primitive neuroectodermal tumor of the cervix: a case report. J Med Case Rep 2011; 5 : 489.
36. Li B, Ouyang L, Han X et al. Primary primitive neuroectodermal tumor of the cervix. Onco Targets Ther 2013; 6 : 707 – 711. doi: 10.2147/ OTT.S45889.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2015 Issue 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Extraoseus Ewing‘s Sarcoma, Primary Affection of Uterine Cervix – Case Report
- Embryonal Tumors with Multilayer Rosettes – Rare Central Nervous System Tumors in Infants
- Potential of Cell‑free Circulating DNA in Diagnosis of Cancer
- Estimated Glomerular Filtration Rate in Oncology Patients before Cisplatin Chemotherapy