
Metabolická plasticita nádorových buněk
Metabolic plasticity of cancer cells
Background: A general characteristic of cancer metabolism is the skill to gain the essential nutrients from a relatively poor environment and use them effectively to maintain viability and create new biomass. The changes in intracellular and extracellular metabolites that accompany metabolic reprogramming associated with tumor growth subsequently affect gene expression, cell differentiation, and tumor microenvironment. During carcinogenesis, cancer cells face huge selection pressures that force them to constantly optimize dominant metabolic pathways and undergo major metabolic reorganizations. In general, greater flexibility of metabolic pathways increases the ability of tumor cells to satisfy their metabolic needs in a changing environment. Purpose: In this review, we discuss the metabolic properties of cancer cells and describe the tumor promoting effect of the transformed metabolism. We assume that changes in metabolism are significant enough to facilitate tumorigenesis and may provide interesting targets for cancer therapy.
Keywords:
Metabolism – Warburg effect – cancer – glutaminolysis – anaplerosis – Krebs cycle – oncogenesis – oncometabolite
Authors:
M. Raudenská 1; B. Peltanová 2; K. Hönigová 2; J. Navrátil 2; M. Masařík 1,2
Authors‘ workplace:
Fyziologický ústav, LF MU Brno
1; Ústav patologické fyziologie, LF MU Brno
2
Published in:
Klin Onkol 2022; 35(3): 195-207
Category:
Review
doi:
https://doi.org/10.48095/ccko2022195
Overview
Východiska: Společným rysem metabolizmu nádorových buněk je schopnost získávat potřebné živiny z poměrně chudého prostředí a využívat je k udržení životaschopnosti a tvorbě nové biomasy. Změny v intracelulárních a extracelulárních metabolitech, které doprovází metabolické přeprogramování spojené s růstem nádoru, mají následně zásadní účinek na genovou expresi, buněčnou diferenciaci a mikroprostředí nádoru. V průběhu kancerogeneze čelí nádorové buňky selekčním tlakům, které je nutí neustále optimalizovat dominantní metabolické dráhy a nádorové buňky tak procházejí zásadními metabolickými reorganizacemi. Obecně platí, že vyšší flexibilita metabolických drah zvyšuje schopnost nádorových buněk sladit metabolické potřeby s měnícím se prostředím. Cíl: V tomto přehledovém článku pojednáváme o metabolických vlastnostech nádorových buněk a popisujeme účinek transformovaného metabolizmu na progresi nádoru. Domníváme se, že metabolické změny jsou pro rozvoj nádorů zásadní a mohly by poskytnout zajímavé cíle pro léčbu.
Klíčová slova:
nádorová onemocnění – metabolizmus – glutaminolýza – Warburgův efekt – anapleróza – Krebsův cyklus – nádorová transformace – onkometabolity
Úvod
Společným rysem nádorových buněk je schopnost získávat potřebné živiny z poměrně chudého prostředí a flexibilně je využívat k udržení životaschopnosti a budování nové biomasy. Bioenergeticky náročné procesy doprovázející maligní transformaci, jako jsou rychlá proliferace a schopnost migrovat, vyžadují zvýšenou produkci adenosintrifosfátu (ATP), nukleových kyselin, proteinů a lipidů. Změny v intracelulárních a extracelulárních metabolitech, které doprovází metabolické přeprogramování spojené s růstem nádoru, mají následně zásadní účinek na genovou expresi, buněčnou diferenciaci a mikroprostředí nádoru [1].
V průběhu kancerogeneze procházejí nádorové buňky častými metabolickými reorganizacemi. Obecně platí, že vyšší flexibilita metabolických drah využitelných danou buňkou nebo skupinou buněk zvyšuje jejich schopnost sladit metabolické potřeby s měnícím se prostředím. Metabolická plasticita je zprostředkována řadou mechanizmů zahrnujících receptory, různé typy signálních drah, pozměněnou aktivitu transkripčních faktorů a mutace ovlivňující expresi či funkci enzymů. Metabolická plasticita není výlučným znakem nádorových buněk, hraje totiž významnou roli v embryogenezi. Časná embrya jsou zpočátku závislá na oxidačním metabolizmu a upřednostňují oxidaci pyruvátu pomocí mateřských mitochondrií z oocytu. Příjem glukózy se postupně zvyšuje v morule a je urychlován ve stadiu blastocysty, kde její příjem převyšuje příjem pyruvátu či laktátu. V tomto období převažuje glykolýza, která je dále akcelerována při implantaci embrya do stěny dělohy. Během pozdějšího vývoje umožňuje mitochondriální replikace opětovné zahájení oxidačního metabolizmu a progresivní pokles glykolýzy [2,3]. Tyto metabolické posuny jsou zásadní; mutace v genech spojených s glykolýzou nebo oxidační fosforylací mohou vést ke zpoždění vývoje nebo k embryonální letalitě [4]. Metabolická plasticita embryonálních buněk může být „odblokována“ v průběhu kancerogeneze a využita nádorovými buňkami.
V tomto přehledovém článku jsme se pokusili shrnout metabolické procesy, které mohou významně přispívat k progresi nádorového onemocnění: Warburgův efekt, význam anaplerózy Krebsova cyklu, glutaminolýzu a oportunistické zdroje nutrientů využívané nádorovými buňkami. Dále jsou rozebrány mutace a změny v expresi metabolických enzymů, které mohou přispívat k nádorové transformaci prostřednictvím produkce onkometabolitů.
Warburgův efekt
Na počátku 20. let minulého století objevil Otto Warburg, že ve srovnání s většinou netransformovaných tkání konzumují nádory obrovské množství glukózy. Většina glukózy spotřebované nádory je fermentována na laktát, a to i v přítomnosti kyslíku [5]. Tento fenotyp se označuje jako aerobní glykolýza. Aerobní glykolýza je charakteristickým znakem proliferativního metabolizmu a je často spojována s nádorovým bujením. V této souvislosti je aerobní glykolýza známá jako Warburgův efekt. Warburg předpokládal, že aerobní glykolýza je příznakem poškození mitochondrií a blokády oxidačního metabolizmu nádorových buněk [6]. Ačkoli mnoho druhů nádorových buněk produkuje ATP prostřednictvím glykolýzy, většina z nich má rovněž funkční a aktivní mitochondrie, které hrají ústřední roli jak v metabolizmu, tak v tumorigenezi. Funkční mitochondrie se zdají být pro nádorovou buňku dokonce nezbytné [7]. Mutace v mitochondriálních genech jsou u nádorů běžné, nicméně většinou neinaktivují energetický metabolizmus, ale spíše mění mitochondriální biosyntézu, což vede ke změnám v komunikaci s jádrem (mitochondriální retrográdní signalizace) za účelem modulace signálních drah, transkripční aktivity a struktury chromatinu [8]. Pro viabilitu nádorové buňky je zásadní zachovat si určitý počet mitochondrií s neporušeným proteomem. Negativní selekci podléhají zejména mutace významně poškozující dýchací řetězec [9]. Nádorové buňky bez mitochondriální DNA (mtDNA) vykazují sníženou proliferaci a k dalšímu rozvoji nádoru je nezbytné získání mtDNA z hostitelských buněk [7]. K překonání zástavy buněčného cyklu není nutná mitochondriální syntéza ATP, je však nutná biosyntéza pyrimidinů závislá na dýchacím řetězci a aktivitě dihydroorotát dehydrogenázy [10].
Provozování aerobní glykolýzy místo dýchání je poměrně energeticky nevýhodné (jsou vytvořeny pouze 2 molekuly ATP, tedy pouze 6 % teoretického maximálního výtěžku z dýchacího řetězce a oxidační fosforylace). Co tedy vede k aerobní glykolýze u nádorů a proč je spojena s proliferací, ačkoli snižuje efektivitu produkce ATP a vede ke ztrátě důležitých biosyntetických prekurzorů? (Většina uhlíků získaných z glukózy se při aerobní glykolýze vylučuje skrze laktát.) Možným vysvětlením je, že aerobní glykolýza usnadňuje produkci kofaktorů a nosičů elektronů potřebných pro redoxní reakce v buňkách (obr. 1). Zvýšená absorpce glukózy umožňuje vyšší produkci nikotinamidadenindinukleotidfosfátu (NADPH) v rámci pentózo-fosfátové dráhy (pentose phosphate pathway – PPP) a fermentace zahrnuje regeneraci oxidované formy nikotinamidadenindinukleotidu (NAD+) působením laktátdehydrogenázy (LDH). NADPH spolupracuje především s enzymy anabolických drah a dodává elektrony pro syntézu energeticky bohatých molekul, zatímco NAD+ působí jako oxidační činidlo v katabolických dějích.

ATP – adenosintrifosfát, c-ACN – cytosolická akonitáza, ETC – dýchací řetězec, Fe – železo, LDH – laktátdehydrogenáza, MCT – monokarboxylátový
transportér, NADPH – nikotinamidadenindinukleotidfosfát, IRP1 – protein regulující železo, PMET – transport elektronů na
plazmatické membráně, PPP – pentózo-fosfátová dráha, UCP – odpřahující proteiny
Proliferující buňky mají vysokou spotřebu NAD+, jehož fatální nedostatek může vést k buněčné smrti typu parthanatos [11]. Hlavními enzymy podílejícími se na regeneraci NAD+ v buňce jsou LDH a mitochondriální komplex I, který je hlavním místem pro aerobní regeneraci NAD+, jelikož katalyzuje oxidaci NADH na NAD+ a zároveň transfer dvou elektronů na koenzym Q. Pokud poptávka po NAD+ překročí rychlost spotřeby ATP, regenerace NAD+ komplexem I bude omezena, jelikož zvýšený potenciál mitochondriální membrány (Dψm) vznikající v důsledku pomalejší syntézy ATP ztěžuje transport elektronů a regeneraci NAD+. Odpojení dýchání od syntézy ATP nebo zvýšení hydrolýzy ATP [12] obnovuje homeostázu NAD+/NADH a proliferaci, a to i při vysoké aktivitě dýchacího řetězce. Aby se zabránilo hyperpolarizaci mitochondriální membrány (vysoká hodnota Dψm), mohou se nádorové buňky spoléhat na indukovaný únik protonů pomocí odpřahujících proteinů (uncoupling proteins – UCP). UCP jsou mitochondriální transportéry přítomné ve vnitřní membráně mitochondrií, které podporují metabolickou flexibilitu tím, že umožňují vysokou rychlost regenerace NAD+ mitochondriálním dýcháním, a to bez tvorby ATP [13]. Zvýšená exprese UCP2 byla popsána u leukemií, nádorů prsu, vaječníků, močového měchýře, jícnu, slinivky břišní, ledvin, varlat, plic, prostaty a kůže [14–17]. Buňky se tedy zapojují do aerobní glykolýzy, pokud poptávka po NAD+ převyšuje poptávku po ATP a aerobní glykolýza je charakterizována přesunem pyruvátu z Krebsova cyklu směrem k fermentaci, která umožňuje snadnější regeneraci NAD+ [13]. Oxidace pyruvátu v mitochondriích vyžaduje komplex pyruvátdehydrogenázy (pyruvate dehydrogenase complex – PDHC), který usnadňuje vstup pyruvátu do Krebsova cyklu. PDHC je negativně regulován pyruvátdehydrogenázovými kinázami (PDK). Transkripční faktory notoricky spojované s kancerogenezí, jako jsou Myc, Wnt a hypoxií indukovatelné faktory (HIF), působí v nádorových buňkách navýšení exprese jedné nebo více izoforem PDK. Aktivované PDK následně fosforylují PDHC, což inhibuje jeho aktivitu a oxidaci pyruvátu [18]. Ke snížení redukčního stresu způsobeného hromaděním intracelulárního NADH využívají nádorové buňky rovněž transport elektronů na plazmatické membráně, jehož hlavní funkcí je recyklace NADH na NAD+ [19].
Široké klinické využití PET s použitím 18F-fluorodeoxyglukózy přispívá k vnímání mnoha nádorů jako glykolytických. Tato metoda však poskytuje informace o absorpci glukózy, ale ne o jejím následném metabolickém osudu. Zvýšený příjem glukózy nemusí být vždy v pozitivní korelaci s mírou glykolýzy. Maligní buňky mohou využívat glukózu k produkci buněčné biomasy, podpoře růstu a překonání bariér k metastazování [20]. Z tohoto důvodu je pro nádorové buňky důležitá PPP. Tato dráha poskytuje redukované kofaktory NADPH a pětiuhlíkaté sacharidy (pentózy). NADPH je následně spotřebováván při biosyntéze steroidních látek a mastných kyselin. Vytváření pentóz je nezbytné pro syntézu nukleotidů a nukleových kyselin. Vzhledem k tomu, že PPP a glykolýza sdílejí glukózu-6-fosfát (G6P) jakožto společný meziprodukt, je při rychlé glykolýze G6P zpracována na pyruvát dříve, než se stihne odklonit do PPP. Pyruvát je syntetizován pyruvátkinázou (PK) jako poslední krok glykolýzy. PK se může vyskytovat jako izoforma M1 nebo M2. U mnoha typů nádorů je více exprimovanou formou PK izoforma M2, zatímco netransformované tkáně s vysokou poptávkou po ATP mají tendenci exprimovat spíše izoformu M1, která v porovnání s M2 vykazuje vyšší rychlost [21]. Buňky se zvýšeným poměrem PK-M2/PK-M1 mají potenciál zpomalit produkci pyruvátu v reakci na signály proliferace. Tento jev pravděpodobně umožňuje využití glykolytických meziproduktů pro biosyntézu stavebních bloků vč. nukleotidů a aminokyselin [22–24].
NADPH může být také syntetizován prostřednictvím cytosolické akonitázy (cytosolic aconitase – c-ACN). c-ACN je známější jako protein regulující železo (iron regulatory protein 1 – IRP1). Pokud je intracelulární objem železa nízký, IRP1 se váže na nepřekládané oblasti specifických mRNA a zvyšuje expresi proteinů odpovědných za intracelulární příjem železa. V přítomnosti zvýšených hladin intracelulárního železa tvoří IRP1 klastry 4Fe-4S vykazující aktivitu akonitázy. c-ACN metabolizuje citrát na izocitrát, který se pak transformuje na a-ketoglutarát (a-KG) za současné syntézy NADPH. Snížený tok citrátu přes c-ACN může tedy snížit produkci cytosolického NADPH, což je hlavní zdroj redukčních ekvivalentů pro syntézu mastných kyselin a důležitá obrana proti oxidačnímu stresu [25]. Pokud jsou nádorové buňky schopny importovat dostatečné množství citrátu [26], podporuje přebytek železa přeměnu citrátu na izocitrát pomocí c-ACN s následnou tvorbou NADPH. Inhibice c-ACN vede ke snížení poměru NADPH/NADP a k nižší expresi genů spojených s lipogenezí [27]. Nádorové buňky syntetizují větší množství lipidů než nemaligní buňky a vyžadují vyšší hladinu železa, což zvyšuje jejich náchylnost k ferroptóze (forma buněčné smrti, která je výsledkem akumulace reaktivních forem kyslíku v závislosti na přítomnosti železa) [28], nicméně vysoké hladiny NADPH mohou nádorové buňky před takovým typem smrti ochránit [29].
Důsledkem Warburgova efektu může být akumulace laktátu v nádorovém mikroprostředí (tumor microenvironment – TME) a vyvolání laktátové acidózy. Laktátová acidóza má silný imunosupresivní účinek a v některých případech zprostředkovává únik nádorových buněk před imunitním dohledem (obr. 2). Laktát je molekula schopná potlačovat imunitní reakce prostřednictvím inhibice efektorových T lymfocytů či dendritických buněk a modulací fenotypu makrofágů [30,31]. Naopak diferenciace a aktivita regulačních T lymfocytů, které inhibují protinádorovou imunitní odpověď, je zvýšenou koncentrací laktátu v TME podporována [32]. Acidóza nádorového mikroprostředí významně podporuje rovněž metabolizmus lipidů, indukci autofagie, produkci NADPH v rámci PPP a remodelaci extracelulární matrix [33]. Mírná acidóza může změnit morfologii mitochondrií, což následně umožňuje zachování účinné produkce ATP bez ohledu na hladiny kyslíku a zabraňuje mitochondriální fragmentaci v důsledku hypoxie [34]. Jednou z možných adaptací nádorových buněk na vysoké hladiny laktátu je ustavení metabolické symbiózy mezi buňkami generujícími laktát a buňkami konzumujícími laktát. Laktát se z buňky uvolňuje prostřednictvím monokarboxylátového transportéru 4, jiné buňky přítomné v nádorovém mikroprostředí mohou laktát zachytit pomocí monokarboxylátového transportéru 1 (MCT1) a metabolizovat jej na pyruvát [35]. K přežití nádorových buněk v podmínkách acidózy rovněž přispívá membránový protein asociovaný s lysozomy (lysosome-associated membrane protein 2 – LAMP2), který běžně funguje k ochraně lysozomálních membrán před kyselou proteolýzou. LAMP2 byl nalezen na plazmatické membráně nádorových buněk přizpůsobených acidóze [36].

FDG – fludeoxyglukóza, OXPHOS – oxidační fosforylace, TME – nádorové mikroprostředí
Možné terapeutické cílení Warburgova efektu zahrnuje inhibici transportu glukózy (ritonavir, fasentin, genistein, STF-31 a WZB117) či inhibici fosforylace glukózy a vzniku glukóza-6-fosfátu (3-bromopyruvát, lonidamin, 2-deoxy-D-glukóza) [38,39]. Výsledky klinických studií ukázaly, že lonidamin potencoval terapeutický účinek doxorubicinu u melanomů, nádorů prsu, prostaty, mozku a vaječníků [40,41]. Účinek 2-deoxy-D-glukózy je založen na snížení využitelnosti glukózy, jelikož 2-deoxy-D-glukóza je metabolizována na deoxyglukózu-6-fosfát, kterou nelze dále využít k produkci ATP [42]. Přestože 2-deoxy-D-glukóza samotná má pouze omezený terapeutický potenciál, může být úspěšně kombinována s jinými terapeutickými přístupy (např. radioterapií) [43].
Nadějným přístupem pro terapeutické využití Warburgova efektu vyznačujícího se nadměrným příjmem glukózy je vazba léčiva přímo na glukózu. Příkladem může být glufosfamid, který se během glykolýzy štěpí na glukózu a toxický ifosfamid [44]. Dalšími slibnými látkami, které lze navázat na glukózu, jsou 1,4-naftochinony [45], rozvětvený polyethylenglykol [46], platina [47] či methotrexát [48]. Látky konjugované s glukózou jsou využitelné rovněž pro fotodynamickou terapii. Příkladem fotosenzibilizátoru konjugovaného s glukózou je např. G-chlorin e6 [49].
Jak bylo diskutováno výše, PK-M2 hraje klíčovou roli v metabolizmu a proliferaci nádorových buněk. Mezi inhibitory PK-M2 patří látky TT-232, VK3 a VK5 [38]. Nedávno bylo zjištěno, že mezi inhibitory PK-M2 patří rovněž kyselina oleanolová [50]. Dalším významným enzymem účastnícím se Warburgova efektu je LDH. Zatímco laktát izoforma dehydrogenáza A (LDHA) má vyšší afinitu k pyruvátu a přednostně přeměňuje pyruvát na laktát a NADH na NAD+, izoforma laktát dehydrogenáza B (LDHB) má vyšší afinitu k laktátu a přednostně převádí laktát na pyruvát a NAD+ na NADH [51,52]. LDHA je hlavním enzymem regulujícím Warburgův efekt, a je tedy rovněž důležitým cílem pro vývoj antineoplastických léčiv. Příkladem mohou být 1 - (fenylseleno) -4 - (trifluormethyl) benzen a FX11 [53,54].
Anabolizmus, anapleróza a Krebsův cyklus
Během překotného dělení nádorových buněk je spotřebováváno velké množství lipidů, nukleotidů a aminokyselin. K jejich syntéze používají buňky prekurzory odvozené z meziproduktů Krebsova cyklu (KC). Např. oxaloacetát a a-KG jsou nezbytné pro syntézu neesenciálních aminokyselin a nukleotidů. Během buněčné proliferace se tedy velká část uhlíku, který vstupuje do KC, používá spíše v biosyntetických drahách než k produkci ATP. To má za následek nepřetržitý odliv meziproduktů Krebsova cyklu (katapleróza). Ukázkovým příkladem kataplerózy v proliferujících buňkách je syntéza lipidů (mastných kyselin, cholesterolu a izoprenoidů) z citrátu. Citrát uvolněný do cytoplazmy je metabolizován ATP-citrát lyázou (ACLY) a hraje ústřední roli v syntéze mastných kyselin. Inhibice ACLY může potlačit růst nádorových buněk [55,56]. Metabolické meziprodukty KC musejí být u rychle se dělících buněk neustále doplňovány anaplerotickými reakcemi (oxaloacetát, a-KG či fumarát mohou vznikat z aminokyselin – aspartát může být transaminován na oxaloacetát, glutamát na a-KG; sukcinyl-CoA může být pak syntetizován z propionyl-CoA, jenž se tvoří během degradace mastných kyselin, a acetát může přispívat k navýšení buněčných zásob acetyl-CoA [57]. Zvýšené požadavky na citrát mohou být kromě jeho běžné syntézy z pyruvátu v KC pokryty také a-ketoglutarátem vzniklým z glutaminu (obr. 3). Ten je redukčně karboxylován mitochondriální izocitrátdehydrogenázou 2 za vzniku izocitrátu, který lze poté izomerizovat na citrát [58]. Až donedávna se věřilo, že nádorové buňky nemohou získat citrát z extracelulárního prostoru a vzniká tak jejich závislost na importu glutaminu. To se však ukázalo být chybným předpokladem [26]. Některé nádorové buňky jsou závislé na glutaminu pouze in vitro, zatímco in vivo je jejich míra absorpce glutaminu podobná jako u netransformovaných buněk [59]. Import extracelulárního citrátu může významně podporovat proliferaci nádorových buněk skrze syntézu mastných kyselin a bílkovin [60]. Způsob, jakým je v nádorových buňkách obstaráván citrát, může záviset na okolních metabolických podmínkách (hypoxie, dostupnost glukózy, typ nádorového stromatu). Zvýšená absorpce glutaminu byla pozorována ve stromálních buňkách přítomných v mikroprostředí glioblastomu [61]. Tato zvýšená absorpce glutaminu by byla konzistentní s konceptem metabolické symbiózy, kdy nádorové stroma syntetizuje metabolické substráty podporující růst nádorových buněk; to by mohlo potenciálně zahrnovat také citrát. Syntéza citrátu z glutaminu a jeho uvolňování do TME by mohly být jedním z metabolických důsledků transformace fibroblastů v nádorovém mikroprostředí [62].
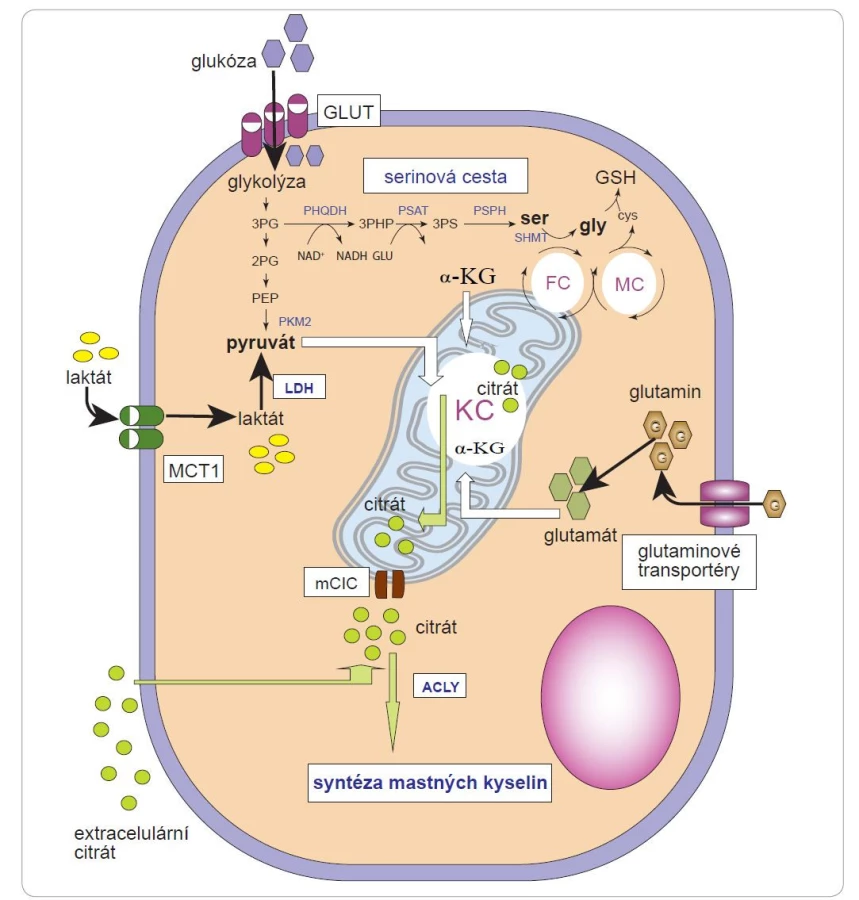
2PG – 2-fosfoglycerát, 3PG – 3-fosfoglycerát, 3PHP – 3-fosfohydroxypyruvát, 3PS – 3-fosfoserin, α-KG – α-ketoglutarát, ACLY – ATP-citrát
lyáza, cys – cystein, FC – folátový cyklus, GLUT – glukózový transportér, GSH – glutathion, gly – glycin, KC– Krebsův cyklus, LDH – laktátdehydrogenáza,
MC – methioninový cyklus, mCIC – mitochondriálního citrátového transportéru, MCT1– monokarboxylátový transportér
1, PEP – fosfoenolpyruvát, PHGDH – fosfoglycerátdehydrogenáza, PKM2 – pyruvátkináza M2, PSAT – fosfoserinaminotransferáza, PSPH –
fosfoserinfosfatáza, ser – serin, SHMT – serinová hydroxymetyltransferáza
Dalším metabolitem, který se může podílet na podpoře mitochondriálního metabolizmu nádorových buněk a v důsledku toho na syntéze citrátu, je laktát, který byl dlouho považován za odpadní produkt. Některé nádorové buňky či buňky přítomné v TME však laktát přijímají a metabolizují [35,63]. Laktát lze do buňky importovat pomocí MCT1. Intracelulárně může být laktát přeměněn na pyruvát a poté na acetyl-CoA, který se přivádí do Krebsova cyklu. Konverze laktátu na pyruvát je navíc doprovázena redukcí NAD+ na NADH, který potlačuje aktivitu glyceraldehyd-3-fosfátdehydrogenázy ve směru odbourávání glukózy, čímž šetří G6P pro převod do PPP. Nadměrná aktivita PPP a zvýšená exprese MCT1 významně podporují metastazování melanomu [64].
U mnoha typů nádorů je navýšena de novo syntéza serinu, při které je relativně velké množství glykolytického uhlíku odváděno pomocí fosfoglycerátdehydrogenázy (PHGDH) do syntetických drah serinu a glycinu a následné tvorby nukleotidů (obr. 3). Limitujícím faktorem využití serinu pro syntézu nukleotidů je přítomnost dostatečného množství NAD+ [65]. Gen pro PHGDH je často amplifikován u melanomů. Zvýšená exprese PHGDH byla také spojena s některými podtypy nádorů prsu [66]. PHGDH a syntéza serinu jsou paradoxně vyžadovány rovněž v přítomnosti nadměrného množství environmentálního serinu, a to i v případě, kdy absorpce serinu přesahuje požadavky na syntézu nukleotidů. Lze tedy předpokládat, že serinová cesta neslouží pouze k syntéze serinu, ale může být alternativním prostředkem k produkci a-KG či glutathionu (GSH). Zvýšená aktivita KC cyklu a PPP může zvrátit důsledky inhibice PHGDH [67,68].
Anapleróza KC umožňuje nádorovým buňkám udržovat vysokou míru proliferace a anabolických reakcí, zároveň se tyto procesy stávají pro nádorovou buňku nezbytnými, a jsou tedy lákavým cílem protinádorové terapie. Možné terapeutické cílení anaplerózy KC a následných anabolických reakcí zahrnuje inhibici ACLY (SB-204990, (-) -kyselina hydroxycitronová, deriváty emodinu, antimycin A2 a A8 či radicicol) [69–72], inhibici LDHB (derivát indolu AXKO-0046 [73]) či nespecifickou inhibici LDH [74], inhibici importu laktátu (syrosingopin, 7ACC2) [75,76] či inhibici PHGDH [77,78].
Glutaminolýza
Mnoho nádorových buněk (obzvláště při hypoxii) konzumuje vysokou rychlostí glutamin a současně vylučuje glutamát. Glutamin je transportován do buněk pomocí transportérů glutaminu v plazmatické membráně, jako jsou proteiny kódované geny SLC1A5, SLC38A1 a SLC38A217. Glutamin je v rámci glutaminolýzy přeměněn na glutamát pomocí glutaminázy (GLS1 či GLS2) [79]. Glutaminolýza má v nádorovém metabolizmu zásadní roli, podílí se na doplňování meziproduktů KC, biosyntéze nukleotidů, GSH a některých aminokyselin. Zvýšená exprese fosforibosylpyrofosfátamidotransferázy, která přenáší g-dusík glutaminu na 5-fosforibosylpyrofosfát (klíčová reakce při biosyntéze purinů), je jedním z nejsilnějších indikátorů špatné prognózy u malobuněčného karcinomu plic [80]. Glutamát hraje ústřední roli v metabolizmu aminokyselin, protože je zásadní pro biosyntézu alaninu, aspartátu, prolinu a serinu, kteréžto aminokyseliny se dále používají pro biosyntézu asparaginu, argininu, cysteinu a glycinu. Glutamát se rovněž využívá jako směnný faktor pro import extracelulárního cystinu prostřednictvím transportéru kódovaného genem SLC7A11 [81]. Asi 30 % glutaminu je metabolizováno v cytosolu [79]. U mitochondriální glutaminolýzy musí být cytosolický glutamin nejprve transportován přes vnitřní mitochondriální membránu prostřednictvím transkripční varianty genu SLC1A5 přepsané z alternativního místa iniciace transkripce [82]. Mitochondriální glutamát lze exportovat z mitochondrií do cytosolu prostřednictvím transportérů kódovaných geny SLC25A18 a SLC25A22 [83].
Spotřeba glutaminu je spojována s doplňováním meziproduktů KC, zvýšenou poptávkou po dusíku a syntézou ATP, ale důvod vylučování glutamátu je spíše nejasný. Předpokládá se, že vylučování glutamátu pomáhá buňce zvýšit rychlost syntézy nukleotidů [79]. Možné je též využití exportu glutamátu v obraně proti oxidačnímu stresu (import cystinu skrze (Xc–) systém) [84]. Nicméně vysoké koncentrace extracelulárního glutamátu mohou blokovat absorpci cystinu systémem (Xc–) a indukovat ferroptózu [85,86]. Některé nádorové buňky vylučující vysoké koncentrace glutamátu mohou být citlivé na ferroptózu v případě, že nezískají podporu dalších buněk nacházejících se v mikroprostředí nádoru. Glutamát může být importován např. fibroblasty asociovanými s nádorem (CAF), a to prostřednictvím transportéru kódovaného genem SLC1A3. Tyto fibroblasty mají často zvýšenou expresi glutamin syntetázy (GS/GLUL) a uvolňují aspartát a glutamin podporující proliferaci nádorových buněk [87]. Nastolená metabolická symbióza umožňuje nádorovým buňkám zbavit se glutamátu podporujícího ferroptózu a získat glutamin. Podobná metabolická výměna může probíhat také mezi nádorovými buňkami a M2-makrofágy [88].
Vzhledem ke kritické roli glutaminolýzy v metabolizmu nádorových buněk a jisté závislosti některých nádorů na glutaminu bylo vyvinuto mnoho účinných inhibitorů tohoto procesu (DON, JHU-083, BPTES, CB-839) [89]. DON je antagonista glutaminu, který široce inhibuje enzymy využívající glutamin. Tato neselektivní inhibice však vyvolává vysoký stupeň toxicity. Aby se minimalizovala toxicita DON, byla vyvinuta strategie proléčiv, která mohou být podávána v inertním stavu a poté aktivována v mikroprostředí nádoru [90]. Na rozdíl od antagonistů glutaminu alosterické inhibitory, jako jsou BPTES a CB-839, selektivně cílí na glutaminázu, aniž by narušily další aspekty metabolizmu glutaminu. Ačkoli BPTES vykazuje vysokou specifičnost a účinnost při inhibici proliferace nádorových buněk in vitro, jeho nevýhodou je špatná rozpustnost ve vodě a nízká biologická dostupnost in vivo omezující jeho další aplikace. Sloučenina CB-839 již vstoupila do klinických studií pro léčbu pokročilých solidních nádorů a hematologických malignit [91]. Dalším slibným přístupem v cílení glutaminolýzy je inhibice vychytávání glutaminu z extracelulárního prostředí. Chemická inhibice transportu glutaminu benzylserinem nebo L - g-glutamyl-p-nitroanilidem vedla k významnému snížení růstu nádorových buněk endometria [92].
Onkometabolity
Pojem onkometabolit označuje metabolit, který je v nádorech nadměrně akumulován a může ovlivňovat progresi onemocnění prostřednictvím epigenetické dysregulace či přepínáním fenotypových vlastností buněk (např. epitelo-mezenchymová tranzice – EMT). Jedním z nejznámějších onkometabolitů je D-2-hydroxyglutarát (D2HG). Jeho akumulace je způsobena mutacemi v izocitrátdehydrogenáze (IDH1 nebo IDH2), která za normálních okolností katalyzuje oxidační dekarboxylaci izocitrátu na a-KG. Tyto mutace se běžně vyskytují u gliomů a akutní myeloidní leukemie. Hladiny D2HG mohou v gliomové tkáni dosahovat až 35 mM koncentrace a tyto vysoké hladiny D2HG byly spojeny s onkogenezí a inhibicí diferenciace buněk. Díky své strukturální podobnosti s a-KG má D2HG inhibiční účinky na enzymy závislé na α-KG [93], a může tedy přispívat k maligní transformaci narušením normálního cyklu metylace a demetylace DNA (prostřednictvím inhibice enzymů závislých na a-KG, jako je Tet metylcytosin dioxygenáza 2 (TET2)). Exprese mutantních alel IDH produkujících D2HG je schopna vyvolat globální hypermetylaci DNA [94], způsobit defekt homologní rekombinace [95], indukovat angiogenní aktivitu prostřednictvím signální dráhy receptoru vaskulárního endotelového růstového faktoru (vascular endothelial growth factor – VEGF) [96] či snížit hladinu glutathionperoxidázy 4, enzymu klíčového pro zvládání oxidačního stresu a peroxidace lipidů. Určité mutantní formy IDH1 mohou tedy vyvolávat senzitivitu k ferroptóze [97]. Kromě D (R) – enantiomeru 2-hydroxyglutarátu může být produkován také jeho L (S) – enantiomer (L2HG). K akumulaci L2HG může vést zvýšená aktivita LDH a malátdehydrogenázy (MDH), nedostatek kyslíku či kyselé pH. Zvýšená produkce L2HG vede ke stabilizaci hypoxií indukovatelného faktoru 1 alfa (HIF-1a), a to i v normoxii [98]. L2HG je onkometabolit významný u nádorů ledvin a funguje jako epigenetický modifikátor, jelikož inhibuje aktivitu TET2 [99].
K produkci onkometabolitů mohou přispívat rovněž mutace či změny v genové expresi enzymů Krebsova cyklu [100]. Mezi onkometabolity odvozené z KC bývají řazeny sukcinát a fumarát (obr. 4). Ztráta funkce sukcinátdehydrogenázy (SDH) doprovázená nadprodukcí sukcinátu byla spojena s iniciací malignit [101]. Akumulace sukcinátu usnadňuje angiogenezi stimulací sukcinátového receptoru 1 (SUCNR1) a způsobuje nadprodukci VEGF [102,103]. Akumulace sukcinátu také přispívá k aktivaci EMT a k epigenetickým modifikacím díky inhibici enzymu TET (snížení oxidace 5-metylcytosinu a následná hypermetylace DNA) [104–106]. Nedostatečná aktivita SDH byla spojena s metabolickým přeprogramováním nádorových buněk směrem ke glykolytickému, pseudohypoxickému fenotypu [107], jelikož sukcinát stabilizuje hypoxií indukovatelný faktor-1a (HIF-1a) a podporuje uvolňování prozánětlivého interleukinu IL-1b imunitními buňkami, jako jsou makrofágy či dendritické buňky [108].

α-KG – α-ketoglutarát, α-KGDH – α-ketoglutarátdehydrogenáza, ACO2 – mitochondriální akonitáza, ADSL – adenylosukcinát lyáza, ASL –
argininosukcinát lyáza, D2HG – D-2-hydroxyglutarát, FH – fumarát hydratáza, KC – Krebsův cyklus, LDH – laktátdehydrogenáza, L2HG – L-
-2-hydroxyglutarát, MDH – malátdehydrogenáza, PNC – purinové nukleotidy, SDH – sukcinátdehydrogenáza
Podobně jako sukcinát je i fumarát důležitým metabolitem Krebsova cyklu. K intracelulární akumulaci fumarátu vede např. ztráta aktivity fumarát hydratázy (FH), která má mnoho rozdílných důsledků vč. aktivace onkogenních drah a vychytávání glutaminu [109]. Ztráta funkce FH přispívá např. k propuknutí dědičné leiomyomatózy, melanomu a nádorů ledvin [101]. Tumorigeneze v FH-deficientních buňkách je zřejmě vícestupňovým procesem. Po ztrátě FH procházejí buňky řadou biochemických adaptací, aby kompenzovaly zkrácení Krebsova cyklu. Tyto kompenzační změny podporují akumulaci intracelulárního fumarátu, což může vést k vyčerpání zásob glutathionu, oxidačnímu stresu a senescenci. Souběžně může fumarát vyvolat epigenetické změny, které umožňují senescenci obejít (např. hypermetylace p16), a aktivaci dalších onkogenních drah vč. těch, které jsou řízeny faktorem 2 souvisejícím s jaderným faktorem erythroid 2 (Nuclear factor erythroid 2-Related Factor – 2NRF2), nereceptorovou tyrozin-protein kinázou (ABL1) a HIF-1a [110]. Akumulovaný fumarát vede k posttranslační modifikaci cysteinových zbytků různých proteinů (proces sukcinace). Sukcinací kelch-like ECH-asociovaného proteinu 1 (KEAP1) a aktivací ABL1 stimuluje fumarát antioxidační odpověď zprostředkovanou transkripčním faktorem NRF2 [110,111]. Jedním z transkripčních cílů NRF2 je rovněž hemoxygenáza 1 (HMOX1), která je nutná pro biosyntézu a degradaci hemu, což je základní cesta pro přežití buněk s deficitem FH. Působením HMOX1 vzniká oxid uhelnatý, železnatý iont a biliverdin, který je následně redukován na bilirubin. Sukcinace proteinu regulujícího železo (iron regulatory protein 2 – IRP2) znemožňuje potlačit translaci mRNA pro ferritin. Zvýšené hladiny ferritinu následně podporují expresi promitotického transkripčního faktoru „forkhead box protein M1“, který podporuje proliferaci buněk [112], a způsobují pokles hladiny volného intracelulárního železa. Redukce železa a sukcinace klíčových cysteinových zbytků v katalytickém jádru také inaktivuje mitochondriální akonitázu, což znemožňuje oxidaci citrátu a umožňuje jeho využití pro syntézu lipidů [113]. Fumarát může fungovat také jako inhibitor pyroptotické buněčné smrti, jelikož způsobuje sukcinaci gasderminu D a zabraňuje tak jeho interakci s kaspázami, omezuje jeho oligomerizaci a schopnost indukovat pyroptózu, která úzce souvisí s aktivací protinádorové imunity [114,115]. Akumulace fumarátu v buňkách s deficitem FH vede rovněž k obrácení aktivity enzymu močovinového cyklu argininosukcinát lyázy, což způsobuje produkci argininosukcinátu z fumarátu a argininu. V důsledku toho jsou buňky s deficitem FH pro arginin auxotrofní, a tedy citlivé k jeho nedostatku [109]. Podobně akumulace fumarátu podporuje nadprodukci adenylosukcinátu obrácením aktivity adenylosukcinát lyázy v cyklu purinových nukleotidů [110].
Pro regulaci vzniku onkometabolitů bylo vyvinuto několik typů inhibitorů cílících na mutované formy IDH. Mezi ně patří např. BAY-1436032, AGI-5198, IDH305, FT-2102, HMS-101, MRK-A a GSK321. Mezi léčiva povolená americkým Úřadem pro kontrolu potravin a léčiv patří enasidenib (AG-221) a ivosidenib (AG-120) [116]. K cílení aktivity MDH byl doposud využit např. metyl - -3 - (3 - (4 - (2,4,4-trimethylpentan-2-yl) fenoxy) propanamido) benzoát neboli sloučenina 16c [117] či 4 - (3,4-difluorfenyl) thiazol-2-amin [118]. Možné cílení aktivity LDH bylo popsáno výše.
Oportunistické zdroje nutrientů – kanibalizmus
Neoplastický růst a mnoho charakteristických vlastností nádorů jsou způsobeny rozbitím regulačních molekulárních sítí vytvořených během vzniku mnohobuněčnosti. Narušení komunikace a koordinace mezi různými typy buněk ve tkáních může způsobovat rozvoj nádorů, jelikož přechod od jednobuněčnosti k mnohobuněčnosti zanechal ve struktuře regulačních sítí citlivá místa. Mnoho vlastností typických pro mnohobuněčný organizmus je při onkogenezi utlumeno, zatímco geny sahající až k jednobuněčným předkům jsou během transformace často aktivovány a/nebo jsou vyžadovány pro udržení nádorového fenotypu. Mezi nádorovými buňkami a jednobuněčnými organizmy lze nalézt mnoho fenotypových podobností vč. spíše kompetitivního než kooperativního růstu buněk, závislosti na fermentačních procesech a zvýšené genomové nestability připomínající mutátorové fenotypy jednobuněčných organizmů [119,120]. Předpokládá se, že tato reaktivace starobylých drah je vyvolána závažným environmentálním stresem, jako je deprivace živin, hypoxie, reaktivní formy kyslíku a nízké pH. Selekční priorita se poté přesouvá spíše k přežití jednotlivé buňky než k přežití organizmu, což podporuje maligní transformaci [119]. Známku regrese k primitivnějšímu životnímu stylu podobnému jednobuněčným mikroorganizmům, jako jsou améby, může představovat např. kanibalizmus. Nádorové buňky, stejně jako jednobuněčné organizmy, umí pohlcovat živiny ze svého okolí, a to vč. proteinových shluků či celých buněk. Silnou kanibalistickou aktivitu vykazují např. buňky maligního melanomu, které dokážou pohltit a strávit T lymfocyty i sousední živé nádorové buňky. Kanibalizmus je zřejmě důležitou metabolickou adaptací maligních nádorových buněk [121]. Mechanizmus, který je základem kanibalistické aktivity nádorových buněk, se do určité míry podobá fagocytóze u jednobuněčného organizmu Dictyostelium discoideum, při které je nutná účast fagocytického receptoru Phg1A. Phg1A se v jiných organizmech také nazývá transmembránový protein 9 (TM9) a patří do vysoce konzervativní rodiny transmembránových proteinů. U metastatických buněk melanomu je produkt genu TM9SF4 vyžadován při pohlcování kvasinek a živých lymfocytů a jeho inaktivace potlačuje kanibalistickou aktivitu melanomu. Nádorové buňky mohou pomocí mechanizmů zahrnujících aktin, ezrin a kaveolin 1 aktivně kanibalizovat živé buňky (např. T lymfocyty, neutrofily a NK buňky), mrtvé buňky i neživý materiál (např. umělé mikročástice) [121,122]. Některé studie předpokládají, že by kanibalizmus mohl být formou kompetice mezi nádorovými buňkami a že pohlcené buňky jsou těmi prohrávajícími. Identita pohlcujících („vítězných“) a pohlcených („poražených“) buněk může být diktována aktivitou onkogenu KRAS a mechanickou tuhostí, kdy „měkčí“ nádorové buňky přednostně pohlcují sousední „rigidnější“ buňky [123]. Inhibice ezrinu, kaveolinu či katepsinu B vedla k podstatné inhibici kanibalistické aktivity nádorových buněk [124,125]. Pohlcení a smrt entotických buněk byly významně potlačeny inhibitorem Y27632 [123].
Závěr
Nádorové buňky akumulují metabolické změny, které jim umožňují získat přístup ke konvenčním i méně konvenčním zdrojům živin. Tyto živiny následně využívají k vytváření nové biomasy a k udržení nadměrné proliferace. Při metastazování a kolonizaci vzdálených orgánů čelí nádorové buňky rychle se měnícím podmínkám vč. změn v dostupnosti různých nutrientů. Buňky, které vykazují vyšší metabolickou flexibilitu a mají schopnost provádět glykolýzu i oxidační fosforylaci, mají většinou vyšší fitness.
Některé z meziproduktů nádorového metabolizmu mohou výrazně ovlivňovat výsledný fenotyp samotných nádorových buněk i dalších buněk přítomných v mikroprostředí nádoru. Nejčastější rysy nádorového metabolizmu zahrnují: (1) deregulovanou absorpci glukózy, acetátu, citrátu a některých aminokyselin; (2) použití produktů glykolýzy, fermentace či meziproduktů Krebsova cyklu pro biosyntézu, produkci NADPH a regeneraci NAD+; (3) zvýšenou poptávku po dusíku; (4) epigenetické změny zapříčiněné akumulací onkogenních metabolitů; (5) vznik metabolické symbiózy a využívání metabolitů k manipulaci TME; (6) použití oportunistických způsobů získávání živin. Zatímco některé nádory vykazují všechny výše zmiňované znaky, mnoho typů nádorů jich exprimuje pouze několik. Specifické metabolické znaky, které vykazují jednotlivé typy nádorů, mohou přispět k lepší klasifikaci nádoru a pomoci při výběru vhodné léčby.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential confl icts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Obdrženo/Submitted: 21. 5. 2021
Přijato/Accepted: 18. 10. 2021
prof. RNDr. Michal Masařík, Ph.D.
Ústav patologické fyziologie LF MU
Kamenice 5
625 00 Brno
e-mail: masarik@med.muni.cz
Sources
1. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016; 23 (1): 27–47. doi: 10.1016/j.cmet.2015.12.006.
2. Bradley J, Swann K. Mitochondria and lipid metabolism in mammalian oocytes and early embryos. Int J Dev Biol 2019; 63 (3–4–5): 93–103. doi: 10.1387/ijdb.180355ks.
3. Johnson MT, Mahmood S, Patel MS. Intermediary metabolism and energetics during murine early embryogenesis. J Biol Chem 2003; 278 (34): 31457–31460. doi: 10.1074/jbc.R300002200.
4. Folmes CDL, Dzeja PP, Nelson TJ et al. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012; 11 (5): 596–606. doi: 10.1016/j.stem.2012.10.002.
5. Warburg O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924; 12 (50): 1131–1137.
6. Warburg O. On respiratory impairment in cancer cells. Science 1956; 124 (3215): 269–270.
7. Tan AS, Baty JW, Dong LF et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 2015; 21 (1): 81–94. doi: 10.1016/j.cmet.2014.12.003.
8. Wallace DC. Mitochondria and cancer. Nat Rev Cancer 2012; 12 (10): 685–698. doi: 10.1038/nrc3365.
9. Ju YS, Alexandrov LB, Gerstung M et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014; 3: e02935. doi: 10.7554/eLife.02935.
10. Bajzikova M, Kovarova J, Coelho AR et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab 2019; 29 (2): 399–416.e10. doi: 10.1016/j.cmet.2018.10.014.
11. Huang CT, Huang DY, Hu CJ et al. Energy adaptive response during parthanatos is enhanced by PD98059 and involves mitochondrial function but not autophagy induction. Biochim Biophys Acta 2014; 1843 (3): 531–543. doi: 10.1016/j.bbamcr.2013.12.001.
12. Naguib A, Mathew G, Reczek CR et al. Mitochondrial complex I inhibitors expose a vulnerability for selective killing of pten-null cells. Cell Rep 2018; 23 (1): 58–67. doi: 10.1016/j.celrep.2018.03.032.
13. Luengo A, Li Z, Gui DY et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol Cell 2021; 81 (4): 691–707. doi: 10.1016/j.molcel.2020.12. 012.
14. Li W, Zhang C, Jackson K et al. UCP2 knockout suppresses mouse skin carcinogenesis. Cancer Prev Res (Phila) 2015; 8 (6): 487–491. doi: 10.1158/1940-6207.CAPR-14-0297-T.
15. Kuai XY, Ji ZY, Zhang HJ. Mitochondrial uncoupling protein 2 expression in colon cancer and its clinical significance. World J Gastroenterol 2010; 16 (45): 5773–5778. doi: 10.3748/wjg.v16.i45.5773.
16. Ayyasamy V, Owens KM, Desouki MM et al. Cellular model of Warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS One 2011; 6 (9): e24792. doi: 10.1371/journal.pone.0024792.
17. Baffy G. Mitochondrial uncoupling in cancer cells: liabilities and opportunities. Biochim Biophys Acta Bioenerg 2017; 1858 (8): 655–664. doi: 10.1016/j.bbabio.2017.01.005.
18. Stacpoole PW. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst 2017; 109 (11). doi: 10.1093/jnci/djx071.
19. Herst PM, Berridge MV. Plasma membrane electron transport: a new target for cancer drug development. Curr Mol Med 2006; 6 (8): 895–904. doi: 10.2174/156652406779010777.
20. Kernstine KH, Faubert B, Do QN et al. Does tumor FDG-PET avidity represent enhanced glycolytic metabolism in non-small cell lung cancer? Ann Thorac Surg 2020; 109 (4): 1019–1025. doi: 10.1016/j.athoracsur.2019.10. 061.
21. Desai S, Ding M, Wang B et al. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget 2014; 5 (18): 8202–8210. doi: 10.18632/oncotarget.1159.
22. Lunt SY, Muralidhar V, Hosios AM et al. Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol Cell 2015; 57 (1): 95–107. doi: 10.1016/j.molcel.2014.10.027.
23. Christofk HR, Vander Heiden MG, Harris MH et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008; 452 (7184): 230–233. doi: 10.1038/nature06734.
24. Mazurek S, Boschek CB, Hugo F et al. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol 2005; 15 (4): 300–308. doi: 10.1016/j.semcancer.2005.04.009.
25. Tong W-H, Rouault TA. Metabolic regulation of citrate and iron by aconitases: role of iron–sulfur cluster biogenesis. Biometals 2007; 20 (3): 549–564. doi: 10.1007/s10534-006-9047-6.
26. Peters JM. Flipping a citrate switch on liver cancer cells. J Biol Chem 2017; 292 (33): 13902–13903. doi: 10.1074/jbc.H117.783860.
27. Moreno M, Ortega F, Xifra G et al. Cytosolic aconitase activity sustains adipogenic capacity of adipose tissue connecting iron metabolism and adipogenesis. FASEB J 2015; 29 (4): 1529–1539. doi: 10.1096/fj.14-258996.
28. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther 2020; 5 (1): 108. doi: 10.1038/s41392-020-00216-5.
29. Shimada K, Hayano M, Pagano NC et al. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol 2016; 23 (2): 225–235. doi: 10.1016/j.chembiol.2015.11.016.
30. Stone SC, Rossetti RAM, Alvarez KLF et al. Lactate secreted by cervical cancer cells modulates macrophage phenotype. J Leukoc Biol 2019; 105 (5): 1041–1054. doi: 10.1002/JLB.3A0718-274RR.
31. Fischer K, Hoffmann P, Voelkl S et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007; 109 (9): 3812–3819. doi: 10.1182/blood-2006-07-035972.
32. Watson MJ, Vignali PDA, Mullett SJ et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021; 591 (7851): 645–651. doi: 10.1038/s41586-020-03045-2.
33. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver‘s seat. Nature Rev Cancer 2017; 17 (10): 577–593. doi: 10.1038/nrc.2017.77.
34. Khacho M, Tarabay M, Patten D et al. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nature Commun 2014; 5 : 3550. doi: 10.1038/ncomms4550.
35. Faubert B, Li KY, Cai L et al. Lactate metabolism in human lung tumors. Cell 2017; 171 (2): 358–371.e9. doi: 10.1016/j.cell.2017.09.019.
36. Damaghi M, Tafreshi NK, Lloyd MC et al. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nature Commun 2015; 6 : 8752. doi: 10.1038/ncomms9752.
37. Siska P, Singer K, Evert K et al. The immunological Warburg effect: can a metabolic-tumor-stroma score (MeTS) guide cancer immunotherapy? Immunol Rev 2020; 295 (1): 187–202. doi: 10.1111/imr.12846.
38. Chen X-S, Li L-Y, Guan Y-D et al. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin 2016; 37 (8): 1013–1019. doi: 10.1038/aps.2016.47.
39. Ganapathy-Kanniappan S, Kunjithapatham R, Geschwind JF. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: specific molecular targeting. Anticancer Res 2013; 33 (1): 13–20.
40. Amadori D, Frassineti GL, De Matteis A et al. Modulating effect of lonidamine on response to doxorubicin in metastatic breast cancer patients: results from a multicenter prospective randomized trial. Breast Cancer Res Treat 1998; 49 (3): 209–217. doi: 10.1023/a: 1006063412 726.
41. Nath K, Nelson DS, Heitjan DF et al. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed 2015; 28 (3): 281–290. doi: 10.1002/nbm.3240.
42. Zhang D, Li J, Wang F et al. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett 2014; 355 (2): 176–183. doi: 10.1016/j.canlet.2014.09.003.
43. Mohanti BK, Rath GK, Anantha N et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: phase I/II clinical trials on human cerebral gliomas. Int J Radiat Oncol Biol Phys 1996; 35 (1): 103–111. doi: 10.1016/s0360-3016 (96) 85017-6.
44. Mazur L, Opydo-Chanek M, Stojak M. Glufosfamide as a new oxazaphosphorine anticancer agent. Anticancer Drugs 2011; 22 (6): 488–493. doi: 10.1097/ CAD.0b013e328345e1e0.
45. Dyshlovoy SA, Pelageev DN, Hauschild J et al. Successful targeting of the Warburg effect in prostate cancer by glucose-conjugated 1,4-naphthoquinones. Cancers 2019; 11 (11): 1690. doi: 10.3390/cancers11111690.
46. Narayanan K, Erathodiyil N, Gopalan B et al. Targeting Warburg effect in cancers with PEGylated glucose. Adv Healthc Mater 2016; 5 (6): 696–701. doi: 10.1002/adhm.201500613.
47. Patra M, Johnstone TC, Suntharalingam K et al. A potent glucose–platinum conjugate exploits glucose transporters and preferentially accumulates in cancer cells. Angew Chem Int Ed Engl 2016; 55 (7): 2550–2554. doi: 10.1002/anie.201510551.
48. Woźniak M, Pastuch-Gawołek G, Makuch S et al. In vitro and in vivo efficacy of a novel glucose–methotrexate conjugate in targeted cancer treatment. Int J Mol Sci 2021; 22 (4): 1748. doi: 10.3390/ijms22041748.
49. Nishie H, Kataoka H, Yano S et al. Excellent antitumor effects for gastrointestinal cancers using photodynamic therapy with a novel glucose conjugated chlorin e6. Biochem Biophys Res Commun 2018; 496 (4): 1204–1209. doi: 10.1016/j.bbrc.2018.01.171.
50. Liu J, Wu N, Ma L et al. Oleanolic acid suppresses aerobic glycolysis in cancer cells by switching pyruvate kinase type M isoforms. PLoS One 2014; 9 (3): e91606. doi: 10.1371/journal.pone.0091606.
51. Urbańska K, Orzechowski A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int J Mol Sci 2019; 20 (9): 2085. doi: 10.3390/ijms20092085.
52. Granchi C, Paterni I, Rani R et al. Small-molecule inhibitors of human LDH5. Future Med Chem 2013; 5 (16): 1967–1991. doi: 10.4155/fmc.13.151.
53. Kim E-Y, Chung T-W, Han CW et al. A novel lactate dehydrogenase inhibitor, 1 - (phenylseleno) -4 - (trifluoromethyl) benzene, suppresses tumor growth through apoptotic cell death. Sci Rep 2019; 9 (1): 3969. doi: 10.1038/s41598-019-40617-3.
54. Le A, Cooper CR, Gouw AM et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010; 107 (5): 2037–2042. doi: 10.1073/pnas.0914433107.
55. Hatzivassiliou G, Zhao F, Bauer DE et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005; 8 (4): 311–321. doi: 10.1016/j.ccr.2005.09. 008.
56. Granchi C. ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem 2018; 157 : 1276–1291. doi: 10.1016/j.ejmech.2018.09.001.
57. Lyssiotis CA, Cantley LC. Acetate fuels the cancer engine. Cell 2014; 159 (7): 1492–1494. doi: 10.1016/ j.cell.2014.12.009.
58. Wise DR, Ward PS, Shay JE et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of a-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 2011; 108 (49): 19611–19616. doi: 10.1073/pnas.1117773108.
59. Davidson SM, Papagiannakopoulos T, Olenchock BA et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab 2016; 23 (3): 517–528. doi: 10.1016/j.cmet.2016.01.007.
60. Mycielska ME, Dettmer K, Rümmele P et al. Extracellular citrate affects critical elements of cancer cell metabolism and supports cancer development in vivo. Cancer Res 2018; 78 (10): 2513–2523. doi: 10.1158/0008-5472.CAN-17-2959.
61. Marin-Valencia I, Yang C, Mashimo T et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 2012; 15 (6): 827–837. doi: 10.1016/j.cmet.2012.05.001.
62. Drexler K, Schmidt KM, Jordan K et al. Cancer-associated cells release citrate to support tumour metastatic progression. Life Sci Alliance 2021; 4 (6): e202000903. doi: 10.26508/lsa.202000903.
63. Raudenská M, Svobodová M, Gumulec J et al. The importance of cancer-associated fibroblasts in the pathogenesis of head and neck cancers. Klin Onkol 2020; 33 (1): 39–48. doi: 10.14735/amko202039.
64. Tasdogan A, Faubert B, Ramesh V et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020; 577 (7788): 115–120. doi: 10.1038/s41586-019-1847-2.
65. Diehl FF, Lewis CA, Fiske BP et al. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nature Metabolism 2019; 1 (9): 861–867. doi: 10.1038/s42255-019-0108-x.
66. Locasale JW, Grassian AR, Melman T et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature Genetics 2011; 43 (9): 869–874. doi: 10.1038/ng.890.
67. Reid MA, Allen AE, Liu S et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nature Commun 2018; 9 (1): 5442–5442. doi: 10.1038/s41467-018-07868-6.
68. Possemato R, Marks KM, Shaul YD et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011; 476 (7360): 346–350. doi: 10.1038/nature10350.
69. Wei X, Shi J, Lin Q et al. Targeting ACLY attenuates tumor growth and acquired cisplatin resistance in ovarian cancer by inhibiting the PI3K–AKT pathway and activating the AMPK–ROS pathway. Front Oncol 2021; 11 : 642229. doi: 10.3389/fonc.2021.642229.
70. Koerner SK, Hanai J-I, Bai S et al. Design and synthesis of emodin derivatives as novel inhibitors of ATP-citrate lyase. Eur J Med Chem 2017; 126 : 920–928. doi: 10.1016/j.ejmech.2016.12.018.
71. Granchi C. ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem 2018; 157 : 1276–1291. doi: 10.1016/j.ejmech.2018.09.001.
72. Ki SW, Ishigami K, Kitahara T et al. Radicicol binds and inhibits mammalian ATP citrate lyase. J Biol Chem 2000; 275 (50): 39231–39236. doi: 10.1074/jbc.M006192200.
73. Shibata S, Sogabe S, Miwa M et al. Identification of the first highly selective inhibitor of human lactate dehydrogenase B. Sci Rep 2021; 11 (1): 21353. doi: 10.1038/s41598-021-00820-7.
74. Fiume L, Manerba M, Vettraino M et al. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med Chem 2014; 6 (4): 429–445. doi: 10.4155/fmc.13.206.
75. Benjamin D, Robay D, Hindupur SK et al. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep 2018; 25 (11): 3047–3058.e4. doi: 10.1016/j.celrep.2018.11.043.
76. Corbet C, Bastien E, Draoui N et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat Commun 2018; 9 (1): 1208. doi: 10.1038/s41467-018-03525-0.
77. Mullarky E, Xu J, Robin AD et al. Inhibition of 3-phosphoglycerate dehydrogenase (PHGDH) by indole amides abrogates de novo serine synthesis in cancer cells. Bioorg Med Chem Lett 2019; 29 (17): 2503–2510. doi: 10.1016/j.bmcl.2019.07.011.
78. Zhao J-Y, Feng K-R, Wang F et al. A retrospective overview of PHGDH and its inhibitors for regulating cancer metabolism. Eur J Med Chem 2021; 217 : 113379. doi: 10.1016/j.ejmech.2021.113379.
79. Nilsson A, Haanstra JR, Engqvist M et al. Quantitative analysis of amino acid metabolism in liver cancer links glutamate excretion to nucleotide synthesis. Proc Nat Acad Sci U S A 2020; 117 (19): 10294–10304. doi: 10.1073/pnas.1919250117.
80. Kodama M, Oshikawa K, Shimizu H et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat Commun 2020; 11 (1): 1320. doi: 10.1038/s41467-020-15136-9.
81. Yoo HC, Yu YC, Sung Y et al. Glutamine reliance in cell metabolism. Exp Mol Med 2020; 52 (9): 1496–1516. doi: 10.1038/s12276-020-00504-8.
82. Yoo HC, Park SJ, Nam M et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab 2020; 31 (2): 267–283.e12. doi: 10.1016/j.cmet.2019.11.020.
83. Stine ZE, Dang CV. Glutamine skipping the Q into mitochondria. Trends Mol Med 2020; 26 (1): 6–7. doi: 10.1016/j.molmed.2019.11.004.
84. Sayin VI, LeBoeuf SE, Singh SX et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 2017; 6: e28083. doi: 10.7554/eLife.28083.
85. Murphy TH, Miyamoto M, Sastre A et al. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989; 2 (6): 1547–1558. doi: 10.1016/0896-6273 (89) 90043-3.
86. Gao M, Monian P, Quadri N et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 2015; 59 (2): 298–308. doi: 10.1016/j.molcel.2015.06.011.
87. Yang L, Achreja A, Yeung TL et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab 2016; 24 (5): 685–700. doi: 10.1016/j.cmet.2016.10.011.
88. Palmieri EM, Menga A, Martín-Pérez R et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep 2017; 20 (7): 1654–1666. doi: 10.1016/j.celrep.2017.07.054.
89. Wang Z, Liu F, Fan N et al. Targeting glutaminolysis: new perspectives to understand cancer development and novel strategies for potential target therapies. Front Oncol 2020; 10 : 589508. doi: 10.3389/fonc.2020.589 508.
90. Lemberg KM, Vornov JJ, Rais R et al. We’re not „DON“ yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Mol Cancer Ther 2018; 17 (9): 1824–1832. doi: 10.1158/1535-7163.MCT-17-1148.
91. Xu X, Meng Y, Li L et al. Overview of the development of glutaminase inhibitors: achievements and future directions. J Med Chem 2019; 62 (3): 1096–1115. doi: 10.1021/acs.jmedchem.8b00961.
92. Marshall AD, van Geldermalsen M, Otte NJ et al. ASCT2 regulates glutamine uptake and cell growth in endometrial carcinoma. Oncogenesis 2017; 6 (7): e367–e367. doi: 10.1038/oncsis.2017.70.
93. Berger RS, Ellmann L, Reinders J et al. Degradation of D-2-hydroxyglutarate in the presence of isocitrate dehydrogenase mutations. Sci Rep 2019; 9 (1): 7436. doi: 10.1038/s41598-019-43891-3.
94. Figueroa ME, Abdel-Wahab O, Lu C et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18 (6): 553–567. doi: 10.1016/j.ccr.2010.11.015.
95. Sulkowski PL, Corso CD, Robinson ND et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 2017; 9 (375): eaal2463. doi: 10.1126/scitranslmed.aal2463.
96. Seok J, Yoon SH, Lee SH et al. The oncometabolite d‑2‑hydroxyglutarate induces angiogenic activity through the vascular endothelial growth factor receptor 2 signaling pathway. Int J Oncol 2019; 54 (2): 753–763. doi: 10.3892/ijo.2018.4649.
97. Wang T-X, Liang J-Y, Zhang C et al. The oncometabolite 2-hydroxyglutarate produced by mutant IDH1 sensitizes cells to ferroptosis. Cell Death Dis 2019; 10 (10): 755. doi: 10.1038/s41419-019-1984-4.
98. Intlekofer AM, Wang B, Liu H et al. L-2-hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol 2017; 13 (5): 494–500. doi: 10.1038/nchembio.2307.
99. Shim EH, Livi CB, Rakheja D et al. L-2-hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 2014; 4 (11): 1290–1298. doi: 10.1158/2159-8290.CD-13-0696.
100. Krejčíř R, Valík D Vojtěšek B. Mitochondrial processes in targeted cancer therapy. Klin Onkol 2018; 31 (Suppl 2): 14–20. doi: 10.14735/amko20182S14.
101. Oermann EK, Wu J, Guan K-L et al. Alterations of metabolic genes and metabolites in cancer. Semin Cell Dev Biol 2012; 23 (4): 370–380. doi: 10.1016/j.semcdb.2012.01.013.
102. Mu X, Zhao T, Xu C et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 2017; 8 (8): 13174–13185. doi: 10.18632/oncotarget.14485.
103. Terra X, Ceperuelo-Mallafré V, Merma C et al. Succinate pathway in head and neck squamous cell carcinoma: potential as a diagnostic and prognostic marker. Cancers 2021; 13 (7): 1653. doi: 10.3390/cancers13071 653.
104. Ko SH, Choi GE, Oh JY et al. Succinate promotes stem cell migration through the GPR91-dependent regulation of DRP1-mediated mitochondrial fission. Sci Rep 2017; 7 (1): 12582. doi: 10.1038/s41598-017-12692-x.
105. Ortiz-Masia D, Gisbert Ferrándiz L, Bauset C et al. Succinate activates EMT in intestinal epithelial cells through SUCNR1: a novel protagonist in fistula development. Cells 2020; 9 (5): 1104. doi: 10.3390/cells9051 104.
106. Xiao M, Yang H, Xu W et al. Inhibition of a-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 2012; 26 (12): 1326–1338. doi: 10.1101/gad.191056.112.
107. Tseng PL, Wu WH, Hu TH et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci Rep 2018; 8 (1): 3081. doi: 10.1038/s41598-018-21361-6.
108. Mills E, O’Neill LAJ. Succinate: a metabolic signal in inflammation. Trends Cell Biol 2014; 24 (5): 313–320. doi: 10.1016/j.tcb.2013.11.008.
109. Sciacovelli M, Frezza C. Oncometabolites: unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med 2016; 100 : 175–181. doi: 10.1016/j.freeradbiomed.2016.04.025.
110. Schmidt C, Sciacovelli M, Frezza C. Fumarate hydratase in cancer: a multifaceted tumour suppressor. Semin Cell Dev Biol 2020; 98 : 15–25. doi: 10.1016/j.semcdb.2019.05.002.
111. Sourbier C, Ricketts CJ, Matsumoto S et al. Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell 2014; 26 (6): 840–850. doi: 10.1016/j.ccell.2014.10.005.
112. Kerins MJ, Vashisht AA, Liang BX et al. Fumarate mediates a chronic proliferative signal in fumarate hydratase-inactivated cancer cells by increasing transcription and translation of ferritin genes. Mol Cell Biol 2017; 37 (11): e00079-17. doi: 10.1128/MCB.00079-17.
113. You X, Tian J, Zhang H et al. Loss of mitochondrial aconitase promotes colorectal cancer progression via SCD1-mediated lipid remodeling. Mol Metab 2021; 48 : 101203. doi: 10.1016/j.molmet.2021.101203.
114. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020; 369 (6511): 1633–1637. doi: 10.1126/science.abb9818.
115. Wang M, Chen X, Zhang Y. Biological functions of gasdermins in cancer: from molecular mechanisms to therapeutic potential. Front Cell Dev Biol 2021; 9 : 638710. doi: 10.3389/fcell.2021.638710.
116. Golub D, Iyengar N, Dogra S et al. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol 2019; 9 : 417. doi: 10.3389/fonc.2019.00 417.
117. Naik R, Ban HS, Jang K et al. Methyl 3 - (3 - (4 - (2,4,4-trimethylpentan-2-yl) phenoxy) -propanamido) benzoate as a novel and sual malate sehydrogenase (MDH) 1/2 inhibitor targeting cancer metabolism. J Med Chem 2017; 60 (20): 8631–8646. doi: 10.1021/acs.jmedchem.7b01231.
118. Romero RA, Lunev S, Popowicz GM et al. A fragment-based approach identifies an allosteric pocket that impacts malate dehydrogenase activity. Commun Biol 2021; 4 (1): 949. doi: 10.1038/s42003-021-02442-1.
119. Trigos AS, Pearson RB, Papenfuss AT et al. How the evolution of multicellularity set the stage for cancer. Br J Cancer 2018; 118 (2): 145–152. doi: 10.1038/bjc.2017.398.
120. Yona AH, Manor YS, Herbst RH et al. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci U S A 2012; 109 (51): 21010–21015. doi: 10.1073/pnas.1211150109.
121. Fais S, Overholtzer M. Cell-in-cell phenomena in cancer. Nat Rev Cancer 2018; 18 (12): 758–766. doi: 10.1038/s41568-018-0073-9.
122. Lozupone F, Fais S. Cancer cell cannibalism: a primeval option to survive. Curr Mol Med 2015; 15 (9): 836–841. doi: 10.2174/1566524015666151026100916.
123. Sun Q, Luo T, Ren Y et al. Competition between human cells by entosis. Cell Res 2014; 24 (11): 1299–1310. doi: 10.1038/cr.2014.138.
124. Lugini L, Matarrese P, Tinari A et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res 2006; 66 (7): 3629–3638. doi: 10.1158/0008-5472.CAN-05-3204.
125. Sharma V, Singh SK. Retrospective analysis of arthrodesis from various options after anterior cervical discectomy. J Neurosci Rural Pract 2018; 9 (1): 14–18. doi: 10.4103/jnrp.jnrp_366_17.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2022 Issue 3
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Meigs’ syndrome
- Analysis of the results of radiotherapy and chemoradiotherapy on the background of immunotherapy of patients with cancer of the oral cavity and oropharynx
- Changes of serum protein N-glycosylation in cancer
- Metabolic plasticity of cancer cells