
Molekulární podstata kancerogeneze epiteliálních ovariálních karcinomů
Molecular Mechanisms of Carcinogenesis of Epithelial Ovarian Cancers
Background:
Epithelial ovarian carcinomas are one of the most common causes of death among gynecologic malignancies in the Czech population. This group of tumors is characterized by considerable heterogeneity in terms of its pathogenesis and response to therapy. It is questionable whether advances in the elucidation of molecular pathogenesis of various types of epithelial ovarian carcinomas can contribute to application of personalized targeted therapy.
Aims:
This work aims to summarize current knowledge on carcinogenesis and molecular basis of epithelial ovarian cancers and point out their potential applications in clinical practice. The characterization of the epithelial ovarian carcinomas is based on a dualistic model, which divides these tumors into two groups based on their different origins and mechanisms of carcinogenesis. Type I includes low-grade serous carcinomas, endometrioid carcinomas, mucinous carcinomas and Brenner tumor. Type II then comprises high-grade serous carcinomas.
Conclusion:
The new findings acquired by next generation sequencing revealed major differences in the genetic alterations in both groups of tumors. Differences in genetic instability between the two groups of tumors determine the mechanisms of their carcinogenesis and show new ways for application of targeted therapy. Deficient homologous recombination and high genetic instability in type II tumors is a prerequisite for efficient application of platinum cytostatics and PARP (poly-ADP ribose polymerase) inhibitors. On the other hand, carcinogenesis of the less aggressive, but often resistant type I tumous is dependent on the activation of signaling pathways PI3K/AKT and RAS/BRAF/MEK/ERK pathway. Targeted inhibition of these pathways could efficiently improve therapy of type I tumors and decrease serious adverse side effects.
Key words:
ovarian cancer – high-grade serous ovarian carcinoma – low-grade ovarian carcinoma – endometrioid carcinoma – mucinous carcinoma – malignant transformation – genetic instability
We would like to thank M.Sc. Eva Michalova for critical reading of the manuscript.
This work was supported by the project MEYS – NPS I – LO1413.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
7. 8. 2016
Accepted:
29. 8. 2016
Authors:
Z. Müllerová 1; T. Müller 1; K. Křivánková 2; B. Vojtěšek 2; P. Müller 2
Authors‘ workplace:
Gynekologicko-porodnické oddělení, Nemocnice Nové Město na Moravě
1; Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
2
Published in:
Klin Onkol 2016; 29(Supplementum 4): 46-53
Category:
Review
doi:
https://doi.org/10.14735/amko20164S46
Overview
Východiska:
Epiteliální ovariální karcinomy jsou v české populaci jednou z nejčastějších příčin úmrtí na gynekologické malignity. Tato skupina nádorů je charakterizována značnou heterogenitou jak z hlediska její patogeneze, tak i odpovědí na terapii. Je otázkou, zda pokroky v objasnění molekulární patogeneze jednotlivých typů epiteliálních ovariálních karcinomů mohou v budoucnu přispět k aplikaci cílené personalizované léčby.
Cíle:
Tato práce se snaží shrnout současné poznatky o kancerogenezi a molekulární podstatě epiteliálních nádorů ovaria a ukázat na možné využití těchto poznatků v klinické praxi. Pro základní charakterizaci epiteliálních ovariálních karcinomů je využit dualistický model, který rozděluje tyto nádory do dvou skupin na základě odlišného původu a mechanizmu kancerogeneze. Typ I zahrnuje low-grade serózní karcinomy, endometrioidní karcinomy, mucinózní karcinomy a Brennerův tumor, typ II pak high-grade serózní karcinomy.
Závěr:
Nově získané poznatky získané sekvenováním nové generace ukazují zásadní odlišnosti v genetických alteracích u obou skupin nádorů. Rozdíly v genetické nestabilitě mezi oběma skupinami nádorů určují způsob jejich kancerogeneze a poukazují na nové cesty pro aplikaci cílené terapie. Porucha homologní rekombinace a vysoká genetická nestabilita u nádorů typu II je hlavní příčinou jejich senzitivity k platinovým cytostatikům a inhibitorům PARP (poly-ADP ribose polymerase). Na druhou stranu karcinogeneze méně agresivních, ale často rezistentních nádorů typu I je závislá na aktivaci signalizačních drah PI3K/AKT a RAS/BRAF/MEK/ERK. Cílená inhibice těchto drah u nádorů typu I by tak v budoucnu mohla představovat efektivnější terapii s nižší toxicitou.
Klíčová slova:
nádor ovaria – high-grade serózní ovariální karcinom – low-grade serózní ovariální karcinom – endometrioidní karcinom – mucinózní karcinom – nádorová transformace – genetická nestabilita
Úvod
Gynekologické zhoubné nádory představují přibližně 15 % všech nádorových onemocnění ženské populace v ČR. Zhoubné novotvary vaječníků tvoří histopatologicky heterogenní skupinu nádorů, z nichž ovariální karcinomy jsou klinicky nejvýznamnější [1]. V současné době existuje celá řada studií zabývajících se molekulárními základy ovariální kancerogeneze a výsledky těchto prací významně napomáhají upřesnit klasifikaci ovariálních karcinomů, zlepšit prognostiku a v konečném důsledku individualizovat léčbu onemocnění. Cílem této práce je přehlednou formou shrnout nejnovější molekulárně genetické poznatky o nádorové transformaci epiteliálních ovariálních karcinomů.
Ovariální karcinomy dlouhodobě zaujímají první místo v mortalitě na gynekologické malignity. I po absolvování adekvátního chirurgického výkonu a následné adjuvantní chemoterapii se časové rozpětí mediánu do progrese nemoci (progression-free survival – PFS) pohybuje mezi 16 a 22 měsíci, medián celkového přežití (overall survival – OS) pak mezi 2 a 4 roky a 5leté OS nedosahuje ani 30 %. V případě tzv. high-grade variant ovariálního karcinomu uvádějí některé práce progresi onemocnění po adekvátním chirurgickém výkonu a standardní adjuvantní chemoterapii s aplikací platinových derivátů v kombinaci s taxány zhruba u 25 % pacientek do šesti měsíců od zahájení léčby [2]. Hereditární výskyt onemocnění je popisován v 10–13 % případech ovariálního karcinomu a je zpravidla spojený se zárodečnou mutací v genech nádorových supresorů BRCA1 a BRCA2 [3]. V rámci klinické manifestace hereditárního ovariálního karcinomu rozlišujeme tři formy: 1. syndrom karcinomu prsu a ovaria, 2. „site-specific“ ovariální karcinom a 3. hereditární nepolypózní kolorektální karcinom (hereditary non-polyposis colorectal cancer – HNPCC), tzv. Lynchův syndrom. První dvě formy charakterizují vrozené výše uvedené mutace genů BRCA1 a BRCA2, HNPCC je spojen s vrozenou mutací genů, tzv. mismatch repair systému (MMR). Celoživotní riziko vzniku karcinomu vaječníků se pohybuje pro nosičky mutací mezi 20 a 40 % a je přibližně 10–20krát vyšší než v běžné populaci. Průměrný věk vzniku dědičných forem karcinomu prsu i vaječníků je navíc snížen oproti sporadickým formám přibližně o 10 let [4].
Klasifikace nádorů ovaria
Podle histopatologické klasifikace a buněčného původu můžeme nádory ovaria dělit do tří základních skupin: 1. ná - dory z povrchového epitelu, 2. stromální nádory a 3. nádory z germinálních buněk. Neepiteliální nádory ovaria jsou reprezentovány tumory z germinálních buněk (3 %) a nádory ze zárodečných pruhů a stromatu ovaria (7 %). Maligní epiteliální nádory ovaria z povrchového epitelu dále členíme na karcinomy serózní, endometrioidní, mucinózní, clear cell karcinomy, Brennerův tumor a karcinomy nediferencované a smíšené [5]. V posledních letech došlo k výrazným změnám v chápání procesu tumorigeneze u ovariálního karcinomu. Skupina epiteliálních ovariálních karcinomů se skládá z různých nádorových typů, které se od sebe liší svojí molekulární patogenezí a rizikovými faktory. V souvislosti s výzkumem patogeneze a analýzami genetických alterací ovariálního karcinomu je obecně přijímáno dualistické dělení epiteliálních ovariálních karcinomů do dvou skupin. Typ I zastupuje low-grade serózní a endometrioidní karcinom, mucinózní karcinom, Brennerův tumor a clear cell karcinomy. Jsou charakteristické postupným vývojem od premaligních lézí v low-grade tumory a jejich typickým znakem je aktivace signalizačních drah zahrnujících protoonkogeny KRAS a BRAF. Jejich proliferace je pomalejší a 5leté OS se udává cca 55 % [6]. Typ II reprezentují nádory s výrazně horší prognózou. Naprostou většinu nádorů typu II tvoří high-grade serózní karcinomy (high-grade serous ovarian carcinoma – HGSC), zbylé patří mezi nediferencované karcinomy, karcinosarkomy a primární peritoneální karcinom. Někteří autoři přiřazují k nádorům typu II i high-grade endometrioidní karcinomy, které jsou typické přítomností mutovaného p53. Nicméně analýza mutací v těchto karcinomech ukazuje, že se v převážné většině případů jedná o vývojovou variantu HGSC [7,8]. Z tohoto důvodu je tento typ označován jako SET (serózní pseudoendometrioidní) varianta HGSC, která je charakterizována vyšší frekvencí mutací v genu BRCA1 a přítomností infiltrujících lymfocytů [9]. Varianta SET rovněž vykazuje lepší odpověď na terapii. Heterogenita v rámci HGSC byla rovněž analyzována pomocí profilování genové exprese. Na základě klastrové analýzy bylo 300 vzorků HGSC rozděleno do čtyř skupin označených jako: 1. imunoreaktivní, 2. diferencovaná, 3. proliferativní a 4. me - zenchymální [10,11]. Toto rozdělení rovněž koreluje s odpovědí na terapii a v případě imunoreaktivního typu podporuje hypotézu o pozitivním vlivu infiltrujících lymfocytů v odpovědi na terapii [12,13]. Mezi obecné molekulární charakteristiky HGSC odlišující tyto nádory od nádorů typu I patří: 1. přítomnost vysoké genetické nestability většinou způsobené poruchou v homologní rekombinaci, 2. časný výskyt mutací genu TP53 a 3. velmi nízká frekvence mutací v genech KRAS a BRAF [11].
Prekurzory epiteliálních nádorů ovaria
Rozdělení epiteliálních ovariálních karcinomů do dvou skupin je založeno zejména na odlišnosti v procesu jejich kancerogeneze a původu v prekurzorových lézích. Nádory typu I jsou charakterizovány nižší genetickou nestabilitou a postupným přechodem z benigních a borderline tumorů k malignímu fenotypu. Přestože nádory typu I vycházejí z různých buněčných typů, je pro všechny subtypy charakteristická aktivace signalizačních drah zahrnující tyrozinkinázový receptor ERBB2, onkoproteiny PI3K, RAS a BRAF vedoucí k aktivaci MAP kináz MEK a ERK.
Předpokládaným primárním prekurzorem low-grade serózních karcinomů (low-grade serous ovarian carcinoma – LGSC) je podobně jako u HGSC epitel vejcovodů. Nicméně vývoj LGSC je na rozdíl od HGSC založen na postupném přechodu z cystadenomů přes premaligní léze označované jako atypický proliferativní tumor (atypical proliferative tumor – APST) nebo též serózní borderline tumor. Domnělým primárním prekurzorem pro APST je pak papilární tubární hyperplazie, která v podobě kortikálních inkluzních cyst přechází na ovarium [14]. Další histologickou variantou premaligních lézí je neinvazivní niLGSC označovaný též jako mikropapilární serózní karcinom. Společným jmenovatelem prekurzorů LGSC je pak aktivace MAP kinázové signalizační dráhy nejčastěji v důsledku mutace KRAS u 33 % nebo BRAF u 25 % [15,16]. Přechod LGSC do high-grade tumorů je ojedinělý zejména z důvodu jejich odlišného průběhu karcinogeneze (obr. 1B).
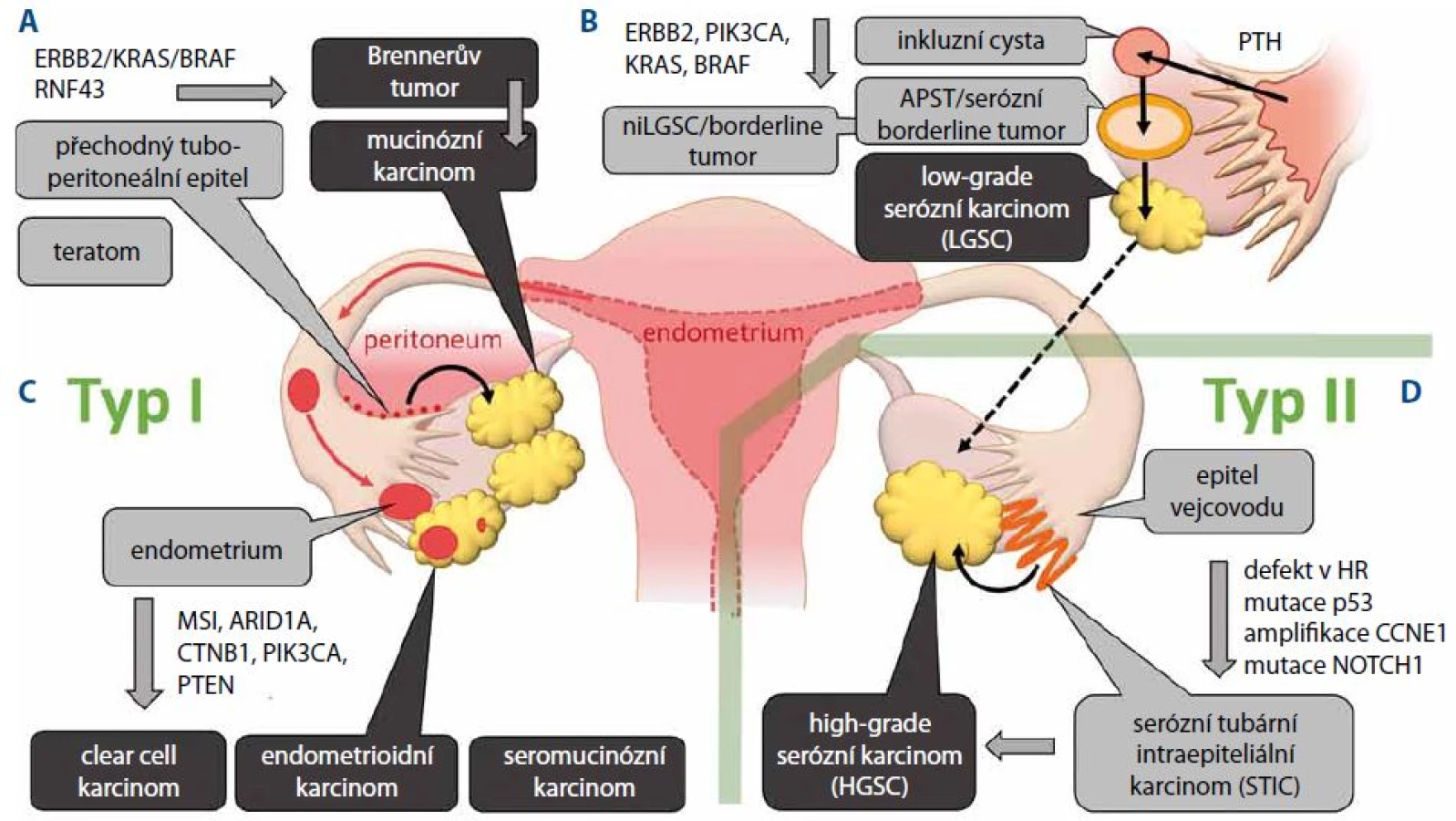
Endometrioidní a clear cell karcinomy mají společné buněčné prekurzory, tj. epitel endometria. Pro endometriální původ obou karcinomů svědčí i molekulární znaky společné s endometriálním endometrioidním karcinomem. Základní charakteristikou těchto nádorů je aktivace PI3K kinázy, která je způsobena aktivační mutací v genu PIK3CA nebo inaktivační mutací fosfatázy PTEN. Frekvence mutací PIK3CA je však častější u clear cell karcinomů, zatímco k mutaci PTEN dochází častěji u karcinomů endometrioidních [17,18]. Další odlišností mezi oběma typy karcinomů je častější výskyt mikrosatelitní nestability (MSI) a aktivace dráhy Wnt β-catenin u endometrioidních karcinomů (obr. 1C).
Mucinózní karcinomy jsou ve většině případů dobře diferencované nádory, které však často vykazují značnou buněčnou heterogenitu a v určitých případech se mohou vyskytovat společně s teratomy. Vzhledem k tomu, že mucinózní karcinomy na rozdíl od ostatních epiteliálních nádorů nevykazují znaky mülleriánského původu (přítomnost estrogenových a progesteronových receptorů), není jejich buněčný původ přesně objasněn. Vedle teratomů, z nichž se mucinózní karcinomy vyvíjí spíše ojediněle, byl jako prekurzor mucinózních karcinomů určen Brennerův tumor. Jedná se obvykle o nádor menšího rozsahu a převážně benigního charakteru. Primárním prekurzorem tohoto nádoru pak byl označen metaplastický epitel v oblasti tubo-peritoneálního spojení označovaný jako Wathardova hnízda [19,20]. Imunohistochemické studie a in situ hybridizace ukázaly, že ztráta exprese nádorového supresoru p16 kódovaného genem CDKN2A je zodpovědná za zvýšenou atypickou proliferaci těchto nádorů a vývoj cystadenomu. Hypotéza o Brennerově tumoru jako prekurzoru mucinózních karcinomů je rovněž podporována nálezem společných klonálních znaků u nádorů obsahujících oba buněčné typy [21]. Typickým molekulárním znakem mucinózních karcinomů je aktivace signální dráhy ERBB2-KRAS-BRAF, která se objevuje u 90 % případů, přičemž aktivační mutace KRAS je při frekvenci 65 % nejčastější genetickou změnou. Oproti ostatním nádorům typu I se mucinózní karcinomy odlišují poměrně častým výskytem mutací v genu TP53, které jsou přítomny až u 50 % případů (obr. 1A).
Objevení prekurzorů HGSC lze datovat do roku 2001, kdy byly histologicky vyšetřovány vzorky tkání pacientek, které preventivně podstoupily salpingo - -ooforektomii z důvodu germinální mutace BRCA1 a BRCA2. U 50 % z nich byly detekovány dysplastické léze vejcovodů, které morfologicky napodobují HGSC a které byly později označeny jako serózní tubární intraepiteliální karcinom (STIC) [21]. U většiny lézí pak byla rovněž detekována akumulace nádorového supresoru p53 v důsledku jeho mutace [22]. Pozdější studie vzorků pacientek nesoucích germinální mutaci v genech BRCA1 a BRCA2 rovněž potvrdily, že k mutaci v genu TP53 dochází v tubárním epitelu, který je primárním prekurzorem STIC a posléze HGSC. Při důkladné analýze fimbrií byl STIC rovněž detekován jako prekurzorová léze u sporadických HGSC [23,24]. Sekvenování genomu HGSC potvrdilo, že mezi základní charakteristiky HGSC patří poruchy v homologní rekombinaci a mutace p53 (obr. 1D).
Využití molekulární charakterizace pro cílenou léčbu ovariálních karcinomů
Všechny nádory vznikají v důsledku somatických mutací DNA, jejichž frekvence je závislá na přítomnosti genetické nestability. To však neznamená, že všechny mutace přítomné v nádorovém genomu jsou příčinou maligní transformace. Ve skutečnosti je díky novým sekvenačním technologiím detekována celá řada mutací, které k nádorovému fenotypu nemusí vůbec přispívat. Z tohoto důvodu byly zavedeny pojmy „driver“ (řídicí) a „passenger“ (doprovodné) mutace [25]. Cílem tohoto konceptu je oddělit mutace, které se přímo podílejí na nádorové transformaci, od těch, které vzniknou pouze v důsledku zvýšené genetické nestability. Driver mutace na rozdíl od passenger mutací poskytují v průběhu maligní transformace buňkám výhodu a vedou ke klonálním expanzím buněk nesoucích právě tyto mutace. Rozlišení driver mutací od passenger mutací je pak klíčovým cílem pro aplikaci nových sekvenačních technologií v klinické praxi. Identifikace driver mutací zodpovědných za nádorovou transformaci by tak v blízké budoucnosti mohla přinést nejen zlepšení v diagnostice ovariálních karcinomů, ale zejména otevřít nové možnosti pro cílenou personalizovanou léčbu.
Geny, jejichž mutace mají zásadní vliv na vývoj epiteliálních ovariálních karcinomů, lze z hlediska jejich funkce rozdělit do tří základních skupin: 1. geny zodpovědné za opravu DNA a udržování integrity genomu, 2. protoonkogeny a 3. geny kódující nádorové supresory.
Genetická nestabilita
Genetická nestabilita je jednou ze základních vlastností nádorových buněk, která katalyzuje sled genetických změn nutných pro maligní transformaci. Genetická nestabilita je charakterizována jako soubor poruch vedoucích k patologickému zmnožení genů nebo částí chromozomů, translokacím genů, aneuploidii nebo zvýšenému počtu mutací. U ovariálních karcinomů se na genetické nestabilitě podílejí zejména poruchy v mechanizmech zodpovědných za opravu poškozené DNA. Jedním z nejdůležitějších opravných mechanizmů je homologní rekombinace, která využívá sesterské chromatidy jako templát k opravě dvouvláknových zlomů DNA. Poškození homologní rekombinace u HGSC je pak příčinou vysoké genomové nestability a abnormálně vysoké frekvence mutací. Příčinou poruchy v homologní rekombinaci u high-grade karcinomů je nejčastěji mutace nebo metylace genů BRCA1 a BRCA2, genu CDK12, CHEK2 nebo BRIP1, přičemž změny genů BRCA1 a BRCA2 jsou nejčastější a z klinického hlediska nejvýznamnější [26].
Na rozdíl od HGSC vykazuje skupina nádorů označovaná jako typ I menší genetickou nestabilitu projevující se zejména menším počtem chromozomálních abnormalit. Klinicky nejvýznamnější poruchou v reparaci DNA ve skupině ovariálních karcinomů typu I je MSI. Tato porucha je způsobena mutacemi nebo metylacemi v genech zodpovědných za opravu chybného párování DNA, tzv. MMR. Ke vzniku MSI pak dochází přednostně u endometrioidních karcinomů.
BRCA1 a BRCA2
BRCA proteiny jsou součástí multiproteinových komplexů, které se účastní rozpoznávání a opravy poškozené DNA. Proteiny BRCA1 a BRCA2 se účastní homologní rekombinace, která využívá sesterské vlákno jako templát k bezchybné opravě dvouvláknového zlomu. V případě ztráty BRCA1 a BRCA2 je porušená DNA opravována alternativním mechanizmem, který vede ke vzniku mutací. Vyšší výskyt dvouvláknových zlomů pak vede k aktivaci nádorového supresoru p53, který jako transkripční faktor spouští expresi řady proapoptotických genů. Kritická role p53 při kompenzaci ztracené funkce BRCA tak může vysvětlit velmi vysoký počet somatických mutací postihujících gen TP53 u ovariálního karcinomu [27,28]. V případě high-grade ovariálních karcinomů jsou hereditární a somatické mutace v genech BRCA1 a BRCA2 popisovány ve 21 % případů, dalších 11 % případů je spojeno se ztrátou exprese BRCA1 v důsledku hypermetylace promotorové oblasti genu [26].
Ztráta schopnosti reparace DNA pomocí homologní rekombinace vede ke zvýšené závislosti buněk na alternativních mechanizmech opravujících vzniklé zlomy. To je pravděpodobně také jeden z důvodů primární citlivosti HGSC k platinovým cytostatikům. Jedním z klíčových proteinů zajišťujících alternativní způsob opravy poškozené DNA je protein poly-ADP ribose polymerase (PARP) a jeho ztráta je pro buňky s poruchou homologní rekombinace letální. Princip této tzv. syntetické letality se proto začal využívat i v terapii HGSC a od roku 2014 je pro tento typ léčby k dispozici specifický inhibitor PARP, olaparib. V současné době jsou v řadě klinických studií testovány nové inhibitory PARP, přičemž řada z nich jako např. niraparib, iniparib nebo talazoparib jsou již ve fázi III.
Porucha opravy chybného párování MMR
Proteiny kódované geny MMR se účastní rozpoznání a opravy určitých typů chyb v DNA, které vznikají během replikace v nově syntetizovaném vláknu DNA. Jde zejména o chyby typu krátkých inzerčních a delečních smyček v úseku krátkých mono - nebo dinukleotidových repetitivních sekvencí, které vznikají sklouznutím řetězce, nebo chybou v párování bází, a to zejména v případě G-T neshody vzniklé zařazením nesprávného nukleotidu. Tyto chyby jsou opravovány komplexem MMR proteinů (tzv. MMR), zahrnující proteiny MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, PMS2. Důsledkem ztráty funkce tohoto opravného systému je vznik krátkých inzercí DNA v postižených buňkách, které mohou být v dceřiných buňkách dále prodlužovány. K těmto změnám dochází nejčastěji v místech nazývaných mikrosatelity, které jsou charakterizovány typickými repeticemi nukleotidů GT/CA, a díky nim je tento typ genetické nestability nazýván MSI. Pro samotný nádor je však porucha opravných mechanizmů MMR důležitá zejména v souvislosti s mutacemi v kódujících oblastech genu. V důsledku MSI dochází k mutacím, které narušují čtecí rámec genu, tzv. frame-shift mutacím. Důsledkem porušení čtecího rámce vznikají proteiny, jejichž sekvence je změněna ve všech aminokyselinách následujících za místem mutace. Přestože jsou takové proteiny nefunkční a jsou v buňkách degradovány, představují významný zdroj nových peptidů, které jsou rozpoznávány imunitním systémem, tzv. neoantigenů. V poslední době se ukazuje, že tvorbu neoantigenů lze velmi dobře využít při cílené imunoterapii nádorů. Přítomnost MSI je také významným prediktorem při terapii kolorektálních karcinomů pomocí checkpoint inhibitorů, terapeutických protilátek blokujících povrchové molekuly PD-1 a PD-L1 [29–31]. Vzhledem k tomu, že použití checkpoint inhibitorů přineslo v posledních letech významný posun v léčbě celé řady nádorových onemocnění, zvažuje se jejich využití i pro terapii ovariálních karcinomů [32,33]. Detekce poruch MMR a MSI by se tak v budoucnu mohla stát jedním z důležitých prediktivních markerů pro terapii využívající checkpoint inhibitory.
K diagnostice MSI se používá analýza pěti specifických míst genomu, která jsou charakterizována vznikem výše popsaných mutací (BAT25, BAT26, D2S123, D5S346 a D17S250). Vedle průkazu MSI může být pro detekci poruchy v MMR použita i analýza jednotlivých genů kódujících proteiny podílející se na MMR (MLH1, MLH3, MSH2, MSH3, MSH6, PMS1 PMS2), a to detekce mutací v těchto genech, metylace jejich promotorů nebo snížení jejich exprese.
ARID1A
Mezi další geny, jejichž mutace přispívá ke genetické nestabilitě ovariálních karcinomů, je gen ARID1A (AT-rich interaction domain 1A). Kóduje protein, který je součástí komplexu SWI/SNF (switch/sucrose non-fermentable), který se podílí na formování nukleozomů, remodelaci chromatinu a regulaci genové exprese. Mutace genu ARID1A byly detekovány zejména u endometrioidních karcinomů dělohy, kolorektálních karcinomů, u endometrioidních a clear cell karcinomů ovaria [34,35]. Mutace v genu ARID1A je jednou z počátečních genetických změn u endometrioidních ovariálních karcinomů a poukazuje tak i na proces jejich karcinogeneze. Přítomnost mutace genu ARID1A byla prokázána v endometrioidním epitelu u endometriomů přiléhajících ke karcinomu, ale nikoli u endometriomů vzdálenějších. Význam mutace genu ARID1A pro tento typ ovariálních karcinomů potvrzují i myší modely. Inaktivace genů ARID1A a PIK3CA vedla k indukci clear cell karcinomů ovaria, zatímco inaktivace PTEN a ARID1A vedla k vytváření endometrioidních karcinomů [36,37]. Současné práce ukazují, že ARID1A může fungovat jako nádorový supresor i jako housekeeping gen. Mutace ARID1A vede k indukci exprese onkogenů PI3K a AKT a naopak ke snížení exprese genu kódujícího protein MLH1, který je zodpovědný za MMR. Přestože souvislost mezi mutací v genu ARID1A a MSI byla popsána u řady nádorů, není jasné, zda mutace ARID1A je příčinou nebo následkem MSI. Naopak výskyt mutací ARID1A negativně koreluje s přítomností mutace p53, což poukazuje na možnost jejich vzájemné spolupráce při potlačování nádorové transformace.
Kromě toho, že mutace v genu ARID1A může být v diagnostice využita pro upřesnění původu endometrioidních a clear cell ovariálních karcinomů, uvažuje se i o jejím využití při cílené terapii. Na myších modelech bylo ukázáno, že nádory nesoucí mutace ARID1A jsou na rozdíl od normálních buněk a nádorů s nemutovaným ARID1A citlivé k inhibici EZH2 metyltransferázy. EZH2 je enzym zodpovědný za metylaci histonů a podobně jako ARID1A se podílí na regulaci chromatinu. Specifické inhibitory EZH2 jsou v současné době testovány v několika klinických zkouškách fáze II. Další terapeutické využití mutace v genu ARID1A představuje aplikace inhibitorů PARP. Inhibitory EZH2 jsou tak příslibem zejména pro léčbu clear cell ovariálních karcinomů, které nesou mutaci ARID1A v 50 % případů a u nichž je odpověď na standardní chemoterapii velmi nízká.
Onkogenní signalizace
Pro přehlednost lze nejvýznamnější onkogeny ovariálních epiteliálních nádorů přiřadit k několika signalizačním drahám: 1. receptorové tyrozinkinázy, 2. PI3K/AKT a mTOR, 3. MAP kinázová dráha RAS/RAF/MEK/ERK, 4. Wnt signalizace a 5. kontrola buněčného cyklu cyklin dependentními kinázami. Z hlediska současných možností a dostupnosti specifických inhibitorů je možné efektivně cílit zejména na MAP kinázovou signalizaci, cyklin dependentní kinázy a receptorové tyrozinkinázy.
Receptorové tyrozinkinázy EGFR1 a ERBB2
EGFR a ERBB2 jsou transmembránové proteiny, které patří do rodiny receptorových tyrozinkináz. Jejich ligandem jsou peptidové růstové faktory, které po vazbě na extracelulární část receptoru vedou k jeho stabilizaci umožňující vytvořit aktivní dimer. Vytváření dimerů následně vede k vzájemné fosforylaci interagujících intracelulárních domén receptoru a transdukci signálu. Fosforylace cytoplazmatické části receptoru umožňuje navázání specifických interakčních partnerů a propagaci signálu. Mezi nejdůležitější signalizační dráhy aktivované receptory EGFR a ERBB2 patří dráhy PI3K/AKT a RAS/BRAF/MEK/ERK. Přestože aktivace obou drah hraje klíčovou roli zejména u ovariálních nádorů typu I, nejsou genetické změny u ERBB2 a EGFR příliš časté. U mucinózních a LGSC nádorů se mutace ERBB2 vyskytuje asi v 5 % [38,39]. Počet nádorů exprimujících zvýšené hladiny EGFR nebo ERBB2 je však podstatně vyšší, což vedlo ke snaze využít tyto receptory jako terapeutické cíle. V klinických zkouškách byly testovány specifické inhibitory EGFR erlotinib, gefitinib a canertinib a terapeutická protilátka cetuximab. Jako inhibitory ERBB2 byly testovány lapatinib a terapeutická protilátka trastuzumab [40–42]. V žádné z těchto studií však nebylo dosaženo požadovaného terapeutického efektu a rovněž nebyla prokázána korelace mezi expresí receptorů a terapeutickým efektem. Tento negativní výsledek lze pravděpodobně vysvětlit tím, že většina ovariálních karcinomů není na těchto receptorech závislá, protože k aktivaci příslušných signálních drah dochází díky mutacím v genech KRAS, BRAF nebo PIK3CA. Dalším důvodem pro nízkou efektivitu inhibitorů EGFR a ERBB2 může být zvýšená exprese jiných receptorových tyrozinkináz, jako jsou např. receptory ERBB3, ERBB4 a MET [43]. Z těchto důvodů bude pro terapii ovariálních karcinomů vhodnější testovat nové inhibitory tyrozinkináz s širším spektrem účinku a selektovat pacienty v závislosti na mutacích KRAS a BRAF. V tomto směru proběhlo několik studií, které prokazují slibný účinek multikinázového inhibitoru sunitinibu u ovariálních clear cell karcinomů [44,45].
KRAS/BRAF/MEK/ERK
Mezi klíčové události v kancerogenezi ovariálních nádorů typu I patří aktivace mitogen aktivovaných proteinkináz (MAP) prostřednictvím kináz KRAS a BRAF. Protein KRAS je GTPáza, která funguje jako molekulární přepínač signalizace mezi membránovými receptory a intercelulárními kinázovými kaskádami. Mutace genu KRAS, ke kterým dochází téměř výhradně v kodonech 12 a 13, vedou k jeho konstitutivní aktivaci, a tím k fosforylaci cílových signálních proteinů. Z hlediska patogeneze ovariálních karcinomů je pro KRAS nejdůležitější cílovou molekulou kináza BRAF, která stejně jako KRAS patří k nejčastěji mutovaným proteinům nádorů typu I. Přestože bylo identifikováno více než 30 onkogenních mutací BRAF, 90 % případů je způsobeno bodovou mutací V600E. Aktivace kinázy BRAF vede ke konstitutivní aktivaci MAP kináz MEK a ERK, které jsou zodpovědné za aktivaci transkripčních faktorů, mezi něž patří protoonkogeny MYC, CREB nebo FOS.
Terapeutické možnosti ovlivnění této signalizační dráhy jsou v současné době omezeny na proteiny BRAF a MEK. Inhibitory BRAF, vemurafenib a dabrafenib, byly schváleny pro léčbu maligního melanomu nesoucí mutaci BRAF V600E. V klinických zkouškách byl vemurafenib testován u LGSC s mutací BRAF V600E s velmi dobrou odpovědí, která ukazuje na potenciální prospěšnost testování této mutace. V roce 2009 proběhla klinická studie fáze II testující účinek inhibitoru MAP kinázy MEK1/2, selumetinibu, na rekurentní LGSC. Selumetinib byl dobře snášen a vykazoval odpověď na léčbu u 15 % pacientek, u 65 % pak vedl ke stabilizaci onemocnění [46]. V současné době jsou schváleny dva inhibitory MEK1/2 kinázy, trametinib a cobimetinib, které jsou testovány i na LGSC.
Geny regulující buněčný cyklus RB1, CCNE1
Buněčný cyklus je v průběhu každé fáze regulován komplexy cyklin dependentních kináz (CDK) a cyklinů. Pro průchod z G1 fáze do S fáze buněčného cyklu je klíčovou fosforylace nádorového supresoru RB1, který funguje jako negativní regulátor přechodu do S fáze. Nefosforylovaný RB1 váže protein E2F a blokuje jeho schopnost aktivovat expresi genů nutných pro zahájení S fáze. Komplex cyklin D-CDK4/6 fosforyluje protein RB1, což vede k uvolnění a aktivaci E2F, který jako transkripční faktor spouští expresi řady genů důležitých pro přechod do S fáze, vč. cyklinu E (CCNE1). Mezi další negativní regulátory přechodu z G1 do S fáze patří protein p16 (CDKN2A) a p21 (CDKN1A), které přímou interakcí inhibují aktivitu CDK.
U ovariálních karcinomů dochází nejčastěji k mutacím proteinu RB1 u nádorů typu II a mutacím CDKN2A u nádorů typu I. Amplifikace nebo aktivační mutace v genu CCNE1 byla zaznamenána v 19 % high-grade ovariálních karcinomů, a to zpravidla v případech, kde nebyla nalezena alterace v genech BRCA1 a BRCA2. CCNE1 je protoonkogenem, jehož produktem je cyklin E. Cyklin E je klíčovým proteinem buněčného cyklu zajišťujícím přechod z G1 do S fáze. Cyklin E v komplexu s cyklin dependentní kinázou CDK2 fosforyluje řadu cílových proteinů, mezi něž patří protein RB1, p27 nebo p21. Důsledkem zvýšené aktivity cyklinu E je pak masivní replikace DNA, které je charakterizována vyšším počtem replikačních počátků a poruchami replikace DNA. Tento replikační stres následně vede k poškození DNA a k aktivaci proteinu RAD51, který zajišťuje opravu poškozené DNA pomocí homologní rekombinace [47,48]. Sekvenováním genomu high-grade ovariálních karcinomů bylo zjištěno, že amplifikace cyklinu E se vylučuje s mutacemi v genech BRCA a poruchou v homologní rekombinaci. Toto zjištění rovněž koreluje s primární odpovědí high-grade ovariálních karcinomů k terapii. Zatímco nádory s mutací v genech BRCA a nebo jinou poruchou v homologní rekombinaci vykazují dobrou primární odpověď k platinovým derivátům, nádory s amplifikací genu CCNE1 patří mezi rezistentní a refrakterní [26]. Stanovení amplifikace genu CCNE1 by se tak v budoucnu mohlo stát užitečným markerem určujícím citlivost k platinovým derivátům a potenciálně otevřít cestu novým cíleným terapeutikům. Novou perspektivu v terapii nádorů nesoucích amplifikaci cyklinu E by tak v budoucnu mohla přinést cílená inhibice cyklin dependentních kináz. V současné době je jako specifický inhibitor CDK schválen palboclib, který je primárně určen k terapii nádoru prsu. Probíhající klinické zkoušky ukazují, že palboclib vykazuje slibné účinky i v terapii rezistentních HGSC. Tyto studie se rovněž zaměřují na korelaci mezi účinkem a genetickými změnami v CDKN2A, RB1 a CCNE1 [49,50].
Závěr
Pochopení základních mechanizmů molekulární patogeneze epiteliálních ovariálních karcinomů vedlo k vytvoření nové klasifikace, která lépe reflektuje buněčný původ nádoru i odlišnosti ve způsobu jejich nádorové transformace. Poznání klíčových genetických změn v patogenezi jednotlivých typů ovariálních karcinomů pak otevírá cestu pro aplikaci nových cílených léčiv, která vykazují nižší toxicitu a jsou příslibem zejména pro léčbu nádorů primárně rezistentních ke klasické terapii. Pro správnou klasifikaci a potenciální aplikaci cílené terapie však bude v budoucnu nutné doplnit histopatologickou klasifikaci o cílenou genetickou analýzu. Z výše uvedeného souhrnu vyplývá, že nejvhodnějšími kandidáty pro sekvenační analýzu jsou geny TP53, BRCA1, BRCA2, KRAS, BRAF, PIK3CA, ARID1A, PTEN, BRIP1, RB1, CDKN2A, CDK12, CHEK2, CTNNB1 a ERBB2. Další genetické analýzy by pak měly být zaměřeny na detekci amplifikace (CCNE1), detekci MSI nebo detekci poruch v homologní rekombinaci.
Poděkování
Děkujeme Mgr. Evě Michalové za formální i věcnou editaci textu.
Práce byla podpořena projektem MŠMT – NPU I – LO1413.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Petr Müller, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: muller@mou.cz
Obdrženo: 7. 8. 2016
Přijato: 29. 8. 2016
Sources
1. Uzis.cz [internetová stránka]. Novotvary 2011 ČR. Ústav zdravotnických informací a statistiky ČR, Česká republika; cÚZIS ČR 2010–2016 [aktualizováno 2016; citováno 7. srpna 2016]. Dostupné z: www.uzis.cz.
2. Miller DS, Blessing JA, Krasner CN et al. Phase II evaluation of pemetrexed in the treatment of recurrent or persistent platinum-resistant ovarian or primary peritoneal carcinoma: a study of the Gynecologic Oncology Group. J Clin Oncol 2009; 27 (16): 2686–2691. doi: 10.1200/JCO.2008.19.2963.
3. Bast RC Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer 2009; 9 (6): 415–428. doi: 10.1038/nrc2644.
4. Boyd J. Specific keynote: hereditary ovarian cancer: what we know. Gynecol Oncol 2003; 88 (1 Pt 2): S8–S10.
5. Cobb LP, Gaillard S, Wang Y et al. Adenocarcinoma of Mullerian origin: review of pathogenesis, molecular biology, and emerging treatment paradigms. Gynecol Oncol Res Pract 2015; 2 : 1. doi: 10.1186/s40661-015-0008-z.
6. Gershenson DM, Bodurka DC, Lu KH et al. Impact of age and primary disease site on outcome in women with low-grade serous carcinoma of the ovary or peritoneum: results of a large single-institution registry of a rare tumor. J Clin Oncol 2015; 33 (24): 2675–2682. doi: 10.1200/JCO.2015.61.0873.
7. Howitt BE, Hanamornroongruang S, Lin DI et al. Evidence for a dualistic model of high-grade serous carcinoma: BRCA mutation status, histology, and tubal intraepithelial carcinoma. Am J Surg Pathol 2015; 39 (3): 287–293. doi: 10.1097/PAS.0000000000000369.
8. Espinosa I, Catasus L, Canet B et al. Gene expression analysis identifies two groups of ovarian high-grade serous carcinomas with different prognosis. Mod Pathol 2011; 24 (6): 846–854. doi: 10.1038/modpathol. 2011.12.
9. Soslow RA, Han G, Park KJ et al. Morphologic patterns associated with BRCA1 and BRCA2 genotype in ovarian carcinoma. Mod Pathol 2012; 25 (4): 625–636. doi: 10.1038/modpathol.2011.183.
10. Tothill RW, Tinker AV, George J et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 2008; 14 (16): 5198–5208. doi: 10.1158/1078-0432.CCR-08-0196.
11. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474 (7353): 609–615. doi: 10.1038/nature10166.
12. Helland A, Anglesio MS, George J et al. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS One 2011; 6 (4): e18064. doi: 10.1371/journal.pone.0018064.
13. Konecny GE, Wang C, Hamidi H et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J Natl Cancer Inst 2014; 106 (10): pii: dju249. doi: 10.1093/jnci/dju249.
14. Kurman RJ, Vang R, Junge J et al. Papillary tubal hyperplasia: the putative precursor of ovarian atypical proliferative (borderline) serous tumors, noninvasive implants, and endosalpingiosis. Am J Surg Pathol 2011; 35 (11): 1605–1614. doi: 10.1097/PAS.0b013e318229449f.
15. Nakayama K, Nakayama N, Kurman RJ et al. Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer Biol Ther 2006; 5 (7): 779–785.
16. Singer G, Oldt R 3rd, Cohen Y et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst 2003; 95 (6): 484–486.
17. Campbell IG, Russell SE, Choong DY et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004; 64 (21): 7678–7681.
18. Kuo KT, Mao TL, Jones S et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol 2009; 174 (5): 1597–1601. doi: 10.2353/ajpath.2009.081000.
19. Auersperg N. The origin of ovarian carcinomas: a unifying hypothesis. Int J Gynecol Pathol 2011; 30 (1): 12–21. doi: 10.1097/PGP.0b013e3181f45f3e.
20. Seidman JD, Khedmati F. Exploring the histogenesis of ovarian mucinous and transitional cell (Brenner) neoplasms and their relationship with Walthard cell nests: a study of 120 tumors. Arch Pathol Lab Med 2008; 132 (11): 1753–1760. doi: 10.1043/1543-2165-132.11.1753.
21. Wang Y, Wu RC, Shwartz LE et al. Clonality analysis of combined Brenner and mucinous tumours of the ovary reveals their monoclonal origin. J Pathol 2015; 237 (2): 146–151. doi: 10.1002/path.4572.
22. Piek JM, van Diest PJ, Zweemer RP et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol 2001; 195 (4): 451–456.
23. Medeiros F, Muto MG, Lee Y et al. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol 2006; 30 (2): 230–236.
24. Kindelberger DW, Lee Y, Miron A et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am J Surg Pathol 2007; 31 (2): 161–169.
25. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009; 458 (7239): 719–724. doi: 10.1038/nature07943.
26. Patch AM, Christie EL, Etemadmoghadam D et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015; 521 (7553): 489–494. doi: 10.1038/nature14410.
27. Hartman AR, Ford JM. BRCA1 and p53: compensatory roles in DNA repair. J Mol Med (Berl) 2003; 81 (11): 700–707.
28. Zheng L, Li S, Boyer TG et al. Lessons learned from BRCA1 and BRCA2. Oncogene 2000; 19 (53): 6159–6175.
29. Lee V, Le DT. Efficacy of PD-1 blockade in tumors with MMR deficiency. Immunotherapy 2016; 8 (1): 1–3. doi: 10.2217/imt.15.97.
30. Blocking PD-1 in tumors with faulty DNA repair. Cancer Discov 2016; 6 (8): OF6. doi: 10.1158/2159-8290.CD-NB2016-082.
31. Le DT, Uram JN, Wang H et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372 (26): 2509–2520. doi: 10.1056/NEJMoa1500596.
32. Budczies J, Bockmayr M, Denkert C et al. Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274) – associations with gene expression, mutational load, and survival. Genes Chromosomes Cancer 2016; 55 (8): 626–639. doi: 10.1002/gcc. 22365.
33. Hamanishi J, Mandai M, Konishi I. Immune checkpoint inhibition in ovarian cancer. Int Immunol 2016; 28 (7): 339–348. doi: 10.1093/intimm/dxw020.
34. Jones S, Wang TL, Shih Ie M et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010; 330 (6001): 228–231. doi: 10.1126/science.1196333.
35. Wiegand KC, Shah SP, Al-Agha OM et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010; 363 (16): 1532–1543. doi: 10.1056/NEJMoa1008433.
36. Guan B, Rahmanto YS, Wu RC et al. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J Natl Cancer Inst 2014; 106 (7): pii: dju146. doi: 10.1093/jnci/dju146.
37. Chandler RL, Damrauer JS, Raab JR et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun 2015; 6 : 6118. doi: 10.1038/ncomms7118.
38. Jones S, Wang TL, Kurman RJ et al. Low-grade serous carcinomas of the ovary contain very few point mutations. J Pathol 2012; 226 (3): 413–420. doi: 10.1002/path.3967.
39. Forbes SA, Beare D, Gunasekaran P et al. COSMIC: exploring the world‘s knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43 (Database issue): D805–D811. doi: 10.1093/nar/gku1075.
40. Teplinsky E, Muggia F. Targeting HER2 in ovarian and uterine cancers: challenges and future directions. Gynecol Oncol 2014; 135 (2): 364–370. doi: 10.1016/j.ygyno.2014.09.003.
41. Makhija S, Amler LC, Glenn D et al. Clinical activity of gemcitabine plus pertuzumab in platinum-resistant ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. J Clin Oncol 2010; 28 (7): 1215–1223. doi: 10.1200/JCO.2009.22.3354.
42. Teplinsky E, Muggia F. EGFR and HER2: is there a role in ovarian cancer? Translational Cancer Research 2015; 4 (1): 107–117.
43. Davies S, Holmes A, Lomo L et al. High incidence of ErbB3, ErbB4, and MET expression in ovarian cancer. Int J Gynecol Pathol 2014; 33 (4): 402–410. doi: 10.1097/PGP.0000000000000081.
44. Stany MP, Vathipadiekal V, Ozbun L et al. Identification of novel therapeutic targets in microdissected clear cell ovarian cancers. PLoS One 2011; 6 (7): e21121. doi: 10.1371/journal.pone.0021121.
45. Anglesio MS, George J, Kulbe H et al. IL6-STAT3-HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin Cancer Res 2011; 17 (8): 2538–2548. doi: 10.1158/1078-0432.CCR-10-3314.
46. Farley J, Brady WE, Vathipadiekal V et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol 2013; 14 (2): 134–140. doi: 10.1016/S1470-2045 (12) 70572-7.
47. Jones RM, Mortusewicz O, Afzal I et al. Increased replication initiation and conflicts with transcription underlie cyclin E-induced replication stress. Oncogene 2013; 32 (32): 3744–3753. doi: 10.1038/onc.2012.387.
48. Teixeira LK, Wang X, Li Y et al. Cyclin E deregulation promotes loss of specific genomic regions. Curr Biol 2015; 25 (10): 1327–1333. doi: 10.1016/j.cub.2015.03.022.
49. Asghar U, Witkiewicz AK, Turner NC et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14 (2): 130–146. doi: 10.1038/nrd4504.
50. Konecny GE. Cyclin-dependent kinase pathways as targets for women‘s cancer treatment. Curr Opin Obstet Gynecol 2016; 28 (1): 42–48. doi: 10.1097/GCO.0000000000000243.
Labels
Paediatric clinical oncology Paediatric radiology Surgery Clinical oncology RadiotherapyArticle was published in
Clinical Oncology

2016 Issue Supplementum 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Úloha PD-1/PD-L1 signalizace v protinádorové imunitní odpovědi
- Nemalobuněčný karcinom plic – od imunobiologie k imunoterapii
- Nádorové buňky jako dynamický systém – molekulární a fenotypové změny v průběhu vzniku, progrese a šíření nádoru
- Nové metody studia metylace DNA – MS-HRM analýza a elektrochemie