
Vícestupňový proces vzniku vzdálených metastáz u karcinomů
Multistep Process of Establishing Carcinoma Metastases
Background:
Dissemination of cancer cells from the primary tumor and establishment of therapy-resistant distant metastases is the most common cause of human cancer deaths. The primary tumor consists of a heterogeneous population of cancer cells that have to overcome activity of the immune system, insufficient delivery of nutrients and oxygen, chemotherapy, radiotherapy etc. that lead to the selection of resistant and plastic cancer cells. Another selection pressure during metastatic spread gives rise to resistant subpopulations of cells, capable of surviving and proliferating in the hostile microenvironment of distant tissues.
Aim:
In this article, individual steps of the metastatic cascade are described as well as the mechanisms and signaling pathways that cancer cells use to deal with them. Metastatic process is generally inefficient and only very few cells released from the primary tumor develop into metastases. This success is enabled by pro-metastatic mutations, accumulated due to the selection pressure and also by cooperation of non-transformed cells that secrete supporting factors.
Conclusion:
Recent advances in research provide deeper insights into the complex processes that lead to formation and dissemination of cancer cells. Deciphering the key points of metastatic cascade and principles of its regulation will perhaps lead to development of efficient therapeutics targeting metastatic cells.
Key words:
metastasis – carcinoma – vascular endothelial growth factor A – epithelial-mesenchymal transitio
This work was supported by the project MEYS – NPS I – LO1413.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
6. 5. 2016
Accepted:
19. 5. 2016
Authors:
E. Ondroušková; L. Sommerová; R. Hrstka
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2016; 29(Supplementum 4): 12-17
Category:
Review
doi:
https://doi.org/10.14735/amko20164S12
Overview
Východiska:
Diseminace nádorových buněk z ložiska primárního nádoru a zakládání vzdálených metastáz, u nichž je úspěšnost terapie již velmi omezená, je nejčastější příčinou úmrtí onkologických pacientů. Samotný primární nádor je tvořen heterogenní populací buněk, na něž působí různé selekční tlaky v podobě aktivity imunitního systému, sníženého přísunu živin a kyslíku, chemoterapie, radioterapie atd., které vedou k postupnému převládnutí rezistentních, přizpůsobivých nádorových buněk. Další selekce nastává při metastatickém procesu a díky ní přežívají ve vzdálených ložiscích, zakládaných v různých tkáních, jen buňky schopné se na nové prostředí úspěšně adaptovat a proliferovat v něm.
Cíl:
Cílem tohoto přehledového článku je představit jednotlivé kroky metastatické kaskády, které musí invazivní buňka překonat, a mechanizmy a signální dráhy, které k tomu využívá. Metastazování je v podstatě velmi neefektivní proces a jen velmi malý zlomek buněk z těch, které opustily primární nádor, v něm uspějí. Důležitou podporu jim při tom poskytují nejen pro-metastatické mutace, které se v nich díky selekčnímu tlaku hromadí, ale také „spolupracující“ nenádorové buňky z jejich okolí a jimi sekretované podpůrné faktory.
Závěr:
Současné pokroky ve výzkumu vedou k hlubšímu porozumění komplexních procesů vedoucích ke vzniku a šíření nádorových buněk. Poznání klíčových bodů metastatické kaskády a principy její regulace snad v budoucnu vyústí ve vývoj účinných chemoterapeutik cílených na metastazující buňky.
Klíčová slova:
metastázy – karcinom – vaskulární endoteliální růstový faktor A – epiteliálně-mezenchymální tranzice
Úvod
Neléčitelné rozšíření metastáz, nikoliv samotný primární nádor, je příčinou více než 90 % úmrtí onkologických pacientů [1]. Metastazování představuje proces přemístění nádorových buněk z místa primárního nádoru, dávající vznik novým ložiskům na vzdálených místech. Jedná se o vícestupňový proces, jehož jednotlivé kroky zahrnují uvolnění buněk z primárního nádoru, intravazaci do krevního nebo lymfatického oběhu, přežití buněk v oběhu, extravazaci v sekundárním místě, tvorbu mikrometastáz a nakonec klinicky detekovatelných, vaskularizovaných a rostoucích makrometastáz. Předmětem intenzivního výzkumu posledních let je taktéž otázka, kdy přesně se metastazující buňky z primárního nádoru uvolní a proniknou do cílové tkáně. Jak naznačují výsledky, u jednotlivých typů nádorů se situace jeví odlišně. Původní představa lineárního vývoje nádoru a pozdního vzniku metastazujících buněk zřejmě odpovídá skutečnosti v případě adenokarcinomů plic a slinivky [2]. U jiných typů karcinomů, obzvláště pak u karcinomů prsu, jsou však metastázy běžně diagnostikovány roky až desítky let po odstranění primárního nádoru. K diseminaci buněk tedy zřejmě dochází velmi brzy, v pre-maligní fázi onemocnění, tyto buňky se po různě dlouhém období dormance v osídlené tkáni dále vyvíjejí nezávisle a od buněk primárního nádoru se svými genetickými změnami významně liší [3].
Experimentální studie ukazují, že zatímco až 80 % buněk, které se uvolnily z primárního nádoru, projde úspěšně prvními kroky až po extravazaci (vč.), jen asi 2–4 % těchto buněk iniciuje růst mikrometastáz a ještě menší část z nich, méně než 0,02 %, v novém prostředí přežijí a dají vznik makrometastázám [4]. Tyto poslední dva kroky jsou tedy při vývoji makrometastáz kritické a jen malá část buněk s vhodnými vyselektovanými vlastnostmi jimi úspěšně projde (obr. 1). Tato hypotéza je podpořena klinickými studiemi ukazujícími, že přestože může být u pacientů v krvi i v tkáních vzdálených od primárního nádoru detekováno velké množství diseminovaných nádorových buněk, jen velmi malému procentu z nich se podaří založit makrometastázy [5,6]. Jednotlivé fáze metastatického procesu a změny chování nádorových či s nádorem asociovaných buněk budou v následujících kapitolách popsány podrobněji.
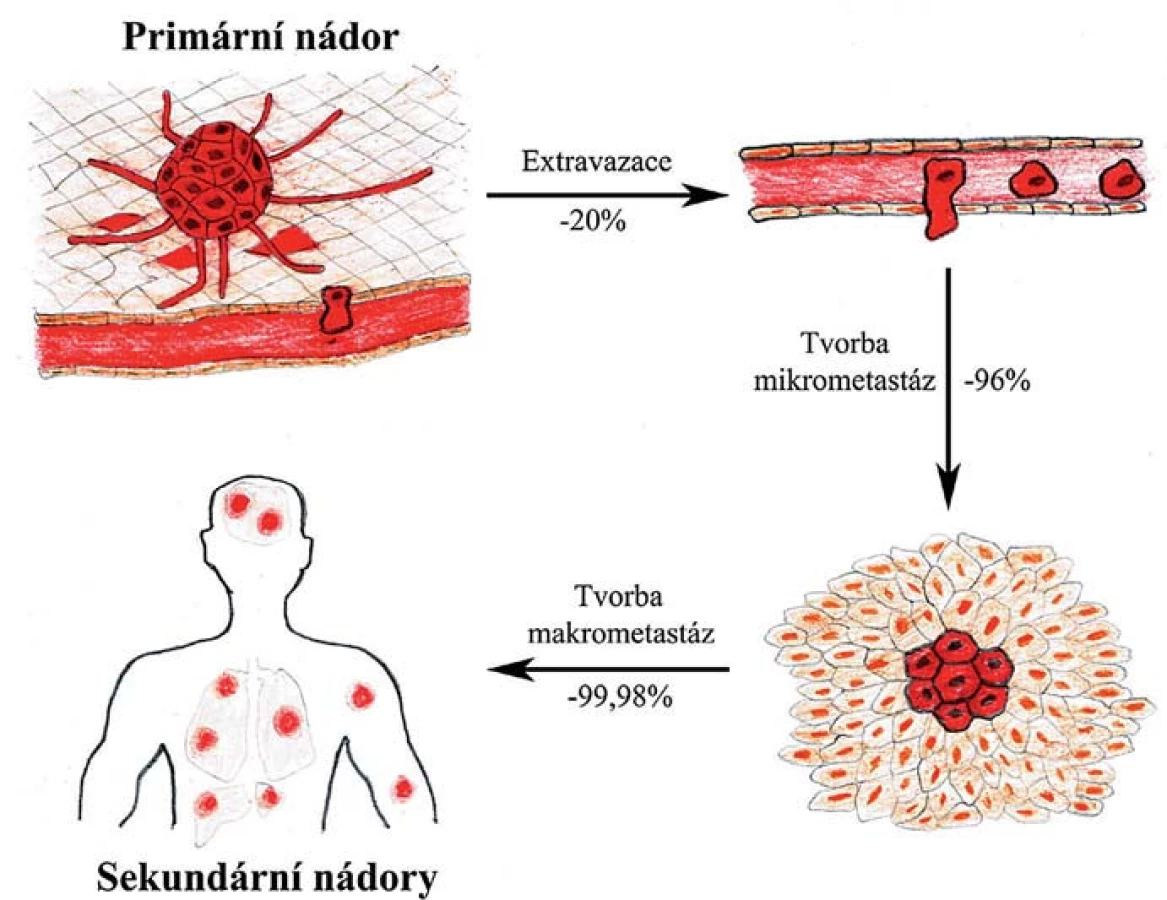
Lokální invaze
Rozrůstající se primární karcinom je obklopen sousedícími nenádorovými buňkami a od pojivové tkáně je oddělen bazální membránou. Invazivní nádorové buňky musí nejprve uvolnit kontakty s okolními buňkami a se složkami extracelulární matrix (ECM). Kontakty s okolními buňkami jsou z podstatné části zajišťovány proteinem E-cadherinem a snížení jeho hladiny je spojeno s progresí nádoru a zhoršeným přežitím např. u pacientek s karcinomem prsu [7]. Snižování hladiny proteinů spojených s epiteliálním fenotypem buněk (E-cadherin, claudin-1, cytokeratiny) a naopak zvyšování hladiny proteinů podporujících mezenchymální charakter (N-cadherin, fibronectin, vimentin) je znakem epiteliálně-mezenchymální tranzice (EMT), tj. procesu, který umožní přeměnu nepohyblivých epiteliálních buněk dlaždicovitého tvaru ve vřetenovité, elastické a migrace schopné buňky mezenchymálního typu [8]. Tyto buňky jsou navíc rezistentní k indukci buněčné smrti způsobené ztrátou kontaktů s ECM a okolními buňkami (tzv. anoikis) [9]. EMT je indukována mnoha signálními drahami, aktivovanými např. ligandy TGF-β (transforming growth factor β), EGF (epidermal growth factor), HGF (hepatocyte growth factor), FGF (fibroblast growth factor), IGF (insulin-like growth factor), Wnt, Notch [10], hypoxií [11], prozánětlivými cytokiny nebo mechanickými faktory, jako je hustota ECM [12]. Tyto signální dráhy vedou k aktivaci některého z transkripčních faktorů TWIST1, SNAI1, SNAI2 (SLUG), ZEB1, ZEB2 (SIP1), Brachyury, Goosecoid, SIX1 a PRRX1, jež přímo nebo nepřímo reprimují expresi E-cadherinu a dalších s EMT asociovaných cílových proteinů [13].
EMT podporuje také aktivaci a sekreci metaloproteináz (MMP), serinových proteáz a katepsinů, a tím narušení kontaktu buněk se složkami ECM zprostředkované nejrůznějšími povrchovými proteiny [14]. Tyto proteinázy jsou produkovány i s nádorem asociovanými stromálními buňkami, především fibroblasty, jež tak přispívají k uvolnění nádorových buněk z místa primárního nádoru [15,16]. Jednotlivé uvolněné buňky pak mohou migrovat dvěma způsoby, buď mezenchymálním pohybem, který je závislý na aktivitě proteáz, nebo améboidním pohybem, který je na aktivitě proteáz závislý v menší míře, případně mezi těmito dvěma způsoby přecházet, jak bylo popsáno např. u buněk melanomu [17]. Kromě jednotlivých buněk migrují nádorové buňky často v klastrech a tento způsob se může dokonce vyznačovat vyšší efektivitou migrace [18].
I v pozdějších stadiích ovlivňuje EMT efektivitu metastatického procesu, protože může vést ke vzniku celé řady přechodných epiteliálně/mezenchymálních fenotypů buněk lišících se svými invazivními a metastatickými schopnostmi, které mohou organizmem cestovat v klastrech, spolupracovat a zvyšovat tak úspěšnost při diseminaci a zakládání vzdálených metastáz [19]. Epiteliální charakter metastáz naznačuje, že proces EMT je reverzibilní a že v pozdějších stadiích diseminace dochází naopak k mezenchymálně-epiteliální tranzici (MET) [20].
Intravazace a přežití v krevním oběhu
Přechod přes stěnu cév, tzv. transendoteliální migrace (TEM), je nezbytným krokem diseminace nádorových buněk. Vstup do cévy (intravazace) je poněkud usnadněn angiogenezí, kterou primární nádor indukuje, protože stěny nových kapilár mají obecně slabší mezibuněčné spoje [21]. Paracelulární prostup do krevního řečiště se děje přes přechodně uvolněné mezibuněčné spoje. Rozvolnění mezibuněčných spojů je indukováno např. TNF-α (tumor necrosis factor α), EGF, VEGF (vascular endothelial growth factor), CSF-1 (colony stimulating factor-1) či TGF-β, které jsou sekretovány s tumorem asociovanými makrofágy nebo samotnými nádorovými buňkami [4]. Rozrušení endoteliálních mezibuněčných spojů je také důsledkem aktivity proteáz, např. MMP1 (štěpící protease-activated receptor 1), MMP2 nebo ADAM12 (štěpící vascular endothelial cadherin a angiopoietin 1 receptor) [22]. In vitro byl pozorován i alternativní typ intravazace, tzv. transcelulární, který je umožněn remodelací membrán a cytoskeletonu endoteliální buňky v místě jejího kontaktu s buňkou nádorovou. Calmodulinovou signalizací je aktivována kináza MLCK (myosin light chain kinase), v důsledku čehož dochází ke kontrakci actomyozinu. Vznikají přechodné pórovité struktury, kterými nádorová buňka pronikne přes endoteliální buňku [23].
Po úspěšném průniku do krevního řečiště se cirkulující nádorová buňka (circulating tumor cell – CTC) ocitne v nepřátelském mikroprostředí, kde se musí vyrovnat s hemodynamickými silami krevního proudu, aktivitou imunitních buněk či s nedostatkem živin. Některé studie ukazují, že maligní buňky jsou k fyzikální zátěži krevním proudem odolnější a dokážou se na něj adaptovat rychleji než netransformované epiteliální buňky [24]. Hlavním obranným mechanizmem nádorových buněk je jejich spolupráce s aktivovanými krevními destičkami, které CTC obklopí a fungují jako ochranný štít před možným mechanickým poškozením turbulencemi v krevním řečišti [25]. Pro-koagulační kaskádu podporují nádorové buňky sekrecí TF (tissue factor) a expresí receptorů Par1, Par2 a phosphatidylserinu na svém povrchu [26,27]. Důsledkem celého procesu je vznik agregátů nádorových buněk a krevních destiček propojených fibrinovými vlákny navázanými prostřednictvím integrinů na povrch obou typů buněk. Tyto agregáty rovněž poskytují nádorovým buňkám ochranu před buňkami imunitního systému, obzvláště NK buňkami [28]. Funkční studie na zvířecích modelech prokázaly, že přítomnost/nepřítomnost destiček v oběhu je pro tvorbu vzdálených metastáz poměrně zásadní [27,29].
Tkáňový tropizmus a extravazace
Mezi dvě základní hypotézy tkáňového tropizmu metastáz patří hypotéza založená na kompatibilitě nádorové buňky a osídlované tkáně a hypotéza hemodynamická/mechanická. První vyslovil již před více než 120 lety Stephen Paget a je známá pod výstižným názvem „seed and soil“ hypotéza. Předpokládá, že konkrétní nádorová buňka se může usídlit a začít proliferovat jen v místě, které ji svým mikroprostředím vyhovuje [30]. „Mechanický“ koncept naproti tomu zdůrazňuje roli fyzického zachycení cirkulujících nádorových buněk v kapilárách optimálního průměru a proudění krve [31]. K extravazaci nádorových buněk totiž dochází typicky v malých kapilárách podobného průměru, jako mají buňky samotné, což naznačuje, že i mechanické zachycení napomáhá pozdější tvorbě stabilních vazeb pomocí povrchových molekul [32]. V některých orgánech (kostní dřeň, játra, slezina) jsou kapiláry sinusoidní, s kolísající průsvitností, širokými štěrbinami mezi endotelovými buňkami a řídkým pojivovým obalem, které jsou pro cirkulující nádorové buňky snadno prostupné [33]. Naopak v dalších orgánech je endotelová vrstva kompaktní a brání snadnému průniku buněk. Přesto fakt, že karcinomy konkrétního typu metastazují přednostně do určitých orgánů (tkáňový tropizmus), ukazuje, že propustnost cév není jediným rozhodujícím faktorem při diseminaci nádorových buněk. Dokonce i podtypy jednotlivých karcinomů se svým tkáňovým tropizmem mohou odlišovat. Kosti jsou nejobvyklejším místem vzniku metastáz u všech subtypů prsních karcinomů s výjimkou bazálního, který nejčastěji metastazuje do plic. Přítomnost HER2 (human epidermal growth factor receptor) je zase asociovaná se zvýšenou frekvencí vzniku metastáz v mozku, játrech a plicích [34,35]. Příklady tkáňového tropizmu nejčastějších typů karcinomů jsou znázorněny na obr. 2.
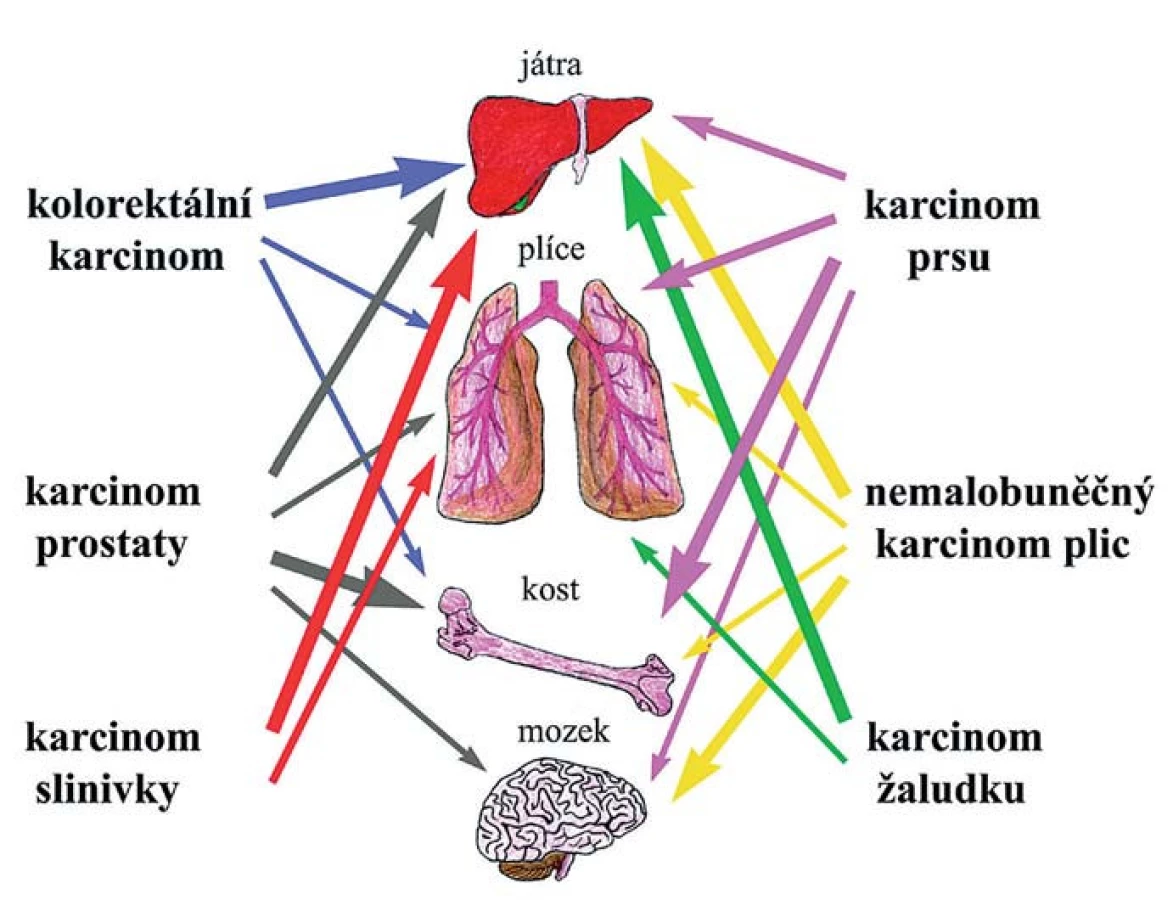
Některé z těchto příkladů preferenčních metastatických míst mohou být vy - světleny kompatibilitou chemokinů v cílové tkáni a expresí odpovídajících receptorů na povrchu nádorových buněk. Chemokiny jsou malé cytokiny, které indukují migraci buněk a aktivují signální dráhy regulující adhezi a remodelaci cytoskeletonu. Zřejmě nejlépe prozkoumaným příkladem je CXC-chemokine ligand 12 (CXCL12) sekretovaný stromálními buňkami, který působí jako chemoatraktant na nádorové buňky exprimující receptory CXCR4 a CXCR7 (s G-proteinem spojené receptory). Tato vzájemná interakce také napomáhá adhezi CTC k endoteliálním buňkám, podporuje extravazaci a následně i ukotvení k ECM a stromálním buňkám. Význam receptoru CXCR4 pro metastazování nádorů prsu, prostaty, plic, kolorekta a mnoha dalších byl potvrzen řadou studií [36–38]. Inhibice interakce CXCR4-CXCL12 in vivo vedla k významnému snížení metastazování nádorových buněk prsu do lymfatických uzlin a plic [39]. Další příklady chemokinů a jejich receptorů ve vývoji metastatického onemocnění, vč. tkáňově specifičtějších interakcí, jsou shrnuty v podrobnějších přehledových článcích [40–42].
Jak již bylo zmíněno, chemokiny a jejich odpovídající receptory napomáhají diseminaci CTC nejen mechanizmem chemoatrakce, ale podporují i následnou adhezi k endoteliálním buňkám, extravazaci a osídlení nového místa v přilehlých tkáních. Pro adhezi CTC na stěny kapilár jsou dále důležité membránové proteiny, především ty interagující s receptory na povrchu endoteliálních buněk, kde se nachází mj. selektiny nebo VCAM1 (vascular cell adhesion molecule) [43]. Mezi tyto proteiny exprimované na povrchu nádorových buněk patří např. E-selectin/L-selectin ligand (HCELL), P-selectin glycoprotein ligand 1 (PSGL1), CD44, CD24, mucin 1 (MUC1) a integriny [44,45]. U buněk karcinomu prsu metastazujících do plic byl detekován povrchový protein metadherin, který zprostředkovává specifickou vazbu mezi nádorovou buňkou a buňkami plicního endotelu [46].
Po vytvoření kontaktů s endoteliálními buňkami musí nádorová buňka znovu přejít přes stěnu cév, přičemž si pomáhá expresí proteinů zvyšujících permeabilitu cév. Rozvolnění mezibuněčných spojů je opět indukováno sekretovanými faktory zvyšujícími permeabilitu cévních stěn, např. VEGF, který vede k disociaci komplexu VE-cadherinu a β-cateninu [47], TGF-β, indukující tvorbu angiopoietin-like 4 (ANGPTL4) [48] nebo SPARC (secreted protein, acidic, rich in cysteine), který vazbou na VCAM1 aktivuje signální dráhu p38-MAPK [49]. Při extravazaci nádorových buněk se často uplatňují i imunitní buňky. Bylo prokázáno, že chemokin CCL2 produkovaný nádorovými buňkami v plicních kapilárách přitahuje zánětlivé monocyty a ty následně sekrecí VEGF podporují extravazaci nádorových buněk [50]. Všechny tyto interakce vedou k reorganizaci cytoskeletonu endoteliálních buněk a k uvolnění jejich mezibuněčných spojů.
Osídlení pre-metastatické niche, dormance a reaktivace
Po úvodních krocích metastatické kaskády, kterými je schopna projít většina z invazivních nádorových buněk, přichází kritické části – osídlení nové tkáně a obnovení proliferace vedoucí ke tvorbě makrometastáz. Ještě předtím, než dorazí první metastatické buňky, může být mikroprostředí v cílové tkáni ovlivněno primárním nádorem. Buňky primárního nádoru podporují tvorbu tzv. pre-metastatické niche tím, že do cirkulace sekretují různé cytokiny či růstové faktory [51]. Mezi jinými k tvorbě pre-metastatické niche u různých typů nádorových buněk přispívají VEGF, placental-growth factor (PlGF), TNF-α, TGF-β, lysyl oxidase (LOX), versican nebo granulocyte-colony stimulating factor (G-CSF). Tyto faktory ovlivňují vlastnosti stromálních buněk a ECM v cílové tkáni, případně mobilizují jiné podpůrné buňky (např. makrofágy z kostní dřeně) k jejich přemístění a přeměně mikroprostředí na příznivější pro diseminované nádorové buňky [52]. Z primárního nádoru jsou uvolňovány také exozomy, detailně byl popsán např. jejich vliv na jaterní metastazování buněk adenokarcinomu slinivky. Exozomy uvolňované z buněk primárního nádoru obsahující faktor MIF (macrophage migration inhibitory factor) byly pohlceny Kupferovými buňkami, které poté začaly sekretovat TGF-β a přitahovat makrofágy podílející se dále na přípravě pre-metastatické niche [53].
Přes tyto předem indukované změny jsou novému prostředí nádorové buňky bezprostředně po jeho dosažení jen málo přizpůsobeny a často je u nich indukována buněčná smrt. U mnoha pacientů s karcinomy prostaty, prsu, ledvin nebo s melanomem však dochází k relapsu onemocnění po letech, někdy i desítkách let od odstranění primárního nádoru. Klinické studie prokázaly, že diseminované nádorové buňky jsou přítomny v kostní dřeni pacientek s prsními karcinomy již v raném stadiu [54]. Toto klinické pozorování bylo vysvětleno zpomalením fyziologických procesů a zastavením proliferace diseminovaných nádorových buněk, tedy navozením tzv. dormance [55]. Zastavení proliferace je důsledkem nedostatečné kompatibility nádorové buňky a mikroprostředí, kdy díky odlišnému složení ECM a jiným typům stromálních buněk, než byly v místě primárního nádoru, není utvořeno s okolím dostatečné množství kontaktů. Přežití buněk ve stadiu dormance je podporováno vyšší aktivitou stresových signálních drah, např. fokální adhezivní kinázy (FAK) [56], Src [57], ERK/p38 či Akt [58]. Tyto dráhy jsou aktivovány prostřednictvím povrchových proteinů a receptorů, např. integrinu β1, CXCR4 nebo VCAM1, v závislosti na tom, v jaké tkáni se buňka nachází [59,60]. Dalším důvodem k tomu, že se mikrometastázy v novém místě nezvětšují, může být i zvýšená frekvence odumírání způsobená nedostatečnou vaskularizací proliferující nádorové tkáně [61]. Rovněž aktivní represe mikrometastáz ze strany stromálních buněk, např. prostřednictvím BMP proteinů, byla popsána v in vivo modelu dormantních buněk prsního karcinomu metastazujících do plic [62].
K reaktivaci proliferace dormantních buněk napomáhá obnovení mezibuněčných kontaktů a parakrinní signalizace se stromálními buňkami. Do plic metastazující buňky prsního karcinomu produkují ve zvýšeném množství protein Tenascin C, složku ECM, která podporuje Notch a Wnt signalizaci [63]. Nádorové buňky dále sekretují TGF-β3, který stimuluje stromální buňky ke zvýšené tvorbě periostinu, jenž po vazbě na Wnt ligand aktivuje Wnt signalizaci [64]. Notch/Wnt signální dráha podporuje průchod buněčným cyklem prostřednictvím proteinů Myc a cyklin D1. TGF-β1 sekretovaný dormantními buňkami prsního karcinomu v plicních mikrometastázách zase u stromálních buněk indukuje tvorbu kolagenu 1, který podporuje přechod z dormantního do proliferačního stavu aktivací integrin β1 signální dráhy [65]. Represe proliferace způsobená vazbou BMP proteinů stromálního původu na receptory nádorových buněk je např. u buněk prsního karcinomu v plicních mikrometastázách zablokována sekrecí BMP inhibitoru Coco [62]. Proliferaci podporuje rovněž zvýšené množství růstových faktorů v prostředí, jejichž důležitým zdrojem jsou infiltrující imunitní buňky původem z kostní dřeně. Infiltraci podporují faktory uvolňované nádorovými buňkami, jako jsou osteopontin nebo CXCL12 [66,67]. Na těchto příkladech jsou demonstrovány některé mechanizmy, kterými může nádorová buňka přejít ze stadia dormance do proliferace a dát vznik klinicky detekovatelným makrometastázám. Jaké faktory přesně rozhodují o čase, kdy se tak stane, a které signální dráhy jsou pro tento proces nejpodstatnější, však ještě není příliš známo, i když výzkum v této oblasti je v posledních letech stále intenzivnější.
Závěr
Přestože jen velmi malé procento nádorových buněk disponuje vlastnostmi, které jsou nezbytné k úspěšné kolonizaci vzdálené tkáně, jsou metastázy jednou z nejčastějších příčin úmrtí onkologických pacientů. V nádorových buňkách se postupně hromadí genetické a epigenetické změny zvyšující jejich invazivitu, rezistenci k buněčné smrti, adaptabilitu a další schopnosti umožňující jim úspěšně projít všemi kroky metastatické kaskády. Nádorové buňky, ať už v místě primárního nádoru, nebo vzdálených metastáz, jsou obklopeny stromálními buňkami, žírnými buňkami, makrofágy a dalšími buňkami imunitního systému. Jejich vzájemná komunikace zprostředkovaná povrchovými molekulami a cytokiny aktivuje různé intracelulární signální dráhy, často dále napomáhající k rozvoji a diseminaci nádorového onemocnění. Intenzivní výzkum těchto komplexních procesů snad povede k identifikaci klíčových molekul, na které by se mohl v budoucnu zaměřit vývoj nových terapeutik.
Poděkování
Děkujeme JUDr. Ivaně Veselé za provedení ilustrací do obr. 1 a 2.
Práce byla podpořena projektem MŠMT – NPU I – LO1413.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Eva Ondroušková, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: eva.ondrouskova@mou.cz
Obdrženo: 6. 5. 2016
Přijato: 19. 5. 2016
Sources
1. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 2006; 127 (4): 679–695.
2. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009; 9 (4): 274–284. doi: 10.1038/nrc2622.
3. Kozłowski J, Kozłowska A, Kocki J. Breast cancer metastasis – insight into selected molecular mechanisms of the phenomenon. Postepy Hig Med Dosw (online) 2015; 69 : 447–451.
4. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011; 147 (2): 275–292. doi: 10.1016/j.cell.2011.09.024.
5. Tarin D, Price JE, Kettlewell MG et al. Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts. Cancer Res 1984; 44 (8): 3584–3592.
6. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2 (8): 563–572.
7. Gould Rothberg BE, Bracken MB. E-cadherin immunohistochemical expression as a prognostic factor in infiltrating ductal carcinoma of the breast: a systematic review and meta-analysis. Breast Cancer Res Treat 2006; 100 (2): 139–148.
8. Heerboth S, Housman G, Leary M et al. EMT and tumor metastasis. Clin Transl Med 2015; 4 : 6–19. doi: 10.1186/s40169-015-0048-3.
9. Rennebeck G, Martelli M, Kyprianou N. Anoikis and survival connections in the tumor microenvironment: is there a role in prostate cancer metastasis? Cancer Res 2005; 65 (24): 11230–11235.
10. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7 (2): 131–142.
11. Zhang L, Huang G, Li X et al. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor-1alpha in hepatocellular carcinoma. BMC Cancer 2013; 13 : 108–117. doi: 10.1186/1471-2407-13-108.
12. Kumar S, Das A, Sen S. Extracellular matrix density promotes EMT by weakening cell-cell adhesions. Mol Biosyst 2014; 10 (4): 838–850. doi: 10.1039/c3mb70431a.
13. Sanchez-Tillo E, Liu Y, de Barrios O et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci 2012; 69 (20): 3429–3456. doi: 10.1007/s00018-012-1122-2.
14. Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 2011; 278 (1): 16–27. doi: 10.1111/j.1742-4658.2010.07919.x.
15. Almholt K, Johnsen M. Stromal cell involvement in cancer. Recent Results Cancer Res 2003; 162 : 31–42.
16. Sloane BF, Sameni M, Podgorski I et al. Functional imaging of tumor proteolysis. Annu Rev Pharmacol Toxicol 2006; 46 : 301–315.
17. Taddei ML, Giannoni E, Morandi A et al. Mesenchymal to amoeboid transition is associated with stem-like features of melanoma cells. Cell Commun Signal 2014; 12 : 24–36. doi: 10.1186/1478-811X-12-24.
18. Kumar S, Kapoor A, Desai S et al. Proteolytic and non-proteolytic regulation of collective cell invasion: tuning by ECM density and organization. Sci Rep 2016; 6 : 19905–19922. doi: 10.1038/srep19905.
19. Jolly MK, Boareto M, Huang B et al. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol 2015; 5 : 155–174. doi: 10.3389/fonc.2015.00155.
20. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013; 27 (20): 2192–2206. doi: 10.1101/gad.225334.113.
21. Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat Rev 2011; 37 (1): 63–74. doi: 10.1016/j.ctrv.2010.05. 001.
22. Reymond N, d‘Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer 2013; 13 (12): 858–870. doi: 10.1038/nrc3628.
23. Khuon S, Liang L, Dettman RW et al. Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: a three-dimensional FRET study. J Cell Sci 2010; 123 (Pt 3): 431–440. doi: 10.1242/jcs.053793.
24. Barnes JM, Nauseef JT, Henry MD. Resistance to fluid shear stress is a conserved biophysical property of malignant cells. PLoS One 2012; 7 (12): e50973. doi: 10.1371/journal.pone.0050973.
25. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer 2011; 11 (2): 123–134. doi: 10.1038/nrc3004.
26. Liu Y, Jiang P, Capkova K et al. Tissue factor-activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res 2011; 71 (20): 6492–6502. doi: 10.1158/0008-5472.CAN-11-1145.
27. Camerer E, Qazi AA, Duong DN et al. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 2004; 104 (2): 397–401.
28. Palumbo JS, Talmage KE, Massari JV et al. Platelets and fibrin (ogen) increase metastatic potential by im - peding natural killer cell-mediated elimination of tumor cells. Blood 2005; 105 (1): 178–185.
29. Karpatkin S, Pearlstein E, Ambrogio C et al. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J Clin Invest 1988; 81 (4): 1012–1019.
30. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 1889; 8 (2): 98–101.
31. Weiss L. Comments on hematogenous metastatic patterns in humans as revealed by autopsy. Clin Exp Metastasis 1992; 10 (3): 191–199.
32. Kienast Y, von Baumgarten L, Fuhrmann M et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010; 16 (1): 116–122. doi: 10.1038/nm.2072.
33. Lorusso G, Rüegg C. New insights into the mechanisms of organ-specific breast cancer metastasis. Semin Cancer Biol 2012; 22 (3): 226–233. doi: 10.1016/j.semcancer.2012.03.007.
34. Kennecke H, Yerushalmi R, Woods R et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010; 28 (20): 3271–3277. doi: 10.1200/JCO.2009.25.9820.
35. Soni A, Ren Z, Hameed O et al. Breast cancer subtypes predispose the site of distant metastases. Am J Clin Pathol 2015; 143 (4): 471–478. doi: 10.1309/AJCPYO5FSV3UPEXS.
36. Furusato B, Mohamed A, Uhlen M et al. CXCR4 and cancer. Pathol Int 2010; 60 (7): 497–505. doi: 10.1111/j.1440 - 1827.2010.02548.x.
37. Cavallaro S. CXCR4/CXCL12 in non-small-cell lung cancer metastasis to the brain. Int J Mol Sci 2013; 14 (1): 1713–1727. doi: 10.3390/ijms14011713.
38. Li YM, Pan Y, Wei Y et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 2004; 6 (5): 459–469.
39. Müller A, Homey B, Soto H et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001; 410 (6824): 50–56.
40. Sarvaiya PJ, Guo D, Ulasov I et al. Chemokines in tumor progression and metastasis. Oncotarget 2013; 4 (12): 2171–2185.
41. Kakinuma T, Hwang ST. Chemokines, chemokine receptors, and cancer metastasis. J Leukoc Biol 2006; 79 (4): 639–651.
42. Géraud C, Koch PS, Damm F et al. The metastatic cycle: metastatic niches and cancer cell dissemination. J Dtsch Dermatol Ges 2014; 12 (11): 1012–1019. doi: 10.1111/ddg.12451.
43. Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol 2012; 2012 : 676731–676741.
44. Miles FL, Pruitt FL, van Golen KL et al. Stepping out of the flow: capillary extravasation in cancer metastasis. Clin Exp Metastasis 2008; 25 (4): 305–324.
45. Kobayashi H, Boelte KC, Lin PC. Endothelial cell adhesion molecules and cancer progression. Curr Med Chem 2007; 14 (4): 377–386.
46. Brown DM, Ruoslahti E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 2004; 5 (4): 365–374.
47. Weis S, Cui J, Barnes L et al. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol 2004; 167 (2): 223–229.
48. Padua D, Zhang XH, Wang Q et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008; 133 (1): 66–77. doi: 10.1016/j.cell.2008.01.046.
49. Tichet M, Prod‘Homme V, Fenouille N et al. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun 2015; 6 : 6993–7008. doi: 10.1038/ncomms7993.
50. Qian BZ, Li J, Zhang H et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011; 475 (7355): 222–225. doi: 10.1038/nature10138.
51. Kaplan RN, Riba RD, Zacharoulis S et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005; 438 (7069): 820–827.
52. Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 2013; 32 (3–4): 449–464. doi: 10.1007/s10555-013-9420-1.
53. Costa-Silva B, Aiello NM, Ocean AJ et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 2015; 17 (6): 816–826. doi: 10.1038/ncb3169.
54. Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer 2008; 8 (5): 329–340. doi: 10.1038/nrc2375.
55. Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst 1999; 91 (1): 80–85.
56. Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 2009; 106 (25): 10290–10295. doi: 10.1073/pnas.0904227106.
57. Barkan D, El Touny LH, Michalowski AM et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res 2010; 70 (14): 5706–5716. doi: 10.1158/0008-5472.CAN-09-2356.
58. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell 2013; 155 (4): 750–764. doi: 10.1016/j.cell.2013.10.029.
59. Zhang XH, Wang Q, Gerald W et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009; 16 (1): 67–78. doi: 10.1016/j.ccr.2009.05.017.
60. Chen Q, Zhang XH, Massagué J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011; 20 (4): 538–549. doi: 10.1016/j.ccr.2011.08.025.
61. Holmgren L, O‘Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med 1995; 1 (2): 149–153.
62. Gao H, Chakraborty G, Lee-Lim AP et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012; 150 (4): 764–779. doi: 10.1016/j.cell. 2012.06.035.
63. Oskarsson T, Acharyya S, Zhang XH et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 2011; 17 (7): 867–874. doi: 10.1038/nm.2379.
64. Malanchi I, Santamaria-Martínez A, Susanto E et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012; 481 (7379): 85–89. doi: 10.1038/nature10694.
65. Barkan D, Chambers AF. Beta1-integrin: a potential therapeutic target in the battle against cancer recurrence. Clin Cancer Res 2011; 17 (23): 7219–7223. doi: 10.1158/1078-0432.CCR-11-0642.
66. McAllister SS, Gifford AM, Greiner AL et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 2008; 133 (6): 994–1005. doi: 10.1016/j.cell. 2008.04.045.
67. Hiratsuka S, Duda DG, Huang Y et al. C-X-C receptor type 4 promotes metastasis by activating p38 mitogen-activated protein kinase in myeloid differentiation antigen (Gr-1) -positive cells. Proc Natl Acad Sci U S A 2011; 108 (1): 302–307. doi: 10.1073/pnas.1016917108.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue Supplementum 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Úloha PD-1/PD-L1 signalizace v protinádorové imunitní odpovědi
- Nemalobuněčný karcinom plic – od imunobiologie k imunoterapii
- Nádorové buňky jako dynamický systém – molekulární a fenotypové změny v průběhu vzniku, progrese a šíření nádoru
- Nové metody studia metylace DNA – MS-HRM analýza a elektrochemie