
Zriedkavý histopatologický nález po pľúcnej resekcii u dieťaťa
A rare histopathological fi nding after lung resection in a child
Background: The authors present a case of a patient with an extremely rare lung tumor in a child. Case: A 9-year-old girl with a 3-day history of dyspnea and stabbing pain in the xiphoid region, irradiating to the area under the left costal margin, both in rest and in physical activities. She was primarily examined in a regional hospital with bounded homogenous focus of the superior right lung lobe on the X-ray. After initial treatment with antibiotics and persistent finding on X-ray, a CT scan of the chest was performed. It revealed an irregular oval lesion of a non-homogenous structure with a contrast dye in the S3 region merged to pericardium and parietal pleura, which the presence of several micronodules. Based on the negative tumor markers, positive PET-CT scan and a negative etiology, biopsy or eventually a lesion exstirpation were indicated. Right-sided thoracotomy, mass enucleation and exstirpation of nodular lesions were performed 2.5 months after the onset of difficulties. Postoperative recovery was uneventful, no sign of recurrence occurred during a follow-up period. The final histological finding was verified as an inflammatory myofibroblastic tumor – an extremely rare pulmonary pathology in the pediatric population. Conclusion: Inflammatory myofibroblastic tumor can be mimicking IgG4 sclerosing disease and inflammatory pseudotumor. It is essential to distinguish between these affections because of different (i.e. surgical vs. conservative) treatment approach.
Keywords:
inflammatory myofibroblastic tumor – inflammatory pseudotumor – IgG4 – lung tumor
Authors:
V. Chrenkova 1; P. Omaník 1; L. Plank 2; J. Babala 1
Authors‘ workplace:
Klinika detskej chirurgie, Národný ústav detských chorôb, Bratislava, Slovenská republika
1; Ústav patologickej anatómie, JLF UK, Martin, Slovenská republika
2
Published in:
Klin Onkol 2021; 34(2): 142-146
Category:
Case Report
doi:
https://doi.org/10.48095/ccko2021142
Overview
Východiská: Autori prezentujú kazuistiku pacientky s veľmi raritným pľúcnym tumorom v detskom veku. Prípad: Deväťročná pacientka s trojdňovou anamnézou dyspnoe a bodavých bolestí v oblasti processus xiphoideus s propagáciou pod ľavý rebrový oblúk v kľude aj pri námahe. Primárne riešená v rajónnej nemocnici, kde na RTG snímke hrudníka nález ohraničeného homogénneho ložiska v hornom pľúcnom poli vpravo. Po preliečení antibiotikami bez rezolúcie v RTG obraze. Následne kontrastná CT hrudníka verifikovala nepravidelne oválne ložisko nehomogénnej štruktúry so škvrnito sa vysycujúcou kontrastnou látkou v oblasti S3, ktoré z časti stenotizuje a obturuje bronchus B3, prechádza na perikard a perietálnu pleuru s prítomnosťou ojedinelých mikronodulov. Na základe negatívnych onkomarkerov, pozitívnej PET-CT a nejasnej etiológie bola indikovaná biopsia, event. exstirpácia ložiska. Dva a pol mesiaca od začiatku ťažkostí bola realizovaná pravostranná torakotómia, enukleácia masy in toto a exstirpácia subpleurálnych nodulárnych lézií. Histopatologickým vyšetrením bol potvrdený zápalový myofibroblastický tumor, ktorého výskyt je u pediatrického pacienta extrémne raritný. Záver: Inflamatórny myofibroblastický tumor môže imitovať skupinu IgG4 sklerotizujúcich ochorení a zápalový pseudotumor. Vzhľadom na diametrálne odlišný spôsob liečby (chirurgická intervencia vs. konzervatívny postup), je jednoznačné rozlíšenie týchto diagnóz pre správnu liečbu rozhodujúce.
Klíčová slova:
zápalový myofibroblastický tumor – zápalový pseudotumor – IgG4 – tumor pľúc
Úvod
Primárne tumory pľúc v detskom veku, na rozdiel od metastazujúcich, sú mimoriadne raritné ochorenia. Môžu postihovať celý dýchací systém, od hrtana, cez tracheu, hlavné a lobárne bronchy až po samotný pľúcny parenchým. Najčastejšie k nim patria karcinoid, mukoepidemoidný karcinóm, Hodgkinov lymfóm a inflamatórny myofibroblastický tumor (IMT). IMT prvýkrát popísal Bunn v roku 1939 [1]. Ide o vzácny benígny nádor s prevalenciou u detí a adolescentov 0,04–0,7 % [1,2]. IMT je tumor mäkkých tkanív, s možným výskytom v pľúcach, mediastíne, bránici, slezine, pankrease, pečeni, prsníkoch, periférnych nervoch, lymfatických uzlinách a iných orgánoch gastrointestinálneho a urogenitálneho traktu [3–5]. Do roku 2019 bolo v literatúre evidovaných okolo 70 prípadov tohto vzácneho tumoru [6]. Etiologicky ide o benígnu neopláziu s vysokým rizikom lokálnej rekurencie a nízkym malígnym potenciálom – len 5 % [5,7,8]. Pio et al popísali vo svojej štúdii, že primárne tracheobronchiálne tumory u detí sú v zastúpení v počte 0,02 % všetkých nádorov detí. IMT je najviac vyskytujúca sa lézia pľúc, a to až v 25 % [9]. Najčastejšie sa zápalový myofibroblastický tumor nachádza u detí práve v pľúcach [3]. V závislosti od lokalizácie sa líši aj klinický obraz, väčšinou je ochorenie sprevádzané kašľom, dýchacími ťažkosťami, febrilitami, nočným potením, bolesťami kĺbov či hrudníka, hemoptýzou a častými respiračnými infekciami [1,3,10,11]. Môže sa nachádzať endobronchiálne alebo v parenchýme pľúc, a preto je v diagnostike, okrem zobrazovacích metód (RTG a CT), potrebná aj bronchoskopia s vizualizáciou lézie a jej následnou biopsiou [1,3,9]. PET/CT je síce vysoko senzitívne, no nízko špecifické vyšetrenie pre IMT, pretože nedokáže odlíšiť, či sa jedná o zápalový proces alebo malignitu [12]. Podľa klasifikácie Svetovej zdravotníckej organizácie, aktualizovanej v roku 2015, patrí medzi epiteliálne mezenchymálne tumory pľúc ako samostatný typ [13]. Histologicky ide o komplexnú štruktúru s prítomnými plazmatickými bunkami, vretenovitými bunkami, proliferujúcimi myofibroblastami s eozinofilnou cytoplazmou spolu s kolagénovými vláknami, fibroblastami, lymfocytmi, eozinofilmi a inými zápalovými bunkami [14–16]. Mikroskopicky nebývajú prítomné nekrózy, atypické mitózy a ani vaskulárna infiltrácia [5]. Imunohistochemicky sa najčastejšie vyskytuje pozitivita na ALK-1 (anaplastic lymphoma kinase – proteín myofibroblastov), menej často býva pozitívny proteín S100, smooth muscle actin (SMA), dezmín, IgG4 a CD34 [1,8,14,17]. Translokácia génu ALK, receptor pre tyrozín kinázu – inzulínový rastový faktor, nachádzajúci sa na krátkom ramienku 2. chromozómu (2p23), ktorý sa normálne vyskytuje v centrálnom nervovom systéme, má pri imunohistochemickej pozitivite silnú onkogenetickú aktivitu, ktorá značí neopláziu [18]. Voľbou liečby je kompletná exstirpácia ložiska, ako prevencia lokálnej rekurencie nádoru [2,9,15,17]. Pred definitívnym operačným riešením sa odporúča aj ihlová biopsia, a následne po obdržaní histopatologického výsledku je možné naplánovanie definitívnej terapie [1,4,9,10]. Pri nemožnosti kompletnej resekcie nádoru, vzhľadom na veľkosť či lokalizáciu, sa predoperačne podáva skôr chemoterapia ako rádioterapia [12,17]. Pre riziko relapsu je dôležité aj dlhodobé sledovanie pacientov [1,4,12]. Po kompletnej resekcii sa 5–10-ročné prežívanie popisuje až v 89 % [1,3,11].
Kazuistika
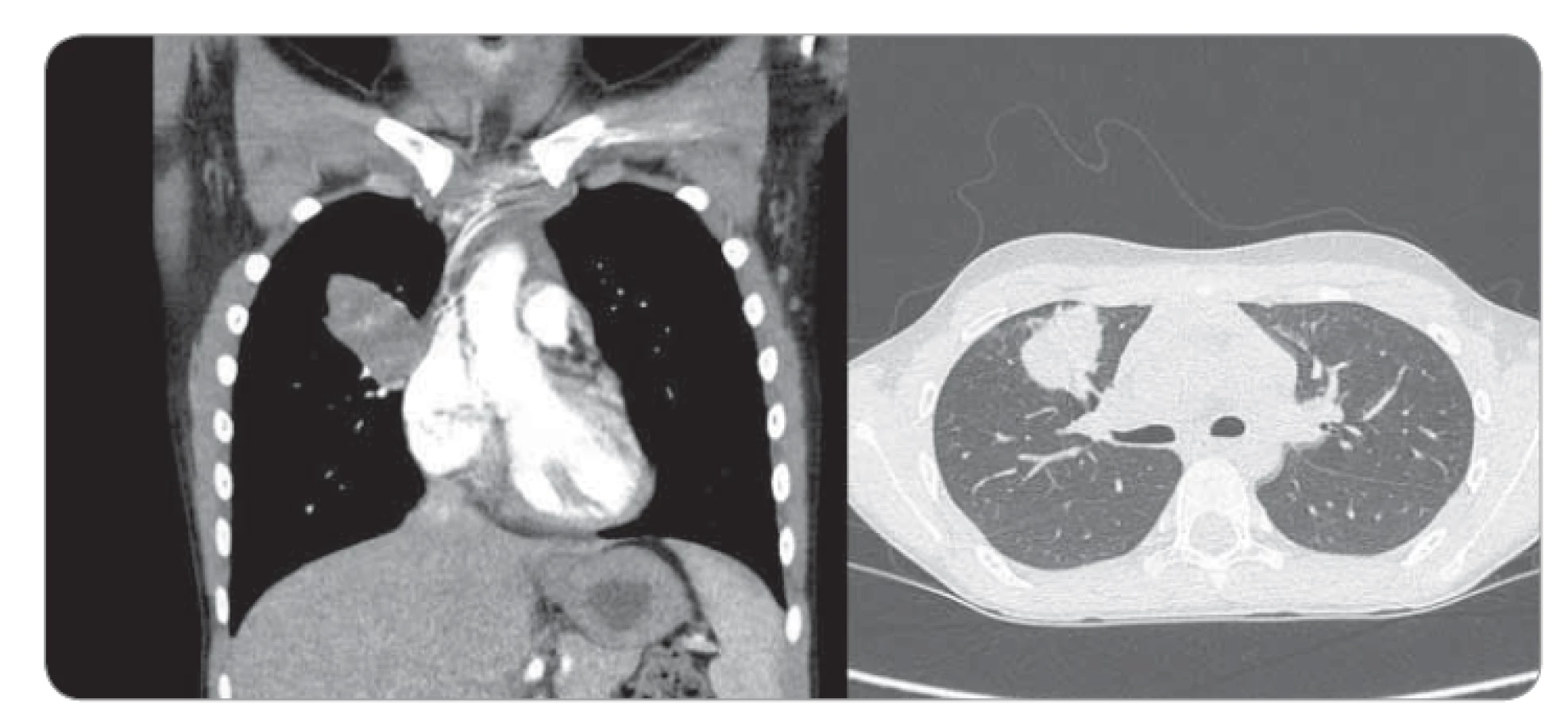
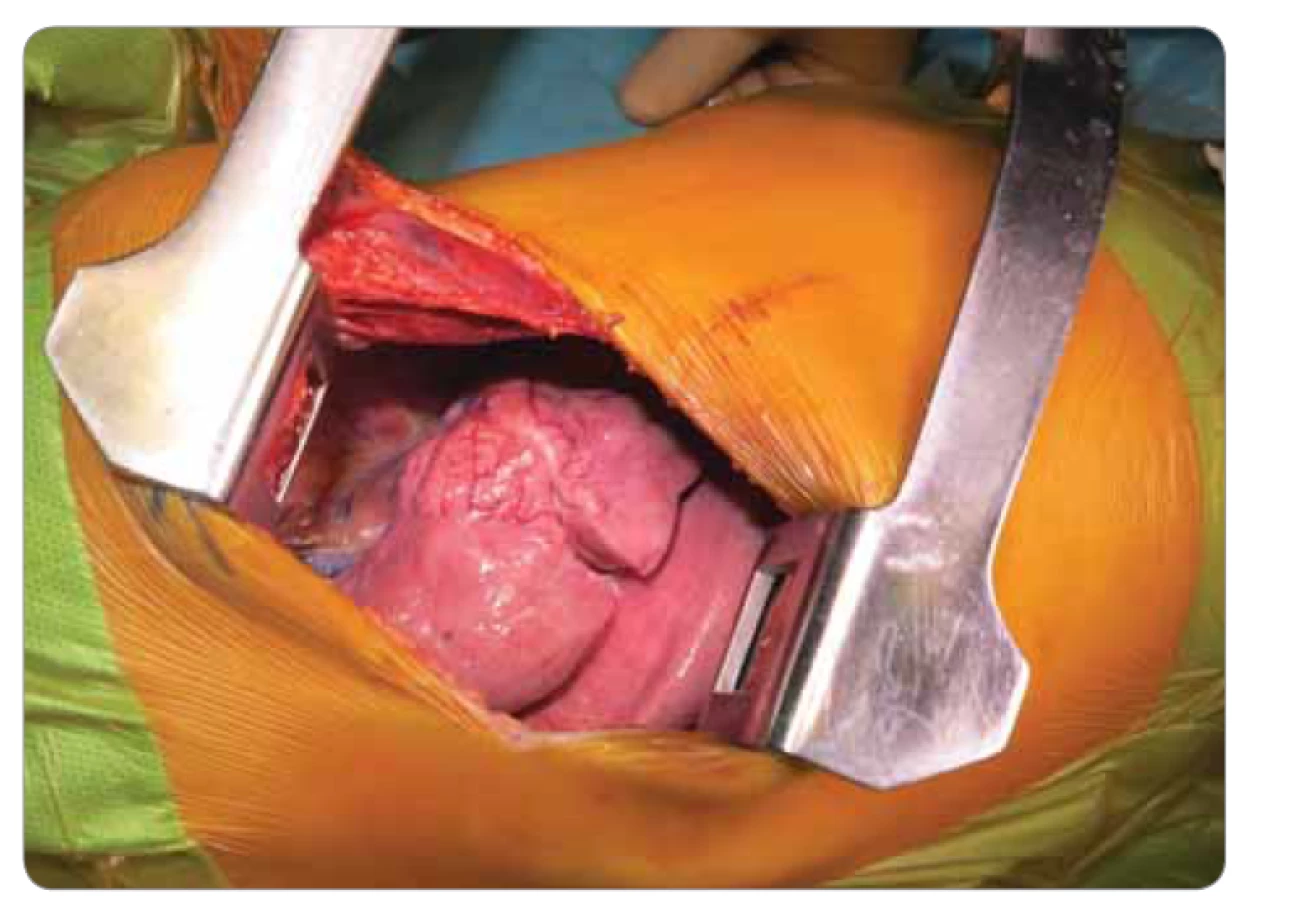
Deväťročná pacientka prichádza do rajónnej nemocnice s 3 dni trvajúcou anamnézou dyspnoe a bodavých bolestí v oblasti processus xiphoideus s propagáciou pod ľavý rebrový oblúk, bez ohľadu na polohu, či aktivitu, so spontánnym ústupom. Konvenčná RTG snímka hrudníka zobrazila ohraničené homogénne ložisko zníženej transparencie (veľkosti 50 × 27 mm) v oblasti horného pľúcneho poľa vpravo. V laboratórnych parametroch bez elevácie zápalových markerov. Bronchoskopicky nález diskrétnych štrukturálnych zmien vpravo v oblasti stredného a dolného lobárneho bronchu. V rámci diferenciálnej diagnostiky vylúčená atypická pneumónia aj tuberkulóza, bronchoalveolárna laváž kultivačne negatívna. Indikované kontrastné CT vyšetrenie hrudníka s obrazom nepravidelného oválneho ložiska nehomogénnej štruktúry (veľkosti 4 × 2,3 × 3 cm) so škvrnito sa vysycujúcimi časťami a s hrubšími spikulami na povrchu v oblasti S3 segmentu horného pravého pľúcneho laloka, nasadajúceho na bronchus B3, ktorý je čiastočne stenotizovaný a obturovaný, a prechádzajúceho na perikard a parietálnu pleuru, prítomné sú aj subpleurálne mikronoduly (obr. 1). Pre nejasnú etiológiu a nemožnosť vylúčenia malignity boli doplnené vyšetrenia (onkomarkery, ultrazvuk brucha a kardiologické vyšetrenie) s fyziologickým nálezom. Následne vykonaná hrubo ihlová punkcia ložiska pod CT kontrolou. Histologicky nález nejednoznačne abnormálne vaskularizovaného myofibroblastického procesu so suspektným reaktívnym ale aj nádorovým charakterom. Preto bola indikovaná rebiopsia, event. kompletná exstirpácia. Pacientka bola preložená na Kliniku detskej chirurgie Národného ústavu detských chorôb a Lekárskej fakulty Univerzity Komenského v Bratislave za účelom operačného riešenia. Na objasnenie bližšieho charakteru patologického útvaru pravých pľúc, vzťahu k okolitým štruktúram a možnosti operability bolo doplnené PET/CT vyšetrenie. Následne v celkovej anestézii posterolaterálna torakotómia, identifikácia útvaru ovoidného tvaru a tmavočervenej farby na rozhraní segmentov S3 a S5 s naliehaním na perikard, ale bez jeho infiltrácie (obr. 2). Tumor je dobre ohraničený, tuhší, s dobre vyvinutou kapsulou, o veľkosti približne 4 × 3 × 3 cm, preto je technicky možná enukleácia in toto. Subpleurálne mikronoduly boli klinovito resekované a spolu s ložiskom boli odoslané na histologické vyšetrenie do Ústavu patologickej anatómie v Martine. Pooperačný priebeh bez komplikácií, pacientka afebrilná, postupne realimentovaná a mobilizovaná, na 11. deň prepustená do domácej a ambulantnej starostlivosti.
Histológia
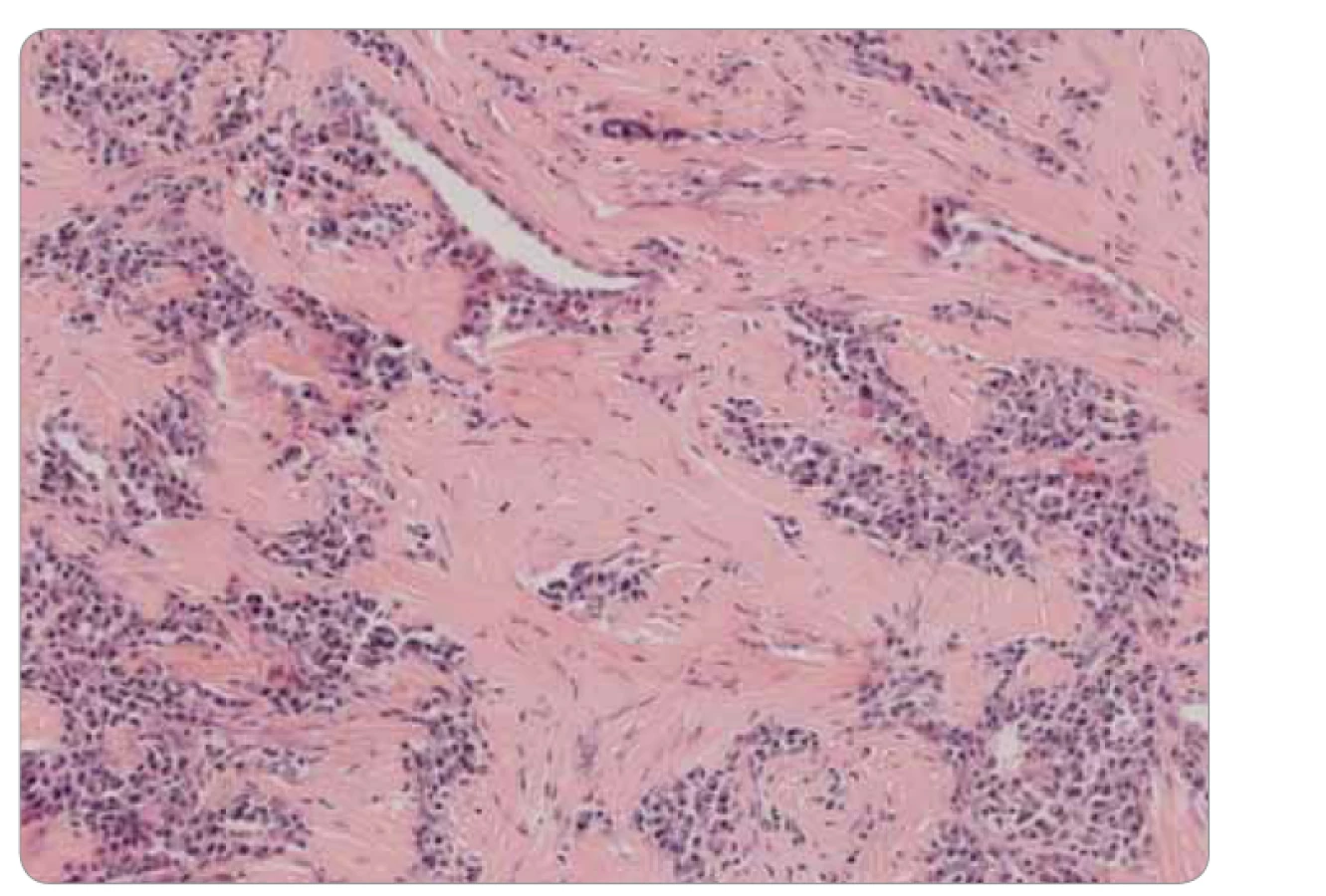
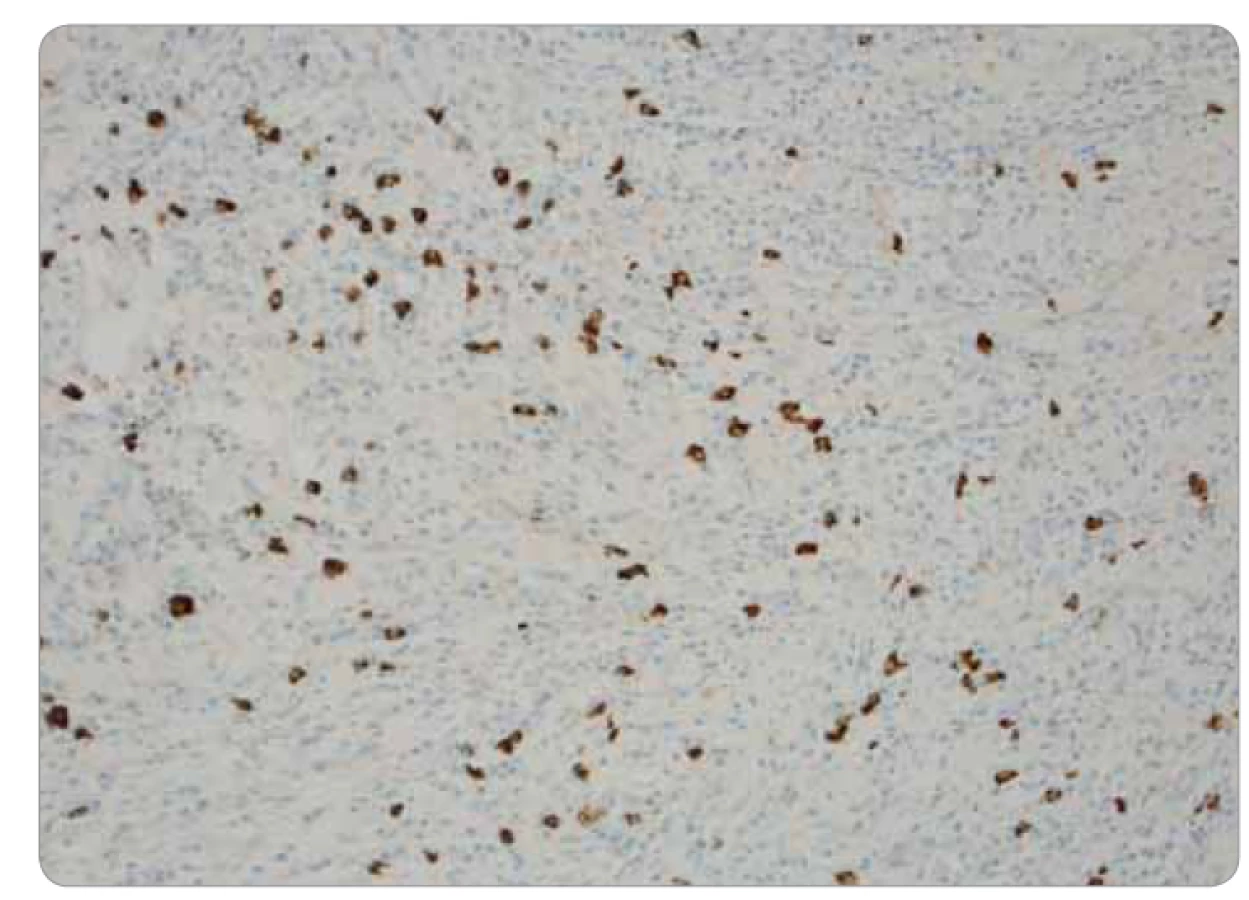
Ide o kompletnú exstirpáciu mikroskopicky polymorfnej až polyfázickej štruktúry s rôznou celularitou, s rôznym zastúpením vretenobunkovej populácie, zápalového plazmocytárneho infiltrátu a s intersticiálnou substanciou. Prevažne ide o chronicky zápalovo a abnormálne vaskularizované a kolagenizované tkanivo s myofibroblastickou populáciou, lymfoplazmocytárnou celulizáciou a výraznou lymfoproliferáciou (obr. 3). Skôr pripomína vzhľad IMT. Pri dôkaze IgG4 je celkový počet týchto plazmocytov difúzne po 33/1 HPF (high-power-field – veľké zorné pole), niekde aj 50/1 HPF. V niektorých oblastiach je nález obturačnej vasopatie a niektoré infiltráty sú typickejšie pre IgG4-patiu (obr. 4). Imunohistochemicky sú vretenné bunky pozitívne na SMA, s výraznejšou expresiou CD68, negatívne na dezmín, proteín S100 a CD246 (ALK). Pomocou fluorescenčnej in situ hybridizácie je dôkaz na translokáciu ALK génu negatívny. Nie sú prítomné nekrózy ani atypické mitózy, mikronoduly sú negatívne. Záver: „Lézia nejasnej biologickej povahy, bez známok malignity, vykazujúca kombinovaný obraz s dominujúcou morfológiou ALK negatívneho zápalového myofibroblastického tumoru a menej aj črty IgG4 ochorenia.“ Následne po doplnení odberu sérového IgG4, ktorého výsledok u pacientky je 0,80 g/l (fyziologické rozmedzie: 0,012–1,699 g/l), je definitívna diagnóza IMT.
Sledovanie a ďalší priebeh
Vzhľadom na vylúčenie malígneho procesu, potvrdenie benígnej lézie v zmysle zápalového myofibroblastického nádoru pľúc a kompletnú exstirpáciu ložiska, nebola indikovaná onkologická liečba a pacientka ostáva v dlhodobej dispenzarizácii. Aktuálne je už vyše roka od operácie, a zatiaľ bez známok recidívy ochorenia.
Diskusia
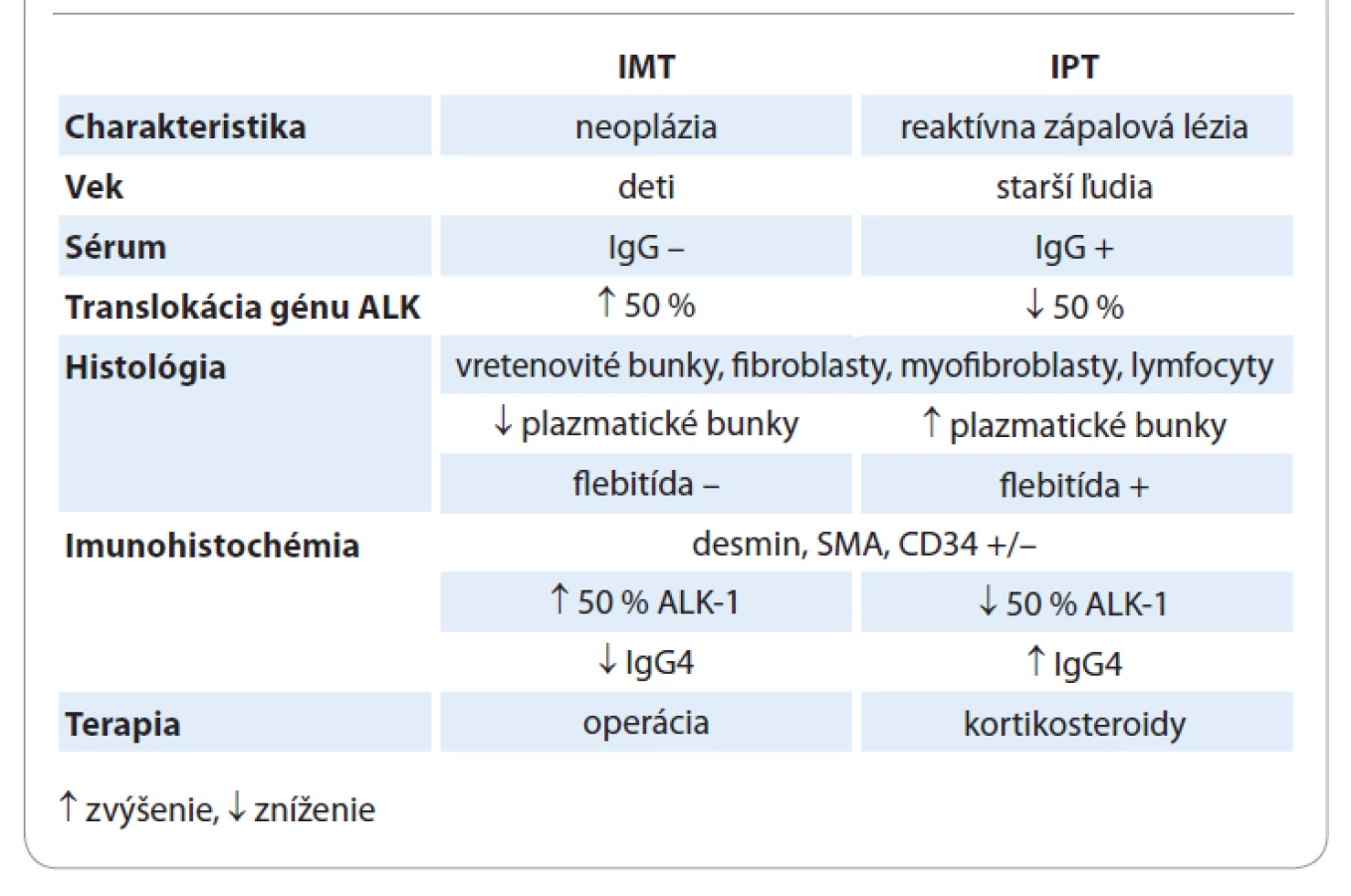
Zápalový myofibroblastický tumor pľúc je u detí najčastejšou neoplázou pľúc benígneho pôvodu [9]. V minulosti bol často zamieňaný alebo synonymicky pomenovaný ako inflamatórny pseudotumor (IPT) [11]. Dnes už poznáme zásadný rozdiel, ktorý je dôležitý nie len v zvolení správnej liečby, ale aj v manažmente ďalšieho sledovania pacienta. [7,14,16,19]. Tab. 1 znázorňuje patognomické odlišnosti medzi IMT a IPT. Zápalový pseudotumor sa zaraďuje do spektra multiorgánových porúch citlivých na steroidy. Ide o zápalový proces zo spektra autoimunitných chorôb, spadajúcich do skupiny tzv. sklerotizujúcich ochorení postihujúcich najmä starších pacientov, kam patrí autoimunitná pankreatitída, sklerotizujúca cholangitída, sklerotizujúce procesy v pľúcach, pleure, pečeni a mnohých iných orgánoch [14,16]. Etiologicky vzniká po úraze, infekcii, zápale, či operácii [8]. IPT sa môže vyskytovať v akejkoľvek lokalizácii, pričom najčastejšie ide o postihnutie pľúc a orbity [21]. Klinické prejavy aj diagnostický postup sú totožné ako pri IMT. Histologicky sa jedná o podobnú štruktúru, s tým rozdielom, že počet plazmatických buniek je vyšší, je prítomný aj infiltrát zápalového fibrosklerotického procesu, či vaskulopatia [14,16]. To dokazuje, že sa nejedná o uniformný proces, ale heterogénnu léziu, základom ktorej je rôzny stupeň fibróznej zápalovej reakcie [21]. Imunohistochemicky je typická produkcia IgG4 protilátok plazmatickými bunkami, ktorých hladina býva zvýšená aj v sére, čo je dôležitá hodnota pri diferenciálnej diagnostike medzi IMT a IPT [14,16,19]. Pozitivita ALK-1, dezmínu, SMA, či CD34 je rovnako nešpecifická ako pri inflamatórnom myofibroblastickom nádore [14]. Vzhľadom ku skutočnosti, že sa jedná o sklerotizujúce ochorenie, s možnou autoimunitnou etiológiou, sú pri liečbe metódou voľby kortikosteroidy, ktoré bývajú efektívne aj pri parciálnych resekciách [8,16,19]. Zápalový pseudotumor nie je neoplázia, nemá malígny potenciál, ale vzhľadom na etiológiu je možná lokálna rekurencia, no nie vždy je potrebná kompletná exstirpácia ložiska [14,19]. K nedoriešeným otázkam sa pripája aj súvis IMT a ALK-pozitivity. Väčšina autorov udáva, že spôsob liečby, riziko rekurencie a malígnej transformácie, žiadnym spôsobom neovplyvňuje fakt, či je tumor ALK-negatívny alebo pozitívny [7,9,18,20]. Coffin et al v roku 2007 otvorili otázku, či bude niekedy možné nájsť vzťah medzi ALK-pozitívnymi génovými mutáciami a liečbou, v zmysle nájdenia inhibítora ALK, a tým pádom primárne IMT liečiť konzervatívne [20]. Mnohí autori zistili, že pri zápalovom myofibroblastickom tumore, s dokázanou translokáciou génu ALK, je pri neresekabilných nádoroch metódou voľby práve inhibítor tyrozínkinázy – krizotinib, ktorý môže predĺžiť interval rizika rekurencie a docieliť kompletnú remisiu ochorenia [2,19,22]. Táto nová možnosť liečby je však stále predmetom diskusie a štúdií.
Záver
Inflamatórny myofibroblastický tumor pľúc ako benígna neoplázia nejasnej etiológie, s možnou lokálnou agresivitou, avšak s nižším metastatickým potenciálom, sa vyskytuje najčastejšie u detí a adolescentov. Vzhľadom k diametrálne odlišnému spôsobu liečby hrá jeho oddiferencovanie od zápalového pseudotumoru kľúčovú úlohu v diagnosticko-terapeutickom algoritme. Metódou voľby je kompletná chirurgická resekcia, s možnosťou neoadjuvantnej liečby v prípade topograficky nepriaznivej lokalizácie. Pre riziko lokálnej rekurencie ochorenia je dlhodobá dispenzarizácia pacientov nevyhnutná.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential confl icts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
MUDr. Veronika Chrenková
Klinika detskej chirurgie
Národný ústav detských chorôb
Limbová 1
833 40 Bratislava
Slovenská republika
Obdŕžané/Submitted: 19. 9. 2020
Prijaté/Accepted: 26. 11. 2020
Informace z České onkologické společnosti
Zápisy ze schůzí výboru České onkologické společnosti konaných 23. 2. a 16. 3. 2021 ve FN Motol v Praze naleznete na www.linkos.cz.
Sources
1. Liu L, Kong X, Lu X, Cao D. Pediatric endobronchial inflammatory myofibroblastic tumor: a case report and review of literature. Clin Pract 2016; 6 (4): 853. doi: 10.4081/cp.2016.853.
2. Soyer T, Talim B, Karnak I et al. Surgical treatment of childhood inflammatory myofibroblastic tumors. Eur J Pediatr Surg 2017; 27 (4): 319–323. doi: 10.1055/s-0036-1593380.
3. Camela F, Gallucci M, di Palmo E et al. Pulmonary inflammatory myofibroblastic tumor in children: a case report and brief review of literature. Front Pediatr 2018; 6 : 35. doi: 10.3389/fped.2018.00035.
4. Dalton BGA, Thomas PS, Sharp NE et al. Inflammatory myofibroblastic tumors in children. J Pediatr Surg 2015; 51 (4): 541–544. doi: 10.1016/j.jpedsurg.2015.11.015.
5. Gleason BC, Hornick JL. Inflammatory myofibroblastic tumour: where are we now? J Clin Pathol 2008; 61 (4): 428–437. doi: 10.1136/jcp.2007.049387.
6. Štiková Z, Ptáková N, Horáková M et al. Inflamatorní myofibroblastický tumor dělohy – kazuistika. Cesk Patol 2019; 55 (4): 239–243.
7. Siminovich M, Galluzzo L, López J et al. Inflammatory myofibroblastic tumor of the lung in children: anaplastic lymphoma kinase (ALK) expression and clinico-pathological correlation. Pediatr Dev Pathol 2012; 15 (3): 179–186. doi: 10.2350/11-10-1105-OA.1.
8. SurabhiVR, Chua S, Patel RP. Inflammatory myofibroblastic tumors: current update. Radiol Clin North Am 2016; 54 (3): 553–563. doi: 10.1016/j.rcl.2015.12.005.
9. Pio L, Varela P, Eliott MJ et al. Pediatric airway tumors: a report from the international network of pediatric airway teams (INPAT). Laryngoscope 2019; 130 (4): E243–E251. doi: 10.1002/lary.28062.
10. Giuseppucci C, Reusmann A, Giubergia V et al. Primary lung tumors in children: 24 years of experience at a referral center. Pediatr Surg Int 2016; 32 (5): 451–457. doi: 10.1007/s00383-016-3884-3.
11. Fabre D, Fadel E, Singhal S et al. Complete resection of pulmonary inflammatory pseudotumors has excellent long-term prognosis. J Thorac Cardiovasc Surg 2009; 137 (2): 435–440. doi: 10.1016/j.jtcvs.2008.07.009.
12. Oguz B, Ozcan HN, Omay B et al. Imaging of childhood inflammatory myofibroblastic tumor. Pediatr Radiol 2015; 45 (11): 1672–1681. doi: 10.1007/s00247-015-3377-x.
13. Pešek M. Nová WHO klasifikace nádorů plic. Onkologie 2016; 10 (1): 20–24.
14. Bhagat P, Bal A, Das A et al. Pulmonary inflammatory myofibroblastic tumor and IgG4-related inflammatory pseudotumor: a diagnostic dilemma. Virchows Arch 2013; 463 (6): 743–747. doi: 10.1007/s00428-013-1493-2.
15. Karnak BI, Șenocak ME, Ciftci AO et al. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 2001; 36 (6): 908–912. doi: 10.1053/jpsu.2001.23970.
16. Saab ST, Hornick JL, Fletcher CD et al. IgG4 plasma cells in inflammatory myofibroblastic tumor: inflammatory marker or pathogenic link? Mod Pathol 2011; 27 (7): 606–612. doi: 10.1038/modpathol.2010.226.
17. Chun YS, Wang L, Nascimento AG et al. Pediatric inflammatory myofibroblastic tumor: anaplastic lymphoma kinase (ALK) expression and prognosis. Pediatr Blood Cancer 2005; 45 (6): 796-801. doi: 10.1002/pbc.20294.
18. Coffin CM, Patel A, Perkins S et al. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol 2001; 14 (6): 569–576. doi: 10.1038/modpathol.3880352.
19. Watanabe H, Yamasaki N, Miyazaki T et al. Successful treatment based on molecular biological assessment of invasive anaplastic lymphoma kinase-positive inflammatory myofibroblastic tumor of the lung. Surg Case Rep 2019; 5 (1): 118. doi: 10.1186/s40792-019-0674-x.
20. Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, ad immunohistochemical features including ALK expression in atypical and aggressive case. Am J Surg Pathol 2007; 31 (4): 509–520. doi: 10.1097/01.pas.0000213393.57322.c7.
21. Adamkov J, Kašparová P, Česák T. Zánětlivý pseudotumor imitující intrakraniální, konvexitární meningeom – kazuistika. Cesk Slov Neurol N 2015; 78/111 (2): 233–236.
22. Butrynski JE, D’Adamo DR, Hornick JL et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 2010; 363 (18): 1727–1733. doi: 10.1056/NEJMoa1007056.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2021 Issue 2
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Onemocnění asociované s IgG4
- Postižení dutiny ústní onkologických a hematoonkologických pacientů
- FDG-PET/ CT v diagnostice a hodnocení léčebné odpovědi Castlemanovy choroby – retrospektivní studie 29 případů z jednoho centra
- Význam 18F-FDG-PET vyšetření v léčbě adenokarcinomu jícnu a gastroezofageální junkce – přehled