
Mechanizmy regulace proteinové homeostázy ve vývoji nádorových onemocnění
Mechanisms of Protein Homeostasis Regulation in Cancer Development
Background:
The proteome of eukaryotic cells represents a complex system. Its components are exposed to various intrinsic and extrinsic stresses. Therefore, the function of the cellular proteome is dependent on the existence of compensatory mechanisms balancing the inner protein homeostasis – proteostasis. These mechanisms involve the network of molecular chaperones and transcriptional program regulating their expression. The process of cancerogenesis is accompanied by significant changes in the intracellular milieu of cancer cells – temperature, pH, availability of nutrients. On the one hand, these changes represent a consequence of the deregulated growth of the tumor tissue; on the other hand, they can be a source of selection pressure, which allows the emergence of resistant and aggressive tumor cell populations. Description of the proteostatic apparatus components and the mechanism of their involvement in the tumor tissue development is provided in this review article.
Aim:
This review focuses on the description of two causally linked groups of proteostatic events; their mutual coordination is crucial to the process of tumor cell and by extension the entire tumor tissue response to environmental and internal stress factors. The first group of these processes is represented by the “executory” role of molecular chaperones from HSP70, HSP90 and so-called small molecular chaperone protein families. These proteins are involved in maintaining stability of cellular proteins essential for proliferation, apoptosis, senescence, migration and phenotypic plasticity of tumor cells. The second group of the described processes comprises the posttranslational control of the “systemic” role of the transcription factor HSF1 in regulating the gene expression of molecular chaperones and other genes specifically regulated by this transcription factor in the tumor and stromal cells.
Key words:
molecular chaperones – heat-shock factor 1 – cancer – protein homeostasis
This work was supported by the project MEYS – NPS I – LO1413.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
15. 5. 2016
Accepted:
25. 5. 2016
Authors:
F. Trčka; P. Müller; B. Vojtěšek
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2016; 29(Supplementum 4): 18-24
Category:
Review
doi:
https://doi.org/10.14735/amko20164S18
Overview
Východiska:
Proteom eukaryotické buňky představuje komplexní systém, jehož složky jsou vystaveny nepříznivým vlivům vnitřního a vnějšího prostředí. Funkce buněčného proteomu je tudíž závislá na existenci kompenzačních mechanizmů udržujících vnitřní proteinovou homeostázu – proteostázu. K těmto mechanizmům náleží síť molekulárních chaperonů a transkripční program řídící jejich syntézu. Proces kancerogeneze je provázen výraznými změnami faktorů vnitřního prostředí nádorových buněk – teplota, pH, dostupnost živin. Tyto změny představují na jedné straně důsledek deregulovaného růstu nádorové tkáně a na straně druhé mohou být zdrojem selekčního tlaku, který umožňuje vznik rezistentních a agresivních populací nádorových buněk. Popis složek proteostatického aparátu a mechanizmus jejich zapojení ve vývoji nádorové tkáně jsou předmětem tohoto přehledového článku.
Cíl:
Tento přehledový článek se věnuje popisu dvou kauzálně propojených skupin proteostatických dějů, jejichž vzájemná koordinace je klíčová pro průběh odpovědi nádorové buňky a potažmo i celé nádorové tkáně na environmentální i vnitřní stresové faktory. První skupina těchto dějů je zastoupena „vykonavatelskou“ úlohou molekulárních chaperonů z rodiny HSP70, HSP90 a tzv. malých molekulárních chaperonů. Tyto proteiny se podílejí na udržování stability buněčných proteinů nezbytných pro regulaci proliferace, apoptózy, senescence, migrace a fenotypové plasticity nádorových buněk. K druhé skupině popisovaných dějů pak náleží posttranslační řízení „systémové“ úlohy transkripčního faktoru HSF1 při regulaci exprese genů pro molekulární chaperony a dalších genů specificky regulovaných tímto transkripčním faktorem v nádorových a stromálních buňkách.
Klíčová slova:
molekulární chaperony – heat-shock factor 1 – rakovina – proteinová homeostáza
Úvod
Proteinová homeostáza nebo také proteostáza je souhrnné označení molekulárních dějů podílejících se na udržení balancovaného stavu buněčného proteomu [1]. Mezi procesy, jejichž průběh je regulován proteostatickým buněčným aparátem, patří zejména syntéza proteinů na ribozomech, jejich skládání, intracelulární transport a konečně degradace. Homeostáza intracelulárního proteinového prostředí je za fyziologických podmínek udržována konstitutivní syntézou proteinů tepelného šoku (heat-shock proteins – HSP). HSP proteiny představují molekulární chaperony („gardedámy“, doprovod) napomáhající skládání sekundárních, terciárních i kvarterních proteinových struktur, umožňují transport proteinů, jejich modifikace typu ubikvitinace a také jejich řízenou degradaci v proteazomu [2]. Překročení proteostatické kapacity HSP proteinů vlivem vnějších či vnitřních faktorů vyvolává v buňce stav proteotoxického stresu provázený akumulací/agregací poškozených a nedegradovaných proteinů [3]. Proteotoxický stres aktivuje evolučně konzervovaný transkripční program, v jehož důsledku dochází k silné indukci exprese molekulárních chaperonů [4,5]. Tento transkripční program se nazývá heat-shock response (HSR) a je řízen skupinou sekvenčně příbuzných transkripčních faktorů HSF (heat-shock factor). Klíčovým členem (master regulator) této rodiny transkripčních faktorů je protein HSF1 [6]. Inhibice aktivity HSF1 vede k narušení indukce HSR a činí buňky vysoce senzitivními k proteotoxickému stresu [7,8]. Buňky s inaktivovaným genem pro protein HSF1 nejsou schopny indukovat expresi HSP70 při zvýšení intracelulární teploty, což vede k jejich snížené termotoleranci a indukci apoptózy [7].
Zvýšená anabolická aktivita, mutační a metabolická zátěž představují významné zdroje proteotoxického stresu provázejícího proces maligní transformace a následné proliferace nádorové tkáně [9–11]. V nádorových buňkách byla prokázána zvýšená hladina HSP proteinů a stejně tak konstitutivní aktivace transkripčního faktoru HSF1 v důsledku chronického narušení homeostázy vnitrobuněčného prostředí [12,13]. Způsob, jakým jsou výše popsané složky proteostatického aparátu (HSP a HSF1) zapojeny v procesu kancerogeneze, bude detailněji popsán v následujících odstavcích.
HSP – molekulární chaperony
V souvislosti se stresem indukovaným v nádorových buňkách se zaměříme na popis dvou skupin molekulárních chaperonů, které se liší mechanizmem svého působení (obr. 1).
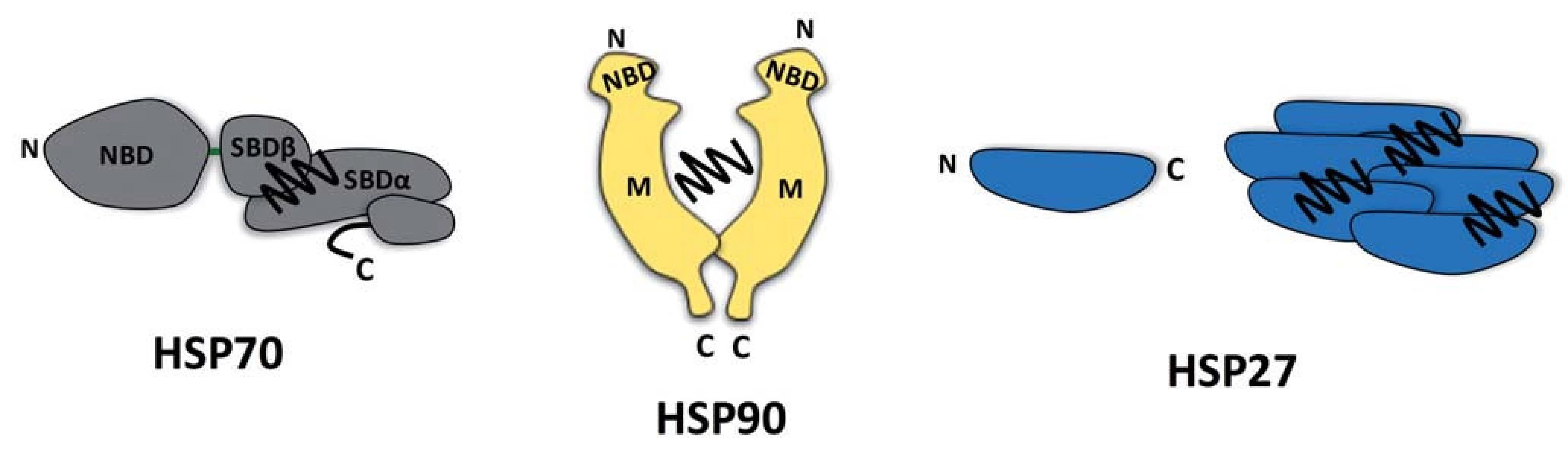
První skupinou jsou HSP proteiny s multidoménovou strukturou vyžadující ke své aktivitě enzymatickou konverzi molekul ATP. Tato skupina chaperonů je reprezentována evolučně konzervovanými proteiny HSP70 a HSP90 (výskyt už v prokaryotech, sekvenční konzervovanost s lidskými proteiny je až 50 %) [14,15], jejichž klidová hladina v buňce dosahuje v případě HSP90 až 1–2 % celkového proteomu [16,17]. Strukturně jsou proteiny HSP70/HSP90 definovány přítomností nukleotid a substrát vázajících domén, jejichž vazebná aktivita je obousměrně alostericky regulována [18,19]. Výrazná konformační flexibilita těchto alostericky aktivních proteinů řízená hydrolýzou ATP představuje mechanistickou podstatu jejich úlohy jakožto molekulárních chaperonů. Avšak zatímco protein HSP70 váže svou substrátovou doménou krátké hydrofobní sekvence existujících či nově tvořených proteinů, čímž zabraňuje jejich denaturaci a agregaci [20], molekulární chaperon HSP90 postrádá přesně definovaný vazebný motiv ve struktuře svých substrátů a přednostně kontaktuje konformačně nestabilní proteiny typu receptorů pro steroidní hormony nebo protein kináz [21–23]. Je nutné poznamenat, že ATP závislý chaperonový cyklus, selekce substrátů a také lokalizace HSP70 a HSP90 v buňce jsou významně ovlivňovány přítomností celé řady tzv. ko-chaperonů, tedy proteinů asociovaných s těmito molekulárními chaperony [24].
Druhou zde popisovanou skupinou molekulárních chaperonů jsou tzv. malé HSP proteiny. Na rozdíl od HSP70/HSP90 představují malé HSP proteiny molekulární chaperony se značně odlišnou aminokyselinovou sekvencí, strukturou i velikostí. Jejich společným rysem je přítomnost asi stoaminokyselinové domény homologní s doménou kanonického člena malých HSP proteinů αB-krystalinu [25]. Tyto proteiny se nezávisle na ATP vážou na denaturované nebo nestrukturované oblasti proteinů a zabraňují jejich agregaci, čímž je udržují ve stavu kompetentním pro jejich zpracovaní ATP závislými chaperonovými systémy HSP70/HSP90 [26]. Mechanizmus transferu substrátu mezi těmito skupinami chaperonových systémů je neznámý. Tyto HSP proteiny jsou nazývány malými, jelikož jejich molekulová hmotnost se pohybuje v rozmezí 10–40 kDa. Monomery o této velikosti se však organizují do rozsáhlých oligomerů čítajících až desítky podjednotek v reakci na tepelný šok [27]. Mezi nejznámější stresem indukované malé HSP chaperony patří proteiny HSP27 a αB-krystalin [28].
HSP proteiny a nádory
Proces maligní transformace buňky a vývoj metastazující nádorové tkáně je provázen řadou patologických změn v molekulárních mechanizmech regulace buněčné proliferace, apoptózy, senescence, migrace a genomové stability označovaných souhrnně jako Hallmarks of cancer [29]. Proteostatické mechanizmy řízené HSP proteiny sehrávají v těchto procesech důležitou roli.
Kontrola průchodu buněčným cyklem je u nádorových buněk porušena. Mezi klíčové molekuly řídící proliferační soběstačnost transformovaných buněk patří zejména komponenty mitogenní signální kaskády spouštěné vazbou růstových faktorů k receptorovým tyrozinkinázám [30]. Deregulace některé ze složek mitogenní kaskády (zvýšená míra exprese, zvýšená aktivita, amplifikace genů, aktivační/inaktivační mutace) může mít onkogenní charakter [31,32]. Chaperon HSP90 se podílí na udržování aktivního stavu nukleárních receptorů steroidních hormonů [33,34] a řady signalizačních molekul účastnících se mitogenní signalizace. Aktivační mutace onkogenních kináz EGFR a BRAF vedou k jejich strukturní nestabilitě a činí je výrazně závislými na přítomnosti HSP90 [35–38]. Další onkogenní kinázou stabilizovanou HSP90 je fúzní kináza BCR-ABL [39]. Rovněž virové kinázy v-Src, Fes, Fps a Yes vyžadují asistenci HSP90 [40,41]. Chaperon HSP90 zároveň stabilizuje buněčné kinázy CDK4, HCK a ERBB2 [42–44]. Obdobnou úlohu při regulaci proliferace nádorových buněk pravděpodobně hraje také protein HSP70, jehož inaktivace vede k inhibici proliferace myších nádorových buněk prsní žlázy [45,46]. Stabilizace onkogenních kináz chaperonem HSP90 a proteostatická aktivita HSP70 tak přispívají ke stimulaci mitogenní signalizace v nádorových buňkách.
Dalším znakem maligně transformované buňky je její schopnost nepodléhat programované buněčné smrti – apoptóze [47]. Ukazuje se, že stresem indukované chaperony HSP27 a HSP70 výrazně inhibují apoptotické signální dráhy svou interakcí s jejich proteinovými komponentami [48]. Protein HSP27 ovlivňuje apoptózu na více úrovních – inhibuje uvolňování cytochromu c a proteinu SMAC-Diablo z mitochondrií, tlumí aktivitu kaspázy 3 a 9 a také signalizaci zprostředkovanou ligandy Fas, TNF a TRAIL [49–52]. Chaperon HSP70 inhibuje apoptózu svou vazbou na protein Apaf-1, čímž zabraňuje tvorbě apoptozómu [53]. V nenádorové buňce tak chaperony HSP27 a HSP70 slouží jako homeostatický mechanizmus tolerance určitého stupně proteotoxického stresu dříve, než je nenávratně aktivována programovaná buněčná smrt. Nádorová buňka naopak nadměrnou produkcí HSP27 a HSP70 zvyšuje svou rezistenci k cytotoxickým látkám inaktivací apoptotické dráhy.
Nádorové buňky obcházejí přirozený proces replikativní senescence aktivací telomerázy [54]. Účinnost telomerázy je podmíněna její asociací s HSP90 [55]. Farmakologická inhibice HSP90 vede k inaktivaci telomerázové funkce v nádorových buňkách [56]. HSP90 se v nádorových buňkách podílí také na regu - laci exprese genu kódujícího telomerázu [57]. Těmito mechanizmy přispívá HSP90 v nádorových buňkách k inhibici senescence.
Metastazování nádorových buněk z primárního ložiska je mnohastupňovým procesem zahrnujícím remodelaci nádorové tkáně, extravazaci nádorových buněk do krevního řečiště a jejich usazení v cílové tkáni [29]. Ačkoli spektrum mutací podmiňujících metastatický potenciál nádorových buněk je heterogenní [58,59], zvýšená aktivita molekulárních chaperonů představuje nutnou podmínku metastatické kaskády [60,61]. Stabilizace kináz podílejících se na regulaci signalizace z extracelulární matrix (focal adhesion kinase – FAK, integrin-linked kinase – ILK) a receptorových tyrozinkináz ERBB2 a MET chaperonem HSP90 výrazně přispívá k metastatickému potenciálu nádorových buněk [60]. Inaktivace HSP70 vede ke snížení exprese a aktivity MET kinázy u myších prsních nádorů [46]. Bylo zjištěno, že proces epiteliálně-mezenchymální tranzice, který provází metastazování, je značně závislý na expresi chaperonu HSP27 [61–63].
Hypotéza vysvětlující význam regulátorů proteostázy a zejména pak proteinu HSP90 pro fenotypovou plasticitu nádorové tkáně pokládá HSP90 za „pufr“ genetické nestability [64]. Tato hypotéza vychází z předpokladu, že vzrůstající genetická nestabilita v nádorové tkáni (především mutace strukturních genů) je na úrovni proteinů „pufrována“ chaperonovým systémem HSP90. Limitní hodnoty proteotoxického stresu v buňkách lokalizovaných v centru nádoru vystavených hypoxickému acidifikovanému prostředí [64] mohou vést k inaktivaci chaperonu HSP90 a fenotypové manifestaci nahromaděných genetických změn. Tato „exploze“ fenotypových variant v pokročilejších nádorech by mohla vysvětlovat vznik buněčných klonů vykazujících rezistenci k použité léčbě [65,66].
HSF1 – transkripční regulace HSR
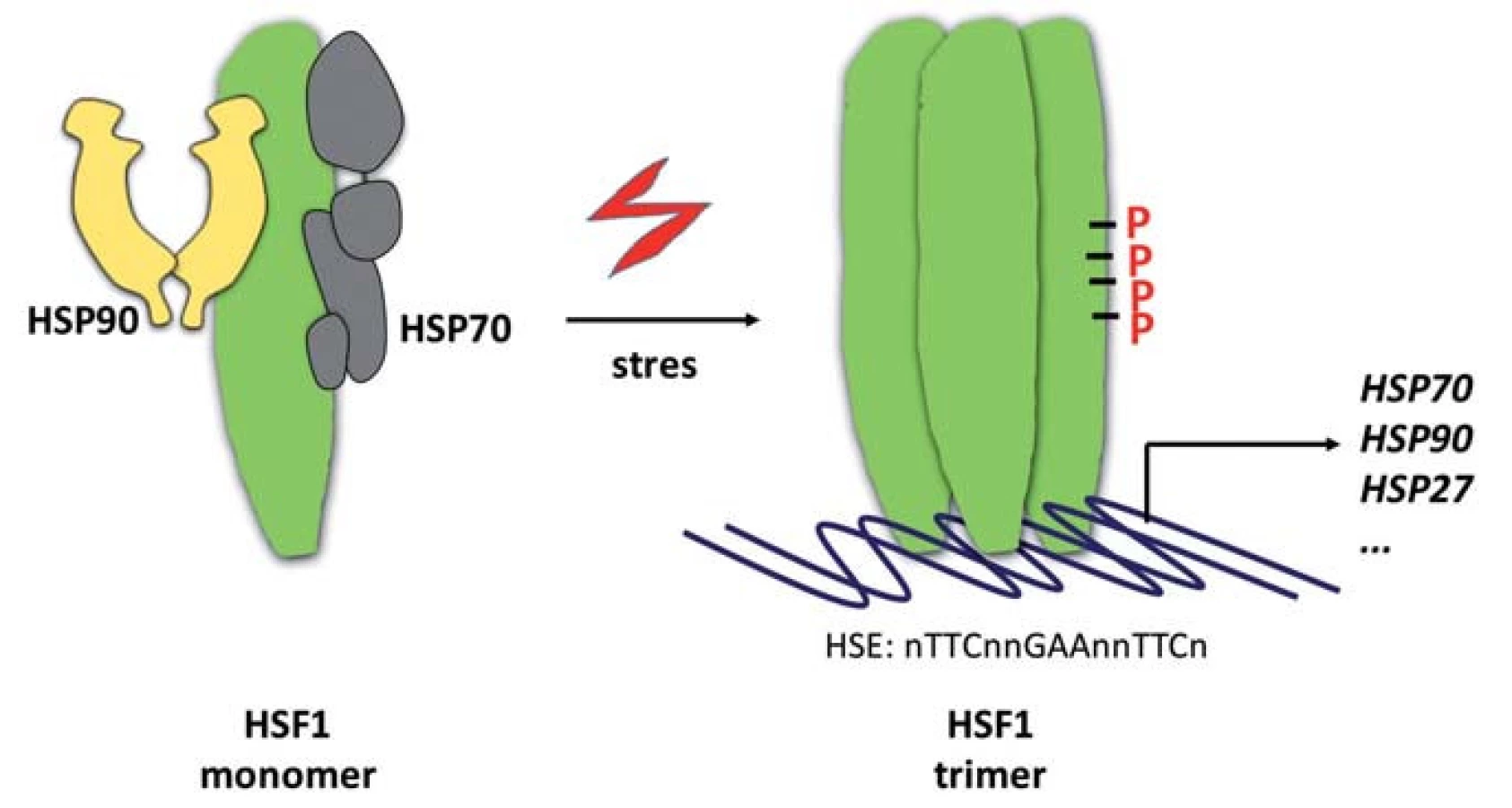
HSP proteiny představují „vykonavatelskou“ složku proteostatického aparátu. Na systémové úrovni je reakce na proteotoxický stres spouštěna robustním transkripčním programem řízeným transkripčním faktorem HSF1. Transkripční faktor HSF1 je v nestresovaných buňkách přítomen v monomerním stavu, který je udržován jeho asociací s chaperony HSP70 a HSP90 (obr. 2) [67,68]. V buňkách vystavených nepříznivým vnitřním či vnějším podmínkám dochází k disociaci HSP70/HSP90 od monomeru HSF1, který následně trimerizuje za vzniku transkripčně aktivní formy lokalizované v jádře, kde se váže ke konsenzuálním sekvenčním motivům označovaným jako heat-shock elements (HSE, sekvence nTTCnnGAAnnTTCn) v promotorových oblastech regulovaných genů [69]. Aktivace HSF1 je provázena rozsáhlým spektrem jeho posttranslačních modifikací. Patří k nim zejména fosforylace [3], acetylace [70] a sumoylace [71]. Tyto modifikace jsou součástí komplexního mechanizmu „vylaďování“ transaktivační aktivity proteinu HSF1.
HSF1 – regulace aktivity fosforylací
Účinná aktivace HSF1 při proteotoxickém stresu je závislá na fosforylaci tohoto faktoru. Modifikaci podléhají zbytky serinu a threoninu v pozicích S230, S320, S419 a T142 katalyzované kinázami CaMKII (calcium/calmodulin-dependent protein kinase II), PKA (protein kinase A), CK2 (casein kinase 2) a PLK1 (polo-like kinase 1) [72–75]. Za klíčovou fosforylaci nutnou pro stresem indukovanou aktivaci HSF1 je považována modifikace serinu S326 [76]. Na modifikaci serinu S326 se podílí kináza mTOR [77] a také kinázy RAS/MAPK kaskády [3].
RAS/MAPK kaskáda (RAS-RAF-MEK-ERK) je centrální signální drahou řídící buněčnou proliferaci a diferenciaci, ale také metabolickou aktivitu [78]. Bylo pozorováno, že zatímco inhibice kinázy MEK vede ke snížení stresem indukované fosforylace HSF1 v pozici S326, inhibice aktivity kinázy ERK naopak tuto fosforylaci zvyšuje [3]. Jelikož je kináza ERK považována za výlučný substrát kinázy MEK a za konečný efektor RAS/MAPK kaskády, bylo toto zjištění překvapující. Navíc byla prokázána existence stresem indukovaného komplexu proteinů HSF1, MEK a ERK, ve kterém dochází k inhibici MEK regulované fosforylace serinu S326 proteinu HSF1 prostřednictvím ERK katalyzovaných inhibičních fosforylací threoninů T292 a T386 kinázy MEK. Tato pozorování dokládají regulační úlohu RAS aktivované kinázy MEK jak v dráze vedoucí ke kináze ERK, tak k transkripčnímu faktoru HSF1 a odhalují vzájemně protikladné efekty kináz MEK a ERK při regulaci aktivity HSF1. Jelikož aktivační mutace komponent RAS/MAPK signalizační dráhy jsou přítomny až u 30 % nádorů, představuje aberantní konstitutivní aktivace transkripčního faktoru HSF1 inherentní proces maligní transformace [79].
Cílená fosforylace se také podílí na inhibici aktivace HSF1. Mezi inhibiční fosforylace HSF1 náleží modifikace serinu S121, která je katalyzována kinázou AMPK (AMP-activated protein kinase) v podmínkách snížené dostupnosti živin vyvolávajících metabolický stres [80]. Tato kináza reaguje na nerovnováhu buněčného metabolizmu projevující se zvýšenou intracelulární koncentrací AMP oproti ATP a fosforyluje řadu buněčných efektorů, které se podílejí na aktivaci energeticky nákladných anabolických procesů (lipogeneze, glykolýza, beta oxidace, proteinová syntéza) [81]. AMPK indukované snížení spotřeby ATP a zvýšení jeho produkce vede k ustanovení metabolické homeostázy [81]. Aktivace AMPK v průběhu metabolického stresu inhibuje HSF1 řízenou HSR a činí nádorové buňky náchylné k proteotoxickému stresu [80]. Tyto antagonistické vztahy mezi úlohou HSF1 (proteotoxický stres) a AMPK (metabolický stres) mohou také vysvětlovat Warburgův efekt, tedy závislost proteotoxicky stresovaných nádorových buněk na glukóze.
HSF1 – translace proteinů
Aktivita mTOR kinázy je spojena s indukcí syntézy proteinů a lipidů v buňkách stimulovaných růstovými faktory a v prostředí s dostatkem živin a kyslíku [82]. Ve stresových podmínkách (energetická disbalance, nedostatek aminokyselin) je naopak mTOR inhibována a dochází k indukci autofágie [83]. Nádorové buňky jsou závislé na robustní proteinové syntéze [9]. Spolu s proteinem p53 tak patří kináza mTOR a její nadřazené regulační komponenty PI3K, PTEN a AKT k nejčastěji mutovaným signálním drahám v nádorech [82]. Již jsme zmínili, že kináza mTOR se podílí na aktivaci HSF1 jeho fosforylací [77]. Inhibice aktivity kinázy mTOR zabraňuje HSF1 indukované expresi HSP proteinů v přítomnosti proteotoxického stresu [84]. Dále bylo popsáno, že k výrazné inaktivaci transaktivační funkce HSF1 dochází při inhibici mTOR aktivovaného [85] eukaryotického iniciačního translačního faktoru 4A (eIF4A) látkou zvanou roglamid A [11]. Pozoruhodné je také zjištění, že alternativní sestřih transkriptu eukaryotického elongačního faktoru eEF1Bδ vede ke vzniku mRNA kódující transkripční faktor kooperující s HSF1 při regulaci exprese genů, jejichž promotory obsahují HSE element [86]. Tyto skutečnosti naznačují, že míra proteinové syntézy regulovaná kinázou mTOR a translačními faktory je koordinována s mírou syntézy složek proteostatického aparátu řízenou HSF1.
V současnosti je intenzivně studován tzv. Ribosome quality complex (RQC) [87]. Jedná se o proteinový komplex asociovaný s ribozomy, které vykazují známky aberantního průběhu syntézy. Je tvořen komponentami RQC1, Linsterin 1 (LTN1), CDC48, TEA2 a dalšími. Vazba RQC k aberantnímu ribozomu vede k ubikvitinaci a proteazomální degradaci syntetizovaných polypeptidů a k recyklaci ribozomálních podjednotek [88]. Protein TEA2 je schopen katalyzovat prodlužování C-konce nedosyntetizovaných polypeptidů o zbytky alaninu a threoninu, což vede k aktivaci HSF1 [87,89]. Mechanizmus této aktivace prozatím není znám. Aktivita HSF1 tak není korelována pouze s mírou translace, ale také s její kvalitou monitorovanou RQC.
HSF1 – nádorově specifická funkce
Transkripční faktor HSF1 neaktivuje pouze geny zapojené do proteostatických mechanizmů, ale také řadu dalších funkčně rozmanitých genů [90,91]. V nádorech je aktivován zcela specifický transkripční program řízený HSF1 [91]. Řada promotorů stresem indukovaných genů není v nádorových buňkách obsazena HSF1. Naopak, HSF1 se v transformovaných buňkách váže k promotorovým sekvencím odlišným od těch, které jsou aktivovány při stresových podmínkách [91]. Nádorově specifický transkripční program proteinu HSF1 je pravděpodobně důsledkem odlišností v typech a míře stresů, kterým je vystavena nádorová buňka na rozdíl od buňky vystavené tepelnému šoku. Mezi geny aktivované HSF1 v nádorových buňkách patří geny zapojené v proteosyntéze (EIF4A2, RPL22), regulaci buněčného cyklu (CDC6, KNTC1, POLA2), reparaci DNA a chromatinové remodelaci (MLH1, CBX3), metabolizmu (FASN, PGK1) a úpravách mRNA (HNRNPA3, RBM23) [91]. Unikátní HSF1 transkripční program je také aktivován ve fibroblastech nádorového stromatu [92]. HSF1 ve stromálních fibroblastech řídí expresi secernovaných cytokinů TGF-β a SDF1, které stimulují růst sousedních nádorových buněk. Tyto skutečnosti ukazují, že transkripční faktor HSF1 je svou transaktivační aktivitou schopen na systémové úrovni ovlivňovat nejen fyziologické děje v nádorových buňkách, ale také ve smíšené nádorové tkáni, čímž výrazně přispívá k fenotypové plasticitě, agresivitě a rezistenci nádorů.
Závěr
„Vykonavatelské“ i „systémové“ složky mechanizmů řídících buněčnou odpověď na proteotoxický stres zastoupené molekulárními chaperony a transkripčním faktorem HSF1 reprezentují evolučně konzervovaný biologický princip, jehož původní funkcí byla reakce na nepříznivé podmínky prostředí. Proteiny HSP jsou známy už u prokaryot. Transkripční faktor HSF1 se objevil u jednobuněčných eukaryot, kvasinek. Vznik mnohobuněčných organizmů přenesl nutnost jednotlivých buněk reagovat na změny prostředí na centralizovanou, organizmální úroveň. Zdá se, že u mnohobuněčných organizmů, u člověka, nezastává osa HSF1-HSP pouze úlohu signální dráhy přežití individuálních buněk, ale koordinuje homeostázu celých tkání [92,93]. Heterogenní populace nádorových a nenádorových buněk v komplexní nádorové tkáni vystavené proměnlivým podmínkám prostředí tak pravděpodobně v deregulované podobě využívají transkripčního programu HSF1 pro své vlastní přežití nezávisle na přežití celého organizmu. Ačkoli systém HSF1-HSP představuje pro nádorovou tkáň efektivní ochranu před cytotoxickou protinádorovou léčbou, cílená a efektivní inhibice tohoto systému v nádorových buňkách by mohla inaktivovat jeden z klíčových mechanizmů vzniku progresivního nádorového onemocnění.
Práce byla podpořena projektem MŠMT – NPU I – LO1413.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Filip Trčka, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: filip.trcka@mou.cz
Obdrženo: 15. 5. 2016
Přijato: 25. 5. 2016
Sources
1. Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem 2015; 84 : 435–464. doi: 10.1146/annurev-biochem-060614-033955.
2. Trcka F, Vojtesek B, Muller P. Protein quality control and cancerogenesis. Klin Onkol 2012; 25 (Suppl 2): 2S38–2S44.
3. Tang Z, Dai S, He Y et al. MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 2015; 160 (4): 729–744. doi: 10.1016/ j.cell.2015.01.028.
4. Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol 2011; 76 : 91–99. doi: 10.1101/sqb.2012.76.010637.
5. Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem 2011; 80 : 1089–1115. doi: 10.1146/annurev-biochem-060809-095203.
6. Vihervaara A, Sistonen L. HSF1 at a glance. J Cell Sci 2014; 127 (Pt 2): 261–266. doi: 10.1242/jcs.132605.
7. Zhang Y, Huang L, Zhang J et al. Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J Cell Biochem 2002; 86 (2): 376–393.
8. Izu H, Inouye S, Fujimoto M et al. Heat shock transcription factor 1 is involved in quality-control mechanisms in male germ cells. Biol Reprod 2004; 70 (1): 18–24.
9. Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer 2003; 3 (3): 179–192.
10. Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspect Biol 2012; 4 (11): pii: a012252. doi: 10.1101/cshperspect.a012252.
11. Santagata S, Mendillo ML, Tang YC et al. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 2013; 341 (6143): 1238303. doi: 10.1126/science.1238303.
12. Dai C, Santagata S, Tang Z et al. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest 2012; 122 (10): 3742–3754. doi: 10.1172/JCI62727.
13. Ciocca DR, Arrigo AP, Calderwood SK. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol 2013; 87 (1): 19–48. doi: 10.1007/s00204-012-0918-z.
14. Hunt C, Morimoto RI. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proc Natl Acad Sci U S A 1985; 82 (19): 6455–6459.
15. Chen B, Zhong D, Monteiro A. Comparative genomics and evolution of the HSP90 family of genes across all kingdoms of organisms. BMC Genomics 2006; 7 : 156.
16. Sreedhar AS, Kalmár E, Csermely P et al. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett 2004; 562 (1–3): 11–15.
17. Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 2003; 112 (1): 41–50.
18. Mayer MP, Schroder H, Rudiger S et al. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat Struct Biol 2000; 7 (7): 586–593.
19. Li J, Buchner J. Structure, function and regulation of the hsp90 machinery. Biomed J 2013; 36 (3): 106–117. doi: 10.4103/2319-4170.113230.
20. Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 2005; 62 (6): 670–684.
21. Taipale M, Tucker G, Peng J et al. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 2014; 158 (2): 434–448. doi: 10.1016/j.cell.2014.05.039.
22. Taipale M, Krykbaeva I, Koeva M et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012; 150 (5): 987–1001. doi: 10.1016/j.cell.2012.06.047.
23. Kirschke E, Goswami D, Southworth D et al. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 2014; 157 (7): 1685–1697. doi: 10.1016/j.cell.2014.04. 038.
24. Calderwood SK. Molecular cochaperones: tumor growth and cancer treatment. Scientifica (Cairo) 2013; 2013: ID 217513. doi: 10.1155/2013/217513.
25. Haslbeck M, Franzmann T, Weinfurtner D et al. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol 2005; 12 (10): 842–846.
26. Cashikar AG, Duennwald M, Lindquist SL. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J Biol Chem 2005; 280 (25): 23869–23875.
27. van Montfort RL, Basha E, Friedrich KL et al. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol 2001; 8 (12): 1025–1030.
28. Arrigo AP, Simon S, Gibert B et al. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett 2007; 581 (19): 3665–3674.
29. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144 (5): 646–674. doi: 10.1016/j.cell.2011.02.013.
30. Momeny M, Saunus JM, Marturana F et al. Heregulin-HER3-HER2 signaling promotes matrix metalloproteinase-dependent blood-brain-barrier transendothelial migration of human breast cancer cell lines. Oncotarget 2015; 6 (6): 3932–3946.
31. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016; 129 (7): 1287–1292. doi: 10.1242/jcs.182873.
32. Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol 2016; 9 : 34. doi: 10.1186/s13045-016-0268-z.
33. Joab I, Radanyi C, Renoir M et al. Common non-hormone binding component in non-transformed chick oviduct receptors of four steroid hormones. Nature 1984; 308 (5962): 850–853.
34. Sanchez ER, Toft DO, Schlesinger MJ et al. Evidence that the 90-kDa phosphoprotein associated with the untransformed L-cell glucocorticoid receptor is a murine heat shock protein. J Biol Chem 1985; 260 (23): 12398–12401.
35. Zhang X, Gureasko J, Shen K et al. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006; 125 (6): 1137–1149.
36. Wan PT, Garnett MJ, Roe SM et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004; 116 (6): 855–867.
37. Shimamura T, Lowell AM, Engelman JA et al. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res 2005; 65 (14): 6401–6408.
38. da Rocha Dias S, Friedlos F, Light Y et al. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res 2005; 65 (23): 10686–10691.
39. Tao W, Chakraborty SN, Leng X et al. HSP90 inhibitor AUY922 induces cell death by disruption of the Bcr-Abl, Jak2 and HSP90 signaling network complex in leukemia cells. Genes Cancer 2015; 6 (1–2): 19–29.
40. Brugge JS, Erikson E, Erikson RL. The specific interaction of the Rous sarcoma virus transforming protein, pp60src, with two cellular proteins. Cell 1981; 25 (2): 363–372.
41. Lipsich LA, Cutt JR, Brugge JS. Association of the transforming proteins of Rous, Fujinami, and Y73 avian sarcoma viruses with the same two cellular proteins. Mol Cell Biol 1982; 2 (7): 875–880.
42. Vaughan CK, Gohlke U, Sobott F et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol Cell 2006; 23 (5): 697–707.
43. Scholz GM, Hartson SD, Cartledge K et al. The molecular chaperone Hsp90 is required for signal transduction by wild-type Hck and maintenance of its constitutively active counterpart. Cell Growth Differ 2001; 12 (8): 409–417.
44. Xu W, Yuan X, Xiang Z et al. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol 2005; 12 (2): 120–126.
45. Meng L, Hunt C, Yaglom JA et al. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene 2011; 30 (25): 2836–2845. doi: 10.1038/onc.2011.5.
46. Gong J, Weng D, Eguchi T et al. Targeting the hsp70 gene delays mammary tumor initiation and inhibits tumor cell metastasis. Oncogene 2015; 34 (43): 5460–5471. doi: 10.1038/onc.2015.1.
47. Ouyang L, Shi Z, Zhao S et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 2012; 45 (6): 487–498. doi: 10.1111/j.1365-2184.2012.00845.x.
48. Lanneau D, de Thonel A, Maurel S et al. Apoptosis versus cell differentiation: role of heat shock proteins HSP90, HSP70 and HSP27. Prion 2007; 1 (1): 53–60.
49. Chauhan D, Li G, Hideshima T et al. Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance. Blood 2003; 102 (9): 3379–3386.
50. Paul C, Simon S, Gibert B et al. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp Cell Res 2010; 316 (9): 1535–1552. doi: 10.1016/j.yexcr.2010.03.006.
51. Zenke G, Strittmatter U, Tees R et al. A cocktail of three monoclonal antibodies significantly increases the sensitivity of an enzyme immunoassay for human granulocyte-macrophage colony-stimulating factor. J Immunoassay 1991; 12 (2): 185–206.
52. Arrigo AP, Gibert B. HspB1 dynamic phospho-oligomeric structure dependent interactome as cancer therapeutic target. Curr Mol Med 2012; 12 (9): 1151–1163.
53. Beere HM, Wolf BB, Cain K et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000; 2 (8): 469–475.
54. Jaskelioff M, Song W, Xia J et al. Telomerase deficiency and telomere dysfunction inhibit mammary tumors induced by polyomavirus middle T oncogene. Oncogene 2009; 28 (48): 4225–4236. doi: 10.1038/onc.2009.268.
55. Toogun OA, Dezwaan DC, Freeman BC. The hsp90 molecular chaperone modulates multiple telomerase activities. Mol Cell Biol 2008; 28 (1): 457–467.
56. Chaklader M, Das P, Pereira JA et al. 17-AAG mediated targeting of Hsp90 limits tert activity in peritoneal sarcoma related malignant ascites by downregulating cyc - lin D1 during cell cycle entry. Exp Oncol 2012; 34 (2): 90–96.
57. Kim RH, Kim R, Chen W et al. Association of hsp90 to the hTERT promoter is necessary for hTERT expression in human oral cancer cells. Carcinogenesis 2008; 29 (12): 2425–2431. doi: 10.1093/carcin/bgn225.
58. Lipsyc M, Yaeger R. Impact of somatic mutations on patterns of metastasis in colorectal cancer. J Gastrointest Oncol 2015; 6 (6): 645–649. doi: 10.3978/j.issn.2078-6891.2015.045.
59. Dhamija S, Diederichs S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int J Cancer 2016; 139 (2): 269–280. doi: 10.1002/ijc.30039.
60. Tsutsumi S, Beebe K, Neckers L. Impact of heat-shock protein 90 on cancer metastasis. Future Oncol 2009; 5 (5): 679–688. doi: 10.2217/fon.09.30.
61. Pavan S, Musiani D, Torchiaro E et al. HSP27 is required for invasion and metastasis triggered by hepatocyte growth factor. Int J Cancer 2014; 134 (6): 1289–1299. doi: 10.1002/ijc.28464.
62. Shiota M, Bishop JL, Nip KM et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res 2013; 73 (10): 3109–3119.
63. Gibert B, Eckel B, Gonin V et al. Targeting heat shock protein 27 (HspB1) interferes with bone metastasis and tumour formation in vivo. Br J Cancer 2012; 107 (1): 63–70. doi: 10.1038/bjc.2012.188.
64. Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature 1998; 396 (6709): 336–342.
65. Burrell RA, McGranahan N, Bartek J et al. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013; 501 (7467): 338–345. doi: 10.1038/nature12625.
66. Turtoi A, Blomme A, Castronovo V. Intratumoral heterogeneity and consequences for targeted ther - apies. Bull Cancer 2015; 102 (1): 17–23. doi: 10.1016/j.bulcan.2014.12.006.
67. Shi Y, Mosser DD, Morimoto RI. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev 1998; 12 (5): 654–666.
68. Ali A, Bharadwaj S, O‘Carroll R et al. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol 1998; 18 (9): 4949–4960.
69. Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 2010; 11 (8): 545–555. doi: 10.1038/nrm2938.
70. Raychaudhuri S, Loew C, Korner R et al. Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell 2014; 156 (5): 975–985. doi: 10.1016/j.cell.2014.01.055.
71. Brunet Simioni M, De Thonel A, Hammann A et al. Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene 2009; 28 (37): 3332–3344. doi: 10.1038/onc.2009.188.
72. Holmberg CI, Hietakangas V, Mikhailov A et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J 2001; 20 (14): 3800–3810.
73. Zhang Y, Murshid A, Prince T et al. Protein kinase A regulates molecular chaperone transcription and protein aggregation. PLoS One 2011; 6 (12): e28950. doi: 10.1371/journal.pone.0028950.
74. Soncin F, Zhang X, Chu B et al. Transcriptional activity and DNA binding of heat shock factor-1 involve phosphorylation on threonine 142 by CK2. Biochem Biophys Res Commun 2003; 303 (2): 700–706.
75. Kim SA, Yoon JH, Lee SH et al. Polo-like kinase 1 phosphorylates heat shock transcription factor 1 and mediates its nuclear translocation during heat stress. J Biol Chem 2005; 280 (13): 12653–12657.
76. Guettouche T, Boellmann F, Lane WS et al. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 2005; 6 : 4.
77. Chou SD, Prince T, Gong J et al. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS One 2012; 7 (6): e39679. doi: 10.1371/journal.pone.0039679.
78. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 2014; 13 (12): 928–942. doi: 10.1038/nrd4 281.
79. Fernández-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer 2011; 2 (3): 344–358. doi: 10.1177/1947601911411084.
80. Dai S, Tang Z, Cao J et al. Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J 2015; 34 (3): 275–293. doi: 10.15252/embj.201489062.
81. Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol 2015; 33 : 1–7. doi: 10.1016/j.ceb.2014.09.004.
82. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12 (1): 21–35. doi: 10.1038/nrm 3025.
83. Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273 (7): 3963–3966.
84. Acquaviva J, He S, Sang J et al. mTOR inhibition potentiates HSP90 inhibitor activity via cessation of HSP synthesis. Mol Cancer Res 2014; 12 (5): 703–713. doi: 10.1158/1541-7786.MCR-13-0605.
85. Showkat M, Beigh MA, Andrabi KI. mTOR signaling in protein translation regulation: implications in cancer genesis and therapeutic interventions. Mol Biol Int 2014; 2014: ID 686984. doi: 10.1155/2014/686984.
86. Kaitsuka T, Tomizawa K, Matsushita M. Transformation of eEF1Bdelta into heat-shock response transcription factor by alternative splicing. EMBO Rep 2011; 12 (7): 673–681. doi: 10.1038/embor.2011.82.
87. Brandman O, Stewart-Ornstein J, Wong D et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 2012; 151 (5): 1042–1054. doi: 10.1016/j.cell.2012.10.044.
88. Brandman O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol 2016; 23 (1): 7–15.
89. Shen PS, Park J, Qin Y et al. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 2015; 347 (6217): 75–78. doi: 10.1126/science.1259724.
90. Trinklein ND, Murray JI, Hartman SJ et al. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell 2004; 15 (3): 1254–1261.
91. Mendillo ML, Santagata S, Koeva M et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012; 150 (3): 549–562. doi: 10.1016/j.cell.2012.06. 031.
92. Scherz-Shouval R, Santagata S, Mendillo ML et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014; 158 (3): 564–578. doi: 10.1016/j.cell.2014.05.045.
93. Douglas PM, Baird NA, Simic MS et al. Heterotypic signals from neural HSF-1 separate thermotolerance from longevity. Cell Rep 2015; 12 (7): 1196–1204.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue Supplementum 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Úloha PD-1/PD-L1 signalizace v protinádorové imunitní odpovědi
- Nemalobuněčný karcinom plic – od imunobiologie k imunoterapii
- Nádorové buňky jako dynamický systém – molekulární a fenotypové změny v průběhu vzniku, progrese a šíření nádoru
- Nové metody studia metylace DNA – MS-HRM analýza a elektrochemie