
MRI zobrazení mozku u pacientů s myotonickou dystrofií DM 1
The Brain MR Imaging in Patients with Myotonic Dystrophy DM 1
Myotonic dystrophies (DM 1 and DM 2) are multisystem disorders with autosomal dominant heredity manifested particularly with muscular weakness, myotonia, cataract, cardiac transmission disturbances and cardiomyopathy. In literature there are given various changes in pictures of magnetic resonance (MRI) of the brain in patients with myotonic dystrophy. A set of 13 patients with DM 1 demonstrated glial changes in the perivascular and deep white matter of the parietal, occipital and frontal lobes (with increasing frequency) in the total of 11 subjects (84%), moreover, even glial changes in the subcortical white matter of the frontal parts of temporal lobes were seen in 8 patients (62%), the cerebral atrophy was described in 8 subjects (62%). Nine patients (69%) showed striking width of Virchow-Robin´s perivascular spaces. The cranium was dilated in 3 patients (23%). Two patients (15%) had a quite normal finding in the MRI picture. The study has demonstrated that MRI of the head and brain in patients with myotonic dystrophy DM 1 shows various frequency of structural and signal changes occurring particularly in the white matter and they are in themselves non-specific. Their cumulation in characteristic locations has confirmed clinical diagnosis of myotonic dystrophy. A negative finding in the brain MRI does not eliminate the disease.
Key words:
magnetic resonance – myotonic dystrophy – diagnostic imaging
:
T. Belšan 1,5; J. Kraus 2; R. Mazanec 3; Z. Mušová 4; Á. Bóday 4; T. Maříková 4; M. Kynčl 1
:
Klinika zobrazovacích metod 2. lékařská fakulta UK a FN v Motole, Praha
přednosta: prof. MUDr. Jiří Neuwirth, CSc.
1; Klinika dětské neurologie 2. lékařská fakulta UK a FN v Motole, Praha
přednosta: doc. MUDr. Vladimír Komárek, CSc.
2; Neurologická klinika 2. lékařská fakulta UK a FN v Motole, Praha
přednosta: doc. MUDr. Martin Bojar, CSc.
3; Ústav biologie a lékařské genetiky 2. lékařská fakulta UK a FN v Motole, Praha
přednosta: prof. MUDr. Petr Goetz, CSc.
4; Radiodiagnostické oddělení ÚVN, Praha
primář: MUDr. František Charvát
5
:
Cesk Slov Neurol N 2007; 70/103(3): 266-271
:
Short Communication
Práce byla podpořena grantem IGA MZ ČR 8052-3, a výzkumnými záměry VZ FNM č. 00000064203 a MZO 00064203.
Myotonické dystrofie (DM 1 a DM 2) jsou multisystémová onemocnění s autozomálně dominantní dědičností projevující se především svalovou slabostí, myotonií, kataraktou, srdečními převodními poruchami a kardiomyopatií. V literatuře jsou uváděny různé změny na obrazech magnetické rezonance (MRI) mozku u pacientů s myotonickou dystrofií. V souboru 13 pacientů s DM 1 autoři prokázali gliové změny v periventrikulární a hluboké bílé hmotě parietálních, okcipitálních a frontálních laloků (se sestupnou četností) celkem u 11 pacientů (84 %). U 8 pacientů (62 %) se navíc zobrazily i gliové změny v subkortikální bílé hmotě předních částí temporálních laloků. U 8 pacientů (62 %) byla popsána atrofie mozku. U 9 pacientů (69 %) byla nápadnější šíře Virchowových-Robinových perivaskulárních prostorů. U 3 pacientů (23 %) bylo prokázáno rozšíření kalvy; 2 pacienti (15%) měli zcela normální nález v MRI obraze. Studie prokazuje, že v MRI obrazech hlavy a mozku pacientů s myotonickou dystrofií DM 1 se s různou četností vyskytují strukturální a signálové změny především v bílé hmotě, které jsou samy o sobě nespecifické. Jejich kumulace v charakteristických lokalitách podporuje klinickou diagnózu myotonické dystrofie. Negativní nález v MRI obraze mozku onemocnění nevylučuje.
Klíčová slova:
magnetická rezonance – myotonická dystrofie – diagnostické zobrazení
Úvod
Myotonické dystrofie (DM 1, DM 2 a DM 3) jsou multisystémová onemocnění s autozomálně dominantní dědičností a s variabilním klinickým fenotypem. Patří k nim především svalová slabost, myotonie, katarakta, srdeční defekty: převodní poruchy, kardiomyopatie. Dalšími postiženími jsou mentální změny, hypogonadizmus, testikulární atrofie, inzulin-dependentní diabetes, tyreopatie, snížení imunoglobulinů v séru a charakteristický nález při elektromyografickém vyšetření. Věk nástupu onemocnění je od narození do dospělosti. U pacientů s DM 1 (88 %) je mutace v genu lokalizovaném na devatenáctém chromozomu v oblasti 19q13.3. Mutace spočívá v amplifikaci repetitivních tripletů CTG na 3´ - konci UTR genu DMPK (DM-protein kinázy). DM 1 je nejčastější svalovou dystrofií dospělého věku s incidencí 1 : 8000. Je pro ní typická anticipace - v následujících generacích se objevují první symptomy v mladším věku a postižení je těžší. U dětí postižených matek se často objevuje těžká kongenitální forma. U části pacientů se sporadickou proximální formou DM2 je mutace na třetím chromozomu v oblasti 3q21 s expanzí CCTG v ZNF9 genu. Nově ohraničená DM3 s frontotemporální demencí se mapuje k lokusu 15q21-24.
V literatuře jsou uváděny vícečetné změny v MRI obrazech mozků u pacientů s myotonickou dystrofií typu 1 se začátkem v dospělosti (dystrophia myotonica typ 1 – DM 1). Jako nejčastější nález jsou popisovány oblasti zvýšeného signálu v T2 vážených obrazech [1, 2,3,4,5,6,7]. Tyto signálové změny jsou ještě nápadnější v sekvenci FLAIR (fluid low-attenuated inversion recovery). Použití sekvence FLAIR však v porovnání s T2 váženými obrazy neumožňuje detekci většího počtu oblastí změněného signálu v bílé hmotě [8). Ložiska změněného signálu mohou být lokalizovaná kdekoli v bílé hmotě mozkových hemisfér. Vyskytují se v periventrikulární či v hluboké bílé hmotě parietálních, okcipitálních a frontálních laloků (se sestupnou četností). Jinou obvyklou lokalizací je subkortikální bílá hmota ventrálních částí temporálních laloků [1,4]. Nález signálových změn v bílé hmotě mozku je popisován přibližně u 70% pacientů s myotonickou dystrofií [1,4]. Patologické nálezy uvádějí závažnou redukci a poruchu uspořádání myelinových pochev axonů a heterotopické neurony v bílé hmotě předních částí temporálních laloků.
Popisovány jsou rozšířené Virchowovy-Robinovy (V-R) perivaskulární prostory v bílé hmotě na konvexitách mozkových hemisfér [2,5], především v počátečních stadiích choroby. Změny v šíři Virchowých-Robinových perivaskulárních prostorů v oblasti bazálních ganglií se dle literárních údajů neliší mezi skupinou pacientů s myotonickou dystrofií a kontrolní skupinou [2].
U pacientů s myotonickou dystrofií 1. typu se často vyskytuje atrofie mozku [1,3], která je výraznější, než atrofie mozku která je patrná u pacientů s příbuznou chorobou – proximální myotonickou myopatií (PROMM či DM 2) [3].

U některých pacientů s DM 1 je popsáno rozšíření lebky, případně kalcifikace ve falx cerebri [1]. Zjištěno bylo prokazatelné snížení anizotropické difuze a zvýšení izotropické difuze v bílé hmotě mozku pacientů s DM 1 v porovnání s kontrolní skupinou [6]. Jedna studie italských autorů porovnává délku trvání choroby se šíři Virchowových-Robinových prostorů a četností signálových změn v centrum semiovale. Uvádějí, že s prodlužující se dobou trvání choroby regreduje rozšíření V-R perivaskulárních prostorů a progreduje četnost ložisek změněného signálu v bílé hmotě mozkových hemisfér [2].
Metodika
Do předkládané studie byli zahrnuti pacienti s klinicky diagnostikovanou a molekulárně genetickým vyšetřením potvrzenou myotonickou dystrofií typu 1 (DM 1), kteří byli klinicky vyšetřeni či kontrolováni na našem pracovišti v roce 2003 a u kterých bylo v průběhu let 2000-2003 provedeno MRI zobrazení mozku.
MRI zobrazení bylo prováděno na supravodivém přístroji Philips Gyroscan ACS 15 NT Power Track 1000 o síle magnetického pole 1,5 Tesla, který byl v roce 2002 upgradován hardwarově i softwarově na přístroj Gyroscan Intera.
Pro vyšetření bylo použito standardního protokolu (tab. 1):

T2 vážené obrazy v turbospinecho (TSE) modu v transversální rovině,
FLAIR (fluid low-attenuated inversion recovery) obrazy v IR-TSE (inversion recovery – TSE) modu v transverzální rovině,
T1 vážení obrazy v modu IR – TSE (inversion recovery – TSE) v koronární rovině.
Při vyšetření nebyla aplikována paramagnetická kontrastní látka.
Vyhodnocení MRI obrazů bylo zaměřeno především na posouzení:
Atrofie mozku – štíhlé subarachnoideální prostory a komorový systém byly posuzovány jako normální nález, rozšířené subarachnoideální prostory a/nebo rozšířené postranní komory byly posuzovány jako atrofie mozku. Subjektivní hodnocení bylo provedeno zkušeným neuroradiologem.
Rozšíření Virchowových-Robinových (V-R) perivaskulárních prostorů – posuzovány byly V-R prostory v hluboké a subkortikální bílé hmotě obou mozkových hemisfér i v oblasti bazálních ganglií. Normální šíře V-R prostorů byly popsána u těch pacientů, u nichž nebyly Virchowovy-Robinovy prostory v MRI obrazech diferencovatelné, nebo měli šíři do 1 mm. Nad touto hranicí byly V-R prostory označeny za rozšířené.

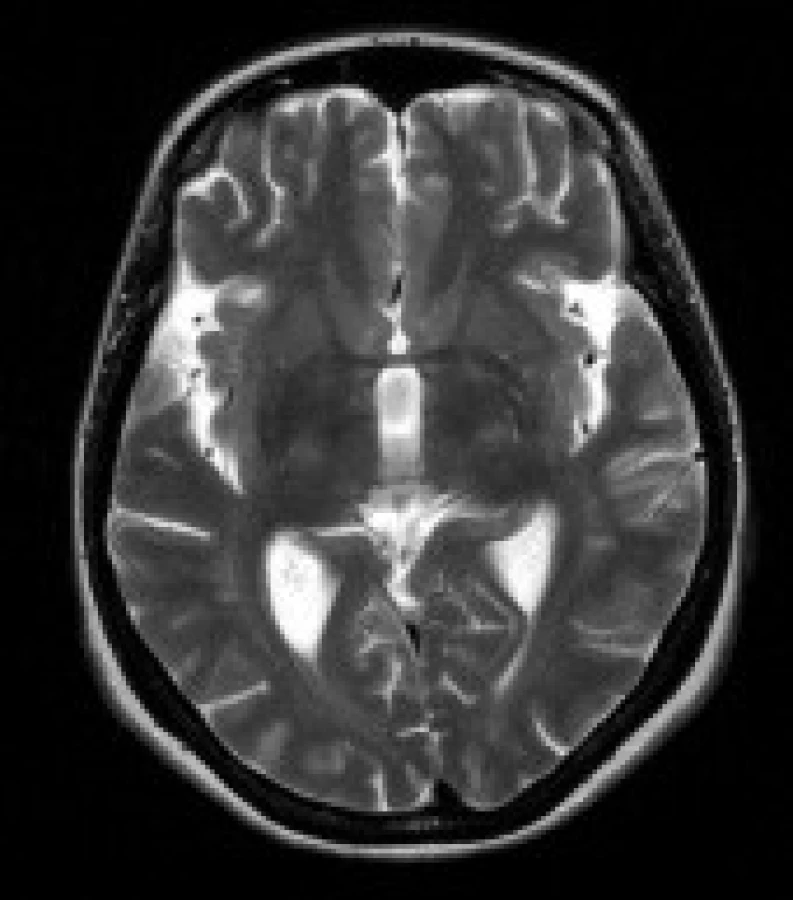
Gliové změny v bílé hmotě předních částí temporálních laloků – jako tyto léze byla označena ložiska změněného signálu v subkortikální bílé hmotě polárních částí temporálních laloků. Ložiska vykazují zvýšení signálu v T2 vážených obrazech a v sekvenci FLAIR. Ložiska mají nízký signál v T1 vážených sekvencích. Léze nemají expanzivní charakter.
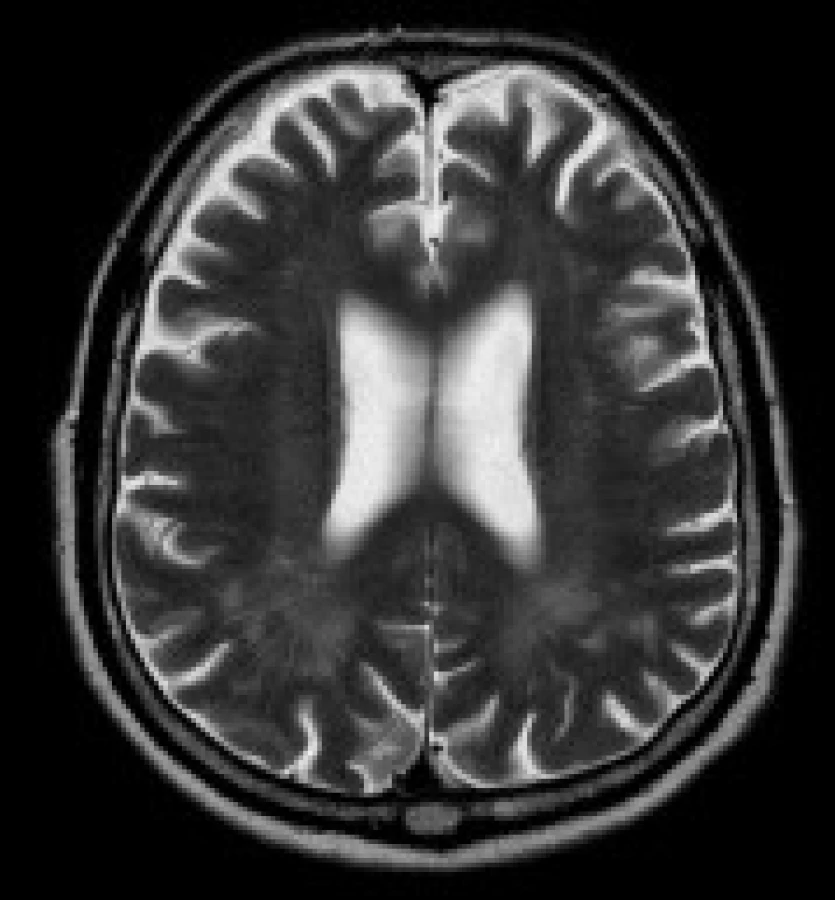
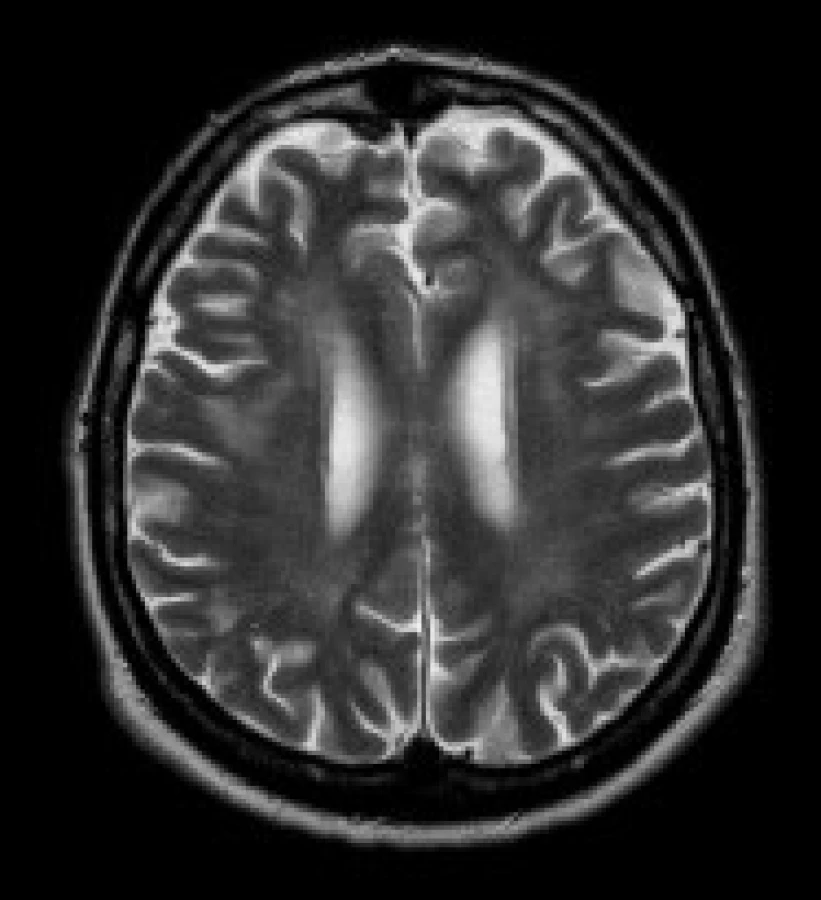
Gliové změny v periventrikulární a hluboké bílé hmotě obou mozkových hemisfér - tato ložiska jsou uložena v periventrikulární a hluboké bílé hmotě obou mozkových hemisfér, především v parietálních, okcipitálních a frontálních lalocích. Ložiska vykazují zvýšení signálu v T2 vážených obrazech a v sekvenci FLAIR. Ložiska mají nízký signál v T1 vážených sekvencích. Léze nemají expanzivní charakter, jejich ohraničení je neostré.
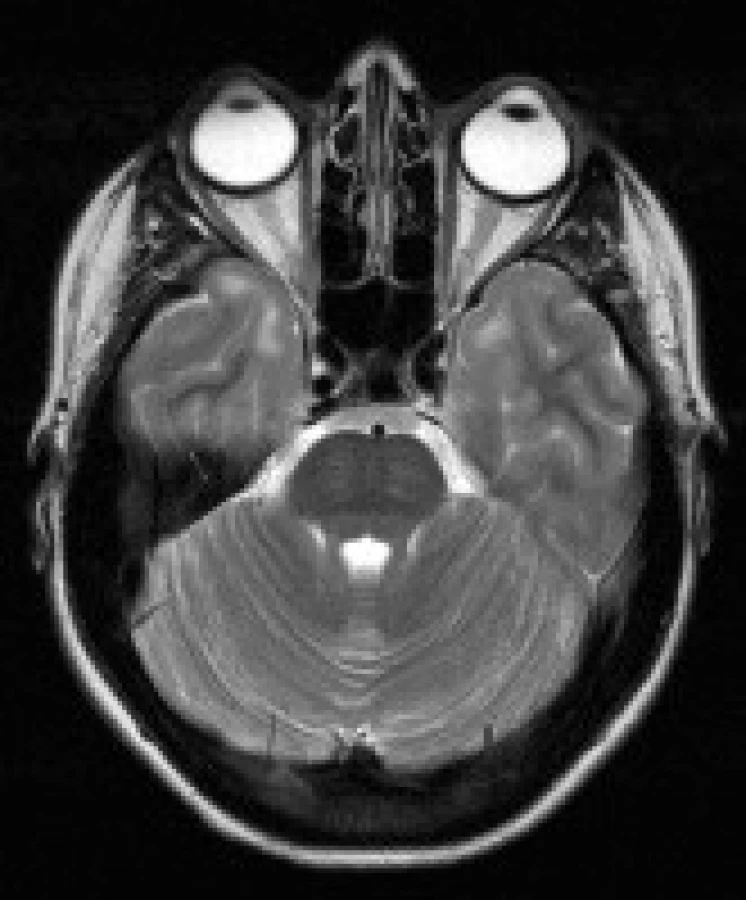
Šíře kalvy – byla posuzována šíře kalvy a hladkost kontury vnitřní laminy lebečních kostí. Subjektivní hodnocení bylo provedeno zkušeným neuroradiologem.
Výsledky
Soubor vyšetřených pacientů je shrnut v tab. 2. V letech 2000-2003 bylo vyšetřeno celkem 13 pacientů s klinicky diagnostikovanou a molekulárně genetickým vyšetřením potvrzenou myotonickou dystrofií typu 1 (DM 1). Pacienti byli ve věku 18-79 let, průměrný věk byl 43,6 roků. V souboru bylo 7 mužů (průměrného věku 45 let) a 6 žen (průměrného věku 42 let).

Nálezy u jednotlivých pacientů jsou uvedeny v tab. 3. Nejčastěji se u našeho souboru pacientů vyskytovaly gliové změny v bílé hmotě – celkem u 11 pacientů (84 %). Tyto gliové změny byly lokalizovány v periventrikulární a hluboké bílé hmotě obou mozkových hemisfér, především v parietálních, okcipitálních a frontálních lalocích. U 8 pacientů (62 %) se nadto zobrazily i gliové změny v subkortikální bílé hmotě předních částí temporálních laloků. U 8 pacientů (62 %) byly rozšířené subarachnoideální prostory a/nebo postranní komory a obraz byl popsán jako atrofie mozku. U 9 pacientů (69 %) jsme prokázali nápadnější šíři Virchowových-Robinových perivaskulárních prostorů. U 3 pacientů (23 %) bylo prokázáno rozšíření kalvy. Zcela normální nález v MRI obraze měli 2 pacienti (15%).



Diskuse
Předkládaná retrospektivní studie hodnotící soubor 13 námi vyšetřených pacientů s myotonickou dystrofií pomocí magnetické rezonance se svým rozsahem neliší od velikosti souborů popisovaných v literatuře. V sestavě jsou přibližně ve stejném poměru zastoupeni muži i ženy.
U našich pacientů jsme nejčastěji prokázali signálové změny v T2 vážených obrazech a v sekvenci FLAIR v periventrikulární a hluboké bílé hmotě frontálních a parietálních laloků – celkem v 84 %. Toto číslo nevýrazně převyšuje literární údaje [1,4]. Signálové změny v subkortikální bílé hmotě ventrálních částí temporálních laloků jsme popsali u 62 % pacientů, což zcela odpovídá i publikovaným údajům [4].
Nápadnější šíři Virchowových-Robinových prostorů jsme posuzovali jak na konvexitách mozkových hemisfér, tak i v oblasti bazálních ganglií. V našem souboru jsme popsali jejich rozšíření u 69 % pacientů. Je obtížné vyjádřit plnou korespondenci s literárním údajem o tom, že v počátečních stadiích myotonické dystrofie jsou nápadněji rozšířené Virchowovy-Robinovy perivaskulární prostory a s pokračující chorobou jejich šíře regreduje [2]. Jednak jsme prokázali rozšíření V-R perivaskulárních prostorů i u starších pacientů s dlouhodobým průběhem nemoci, jednak se domníváme, že by bylo vhodné tento údaj potvrdit opakovaným vyšetřením jednotlivých pacientů v dlouhém časovém odstupu. To však nebylo předmětem naší studie.
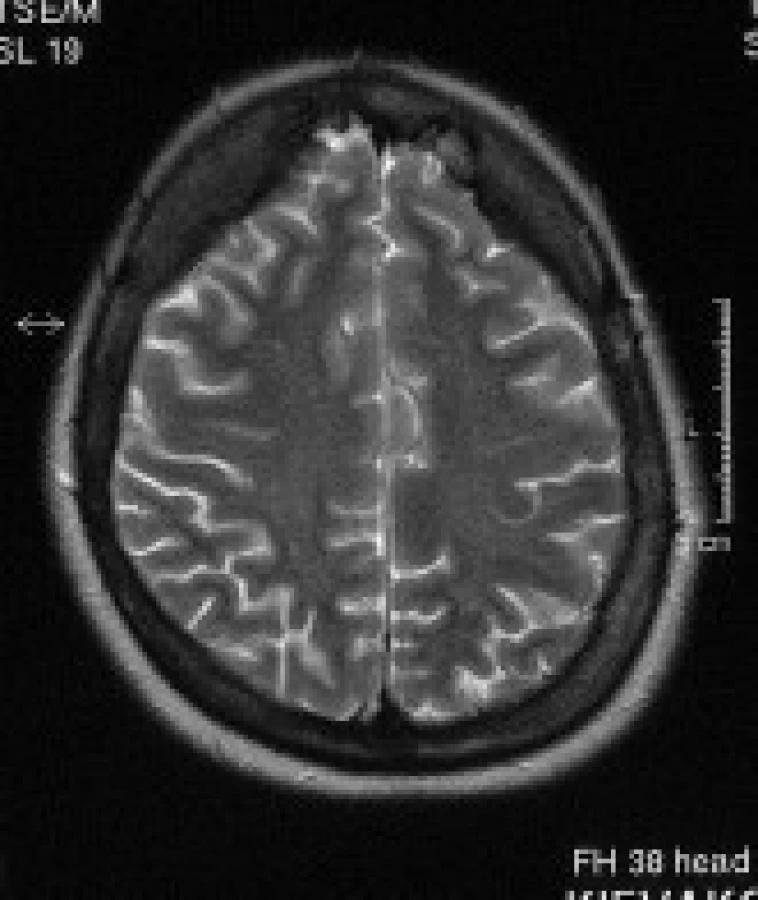
Posuzování atrofie mozku je velmi subjektivní záležitost. Brali jsme v úvahu šíři subarachnoideálních prostorů na konvexitách mozkových hemisfér a šíři subarachnoideálních prostorů v oblasti Sylvijských rýh. Hodnocení bylo prováděno neuroradiology s mnohaletou praxí. Atrofii jsme popsali u 62 % pacientů, tento údaj je ve shodě s dosud publikovanými studiemi [1,3].
Nápadné rozšíření kalvy jsme popsali u 3 pacientů (23 %). Rozšíření se týkalo především diploe. U rozšíření kalvy jsme nezjistili žádnou věkovou predilekci.
Jako vedlejší nálezy jsme ve dvou případech popsali nevelkou arachnoideální cystu – jednou ventrálně od temporálního laloku vlevo, jednou v oblasti cisterna magna. U 2 pacientů vyšší věkové kategorie jsme popsali kortikosubkortikální gliové změny po předchozí nevelké kortikální ischemii, jednou v pravém frontálním laloku, podruhé v levém parietálním laloku, vždy v povodí arteria cerebri media. Vzhledem k věku lze předpokládat, že tyto postischemické změny neměly přímou souvislost s myotonickou dystrofií 1. typu. U 1 pacienta jsme popsali v oblasti inzuly zcela neostře ohraničené signálové změny kortikálně i subkortikálně, které měli charakter hamartomu či low grade gliomu – nález nebyl dosud histologicky ověřen.
U 2 pacientů byl zcela normální nález v MRI obrazech mozku. Tato skutečnost však onemocnění nevylučuje.
Etiologie a patofyziologie abnormalit bílé hmoty je dosud nejasná. Neuropatologické nálezy uvádějí změny svědčící pro pomalu postupující proces demyelinizace [8,9]. Tuto hypotézu také podporují tendence k symetrii a splývání abnormalit [8], progresivní rozšiřování komor [10] a korelace lézí bílé hmoty s trváním choroby [5].
Neuropatologické a MRI nálezy jsou u temporálních lokalizací abnormalit odlišné. Ogata et al [4] uvádí obrazy ukazující na fokální dysplazii. Autoptické nálezy zachytily těžké ztráty a dezorganizaci myelinových pochev a axonů spolu s heterotopickými neurony v bílé hmotě anteriotemporálně.
Léze mohou zprvu zasahovat U-vlákna v anteromediální části temporálních pólů, následně U-vlákna laloků a později celou bílou hmotu pólu. Tuto hypotézu o spíše degenerativním než vývojovém původu lézí také podporuje absence temporálních abnormalit bílé hmoty při kongenitální formě DM 1 [11,12]. Temporální léze mohou být významným faktorem poruch intelektu při DM 1 [4,13].
Etiologii ani patogenezi abnormalit bílé hmoty při DM 1 dosud nepomáhá objasnit ani molekulární genetika. Při expresi mutovaného genu se tvoří inkluze ribonukleonového proteinu v jádrech svalových a jiných buněk. Přítomnost inkluzí ovlivní metabolizmus RNA a proteinů závislých na proteinkináze DM 1.
V lidském mozku při DM 1 zjistil Sergeant et al [14] dysregulaci maturace mRNA microtubule-associated proteinu tau. Prokázal tauopatii (patologie IV. třídy) lokalizovanou v hipokampu, temporálním a frontálním kortexu a v mnoha subkortikálních jádrech [14]. Patologii proteinu tau způsobuje abnormální sestřih mRNA proteinu tau, postihující izoformy obsahující exon 2 a 3.
Z rozdílů mezi nálezy lézí bílé hmoty při DM 1 a DM 2 lze předpokládat, že se na vzniku specifických abnormalit v centrálním nervovém systému podílí mutace svým specifickým účinkem. Temporopolární abnormality při MRI zobrazení, již dříve spojované s dysfunkcí intelektu, se spíše omezují na formu DM1.
Di Costanzo et al [5] se zaměřili na výskyt, rozsah a význam abnormalit bílé hmoty. Nalezli celé spektrum obrazů od normálních až po velké splývající léze. Zjištěnou velkou rozmanitost nálezů autoři vysvětlovali variabilitou fenotypu DM 1. Uvádějí, že na chybění korelace mezi velikostí expandovaných CTG repeatů v leukocytech a MRI nálezy se může také podílet somatický mozaicizmus CTG repeticí při DM 1 [15].
Hyperintenzity bílé hmoty v T2 vážených obrazech nekorelují s atrofií šedé hmoty. Proto se považují léze bílé hmoty i atrofie za nezávislé procesy. Prevalence změn šedé hmoty je frontálně, parietálně, v gyrus temporalis superior a medius a v okcipitálním laloku. Neuropatologické studie popisují ztráty neuronů ve frontálním, parietálním a zejména okcipitálním kortexu [9].
Průřezové studie nemohou spolehlivě rozlišit mezi neurovývojovým charakterem změn, získanou atrofií a progresivní neurodegenerací. Avšak absence korelace s trváním, tíží choroby i molekulárně genetickým nálezem, a naproti tomu vliv věku na objem šedé hmoty poukazují spíše na neurovývojový charakter atrofie s věkovou progresí při DM 1 [16].
U pacientů s DM 1 jsou abnormality bílé hmoty bilaterální, ale ne vždy do detailu symetrické. Temporopolární abnormality bílé hmoty postihuje fibra arcuata, zatímco lobární hyperintenzity a léze bílé hmoty tato vlákna ušetří. Léze také nejsou v mozečku, mozkovém kmeni, capsula interna, basálních gangliích ani v corpus callosum. Atrofie mozku je obvykle difuzní, ale často postihuje subkortikální a posteriorní struktury, capsula externa et extrema. Charakter těchto MRI změn svědčí pro diagnosu DM1.
Závěr
Předložená studie prokazuje, že v MRI obrazech hlavy a mozku pacientů s myotonickou dystrofií typu 1 se s různou četností vyskytují strukturální a signálové změny především v bílé hmotě, které jsou samy o sobě nespecifické. Výskyt alespoň jedné z těchto změn či jejich kumulace v charakteristických lokalitách však podporuje klinickou a molekulárně genetickou diagnózu myotonické dystrofie. Studie neporovnává vztah jednotlivých MRI nálezů ke klinickému obrazu, k délce trváni choroby ani k věku pacienta v době stanovení diagnózy. Tyto skutečnosti budou předmětem dalšího zkoumání.
Přijato k recenzi: 24.2.2005
Přijato do tisku: 25.8.2006
Adresa pro korespondenci:
MUDr. Josef Kraus, CSc.
Klinika dětské neurologie 2. LF UK a FN v Motole
Fakultní nemocnice v Motole
V úvalu 84
150 18 Praha 5 - Motol
e-mail: josef.kraus@lfmotol.cuni.cz
Sources
1. Miaux Y, Chiras J, Eymard B, Lauriot-Prevost MC, Radvanyi H, Martin-Duverneuil N et al. Cranial MRI findings in myotonic dystrophy. Neuroradiology 1997; 39 (3): 166–170.
2. Di Costanzo A, Di Salle F, Santoro L, Bonavita V, Tedeschi G. Dilated Virchow-Robin spaces in myotonic dystrophy: frequency, extent and significance. Eur Neurol 2001; 46 (3): 131–139.
3. Kassubek J, Juengling FD, Hoffmann S, Rosenbohm A, Kurt A, Jurkat-Rott K et al. Quantification of brain atrophy in patients with myotonic dystrophy and proximal myotonic myopathy: a controlled 3-dimensional MRI study. Neurosci Lett 2003; 348 (2): 73–6.
4. Ogata A, Terae S, Fujita M, Tashiro K. Anterior temporal white matter lesions in myotonic dystrophy with intellectual impairment: an MRI and neuropathological study. Neuroradiology 1998; 40 (7): 411–5.
5. Di Costanzo A, Di Salle, F, Santoro L, Tessitore A, Bonavita V, Tedeschi G. Pattern and significance of white matter abnormalities in myotonic dystrophy type 1: an MRI study. J Neurol 2002; 249 (9): 1175–1182.
6. Takaba J, Abe N, Fukuda H. Evaluation of brain in myotonic dystrophy using diffusion tensor MR imaging. Nipp Hosh Gijutsu Gakkai Zasshi 2003; 59 (7): 831–838.
7. Hund E, Jansen O, Koch MC, Ricker K, Fogel W, Niedermaier N et al. Proximal myotonic myopathy with MRI white matter abnormalities of the brain. Neurology 1997; 48 (1): 33–37.
8. Abe K, Fujimura H, Soga F. The fluid-attenuated inversion-recovery pulse sequence in assessment of central nervous systém involvement in myotonic dystrophy. Neuroradiology 1998; 40 (1): 32–35.
9. Mizukami K, Sasaki M, Baba A, Suzuki T, Shiraishi H. An autopsy case of myotonic dystrophy with mental disorders and various neuropathologic features. Psychiatry Clin Neurosci 1999; 53 : 51-55.
10. Refsum S, Lonnum A, Sjaastad O, Engeset A. Dystrophia myotonica. Repeated pneumoencephalographic studies in ten patients. Neurology 1967; 17 : 345–348.
11. Martinello F, Piazza A, Pastorello E, Angelini C, Trevisan CP. Clinical and neuroimaging study of cerebral nervous system in congenital myotonic dystrophy. J Neurol 1999; 246 : 186–192.
12. Tanabe Y, Iai M, Tamai K, Fujimoto N, Sugita K. Neuroradiological findings in children with congenital myotonic dystrophy. Acta Pediatr 1992; 81 : 613–617.
13. Huber SJ, Kissel JT, Shuttleworth EC, Chakeres DW, Clapp LE, Brogan MA. Magnetic resonance imaging and clinical correlates of intellectual impairment in myotonic dystrophy. Arch Neurol 1989; 46 : 536–540.
14. Sergeant N, Sablonnićre B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A et al. Dysregulation of human brain microtubule - associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet 2001; 10 : 2143–2155. 15. Harper PS. Myotonic dystrophy. London: WB Saunders; 2002.
16. Antonini G, Mainero C, Romano A, Giubilei F, Ceschin V, Gragnani F et al. Cerebral atrophy in myotonic dystrophy: a voxel based morphometric study. J Neurol Neurosurg Psychiatry 2004; 75 : 1611–1613.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2007 Issue 3
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine Eases Daily Life for Patients and Caregivers
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
Most read in this issue
- Chiari Malformation: Own Experience
- Osmotic Demyelination Syndrome – MRI Diagnosis: a Case Report
- The Brain MR Imaging in Patients with Myotonic Dystrophy DM 1
- Osteoplastic Decompressive Craniotomy