
Léčba epileptických syndromů u dětí
Treatment of Epileptic Syndromes in Children
The paper provides an overview of the full spectrum of age-related epileptic syndromes in children. The author describes treatment strategies based on semiologic, syndromologic and, in some cases, ethiopathogenetic diagnoses. Traditional pharmacotherapeutic management as well as the potential for epilepsy surgery are described. The paper emphasizes the different approaches to treatment of benign forms and risks related to delayed therapy of catastrophic syndromes. Special attention is paid to paradoxical effects of treatment with unsuitable antiepileptics that have the potential to significantly worsen the progression of some of the age-related syndromes.
Key words:
age-related epileptic syndromes – epileptic encephalopathies – treatment of epileptic syndromes – idiopatic generalized epilepsies – prognosis
Authors:
V. Komárek
Authors‘ workplace:
Klinika dětské neurologie a Centrum pro epilepsie 2. LF UK a FN v Motole, Praha
Published in:
Cesk Slov Neurol N 2007; 70/103(5): 473-485
Category:
Minimonography
Poděkování: autor děkuje MUDr. Katalin Štěrbové a MUDr. Pavlu Krškovi, Ph.D. za pomoc s obrazovou dokumentací.
Overview
Práce přináší základní informace o celém spektru věkově vázaných epileptických syndromů u dětí. Jsou popsány léčebné strategie založené na semiologické, syndromické a v některých případech i etiopatogenetické diagnostice. Je diskutována jak klasická farmakoterapie, tak možnosti epileptochirurgického řešení. V práci jsou zdůrazněny odlišnosti přístupu k léčbě benigních variant a rizika opožděné terapie syndromů s katastrofickým průběhem. Zvláštní pozornost je věnována paradoxnímu efektu nevhodně zvolených antiepileptik, která mohou u některých věkově vázaných syndromů průběh onemocnění signifikantně zhoršit.
Klíčová slová:
věkově vázané epileptické syndromy – epileptické encefalopatie – léčba epileptických syndromů – idiopatické generalizované epilepsie – prognóza
1. Definice onemocnění
Epileptický záchvat je definován jako transientní výskyt příznaků vyvolaných abnormálně rozsáhlou či nadměrně synchronizovanou neuronální mozkovou aktivitou [1,2,3].
Epilepsie je definována jako onemocnění mozku charakterizované jednak jeho trvalou predisposicí generovat epileptické záchvaty a jednak neurobiologickými, kognitivními a psychosociálními důsledky epilepsie [1,2,3]. Pro diagnózu epilepsie jako chronického onemocnění je nutný výskyt nejméně jednoho či spíše dvou spontánních epileptických záchvatů. Termín aktivní epilepsie je vyhrazen pro případy s výskytem alespoň jednoho záchvatu v posledních pěti letech, a to nezávisle na medikaci.
Epileptický syndrom bývá definován jako komplex společně se vyskytujících specifických příznaků a charakteristik [2,3]. Součástí komplexu musí být určení typu záchvatů, popis EEG nálezu, posouzení možné etiologie, věkové vazby a přepokládané prognózy. Epileptické syndromy bývají členěny podle znalosti etiologie na idiopatické (převážně geneticky podmíněné), symptomatické (známá příčina) a tzv. kryptogenní (pravděpodobně symptomatické, ale zatím bez důkazu o vyvolávajícím poškození mozku) a podle znalosti patofyziologie na fokální a generalizované. Třetí možností je klasifikace syndromů podle charakteristické věkové vazby na novorozenecké (do 3 měsíců věku), kojenecké a batolecí (do 3 let), dětské do 12. roku a juvenilní mezi 12. až 18. rokem. Níže uvedené syndromy jsou řazeny do podskupin podle všech zmíněných kritérií.
2. Epidemiologie
Prevalence aktivní epilepsie je asi 8-9 případů na 1000 obyvatel, ve věkové skupině do 15 let prodělá alespoň jeden epileptický záchvat 5 dětí ze 100. Nejčastějším věkově vázaným syndromem jsou febrilní křeče, které postihují 4 % evropské populace (v Japonsku to je až dvojnásobek), ze všech ostatních syndromů tvoří 1/5 případů benigní epilepsie s rolandickými hroty [2,4].
3. Etiopatogeneze
Patofyziologické mechanizmy vzniku epileptických záchvatů nejsou stále dostatečně objasněny a ani nejnovější poznatky základního výzkumu nepřispěly k úplnému pochopení mysteria náhlého a neočekávaného vzplanutí epileptického ohniska. Ukazuje se, že starší koncepce tří hlavních komponent (pohotovost k záchvatu, epileptické ohnisko a spouštěcí podnět) publikovaná u nás Servítem, je kompatibilní s moderními hypotézami, které vycházejí z geneticky podmíněné seizures susceptibility (zvýšené náchylnosti k záchvatům), epileptogenní sítě ohnisek a různých endo - či exogenních faktorů uvolňujících stavidla bezprostředně před záchvatem [1,3].
V zásadě lze rozlišit dva hlavní patofyziologické mechanizmy iktogeneze, které mají rovněž odlišný iktální EEG vzorec. Konvulzivní záchvaty se v EEG manifestují repetitivními hroty s postupně narůstající amplitudou odpovídající rychlému pálení dalších a dalších depolarizovaných excitačních neuronů (náborová aktivita) a inhibiční neurony jsou aktivovány až ve fázi ukončování záchvatu. Antiepileptika tlumící excitaci či posilující inhibici mohou vznik těchto záchvatů potlačit. Naproti tomu záchvaty, jako jsou absence a syndromy s bilaterálně synchronními SW-komplexy jsou generovány inhibičními talamokortikálními okruhy (tendence ke spontánním oscilacím je typická pro talamické neurony s nízkoprahovými vápníkovými kanály T-typu) . Extrémní synchronizace těchto neuronů aktivuje nadměrné pálení kortikálních neuronů (ostrá složka SW-komplexu) a zpětnovazebná kortikotalamická excitace retikulárních talamických GABA-neuronů vede k hyperopolarizaci s EEG korelátem pomalé vlny. Po jejím odeznění se opět aktivují vápníkové T-kanály a oscilace SW-výbojů pokračují. Z výše uvedeného vyplývá, že tento typ záchvatů s nadměrnou aktivitou inhibičních neuronů GABA se může zhoršit po aplikaci gabaergních antiepileptik a efektivní jsou léky potlačující aktivitu neuronů s T-typem vápníkových kanálů (např. etosuximid). Na rozmanitosti věkově vázaných epileptických syndromů se podílí i postupné (zadopřední) vyzrávání jednotlivých kortikálních oblastí. V prvních 3 letech života jsou proto relativně nejméně stabilní a k výbojům predisponované posteriorní (okcipito-parietální) oblasti, následně mezi 4.-9. rokem to je centrotemporální (rolandický) region, a konečně v dospívání bouřlivě dozrávající oblasti anteriorní (frontální). Mohutný rozvoj inhibičních systémů sice snižuje tendenci k rozvoji sekundárně generalizovaných konvulzivních záchvatů, ale zejména v předškolním a mladším školním věku zároveň zvyšuje náchylnost k nekonvulzivním záchvatům charakteru absencí. Epileptologickou zvláštnostní v dospívání je přechodná perimenarcheální instabilita zapříčiněná dřívějším nástupem estrogenů oproti gestagenům [4].
4. Klinický obraz
Součástí klinického obrazu epileptického onemocnění jsou kromě záchvatů i neparoxyzmální projevy (např. kognitivní a behaviorální poruchy), které mohou výrazně poznamenat klinický průběh onemocnění a ovlivnit prognózu.
4.1 Semiologické spektrum záchvatů
Prapůvodní členění záchvatů (např. Gowers 1881) spočívalo v rozlišení velkých (později grand mal) a malých záchvatů (později petit mal), mezinárodní klasifikace záchvatů (ILAE 1981 resp. 1989) dělí záchvaty podle jejich původu na parciální/fokální (lze nalézt ohnisko - fokus, odkud záchvaty vycházejí) a generalizované, které začínají současně v obou hemisférách (zde hrají významnou roli již zmíněné thalamokortikální okruhy). I když tato klasifikace byla vypracována se snahou po postižení všech semiologických variant, klinická praxe ukázala, že zejména v dětském věku existuje velké množství semiologicky rozmanitých forem záchvatů. Nezralost mozku v prvních letech života se nepochybně podílí na „globálnějších“ klinických projevech fokálního záchvatu (např. záchvat vycházející z temporálníha ohniska se může manifestovat hemikonvulzemi nebo dokonce flekčními spazmy), nonkonvulzivní záchvaty u kojenců se mohou projevit zástavou fyziologické nekoordinované fidgety motoriky (tzv. hypomotorický záchvat) a naopak eratické „fokální“ záškuby u novorozenců mohou souviset s difuzním metabolickým postižením mozku, který není dostatečně vyzrálý pro genezi synchronizovaného generalizovaného záchvatu. V tomto smyslu je třeba jak klinické, tak elektroncefalografické (skalpové) známky fokality či generalizace hodnotit velmi obezřetně.
4.2 Neparoxyzmální projevy epilepsie
Zvláštností epileptologie dětského věku jsou syndromy, u kterých nedominuje problematika epileptických záchvatů, ale subklinické EEG výboje s korelátem vývojových kognitivních a behaviorálních poruch. To se týká především rolandické epilepsie (nejčastěji její atypické varianty - viz dále), u které až 30 procent dětí může mít kognitivní poruchy. Extrémním příkladem specificky věkově vázaného epileptického syndromu je LKS a CSWS, v obou případech nalézáme kontinuální výboje hrotů a vln v pomalém spánku, dominantou onemocnění jsou kognitivně behaviorální problémy a epileptické záchvaty se vyskytují u necelé 1/4 postižených [5].
4.3 Průběh a prognóza
Specifickým aspektem věkově vázaných epilepsií jsou velké rozdíly ve výsledném vyznění onemocnění - na jedné straně existují syndromy s velmi dobrou prognózou, jako jsou benigní novorozenecké křeče, febrilní křeče či rolandická epilepsie, a na straně druhé syndromy s potenciálně katastrofickým (Westův syndrom) či s jednoznačně nepříznivým (Ohtahara či Lennox-Gastautův syndrom) průběhem. Jiří Dolanský v závěru své monografie zmiňuje koncept členění epileptických záchvatů na soft a hard varianty, z nichž každá vyžaduje odlišný přístup [6]. Prognóza bývá rovněž ovlivněna řadou dalších faktorů, mezi které patří i dobře vedená léčebná strategie. U dlouhodobě léčených epileptických syndromů je nutno počítat s přirozenou fluktuací četnosti záchvatů a proměnami semiologie během vývoje dítěte. Často lze pozorovat různě dlouhé seizmické období vzdorující veškeré medikaci a následné periody klidu po bouři nezávisle na léčebné taktice.
5. Diagnóza a diferenciální diagnóza epileptických syndromů
Diagnostické schéma doporučené ILAE se sestává z 5 základních součástí:
Krok 1. Posouzení iktální fenomenologie (semiologie) a rozhodnutí o povaze záchvatu (pravý epileptický nebo některý z mnoha epilepsii imitujících záchvatů). Optimální popis lze získat pouze z video-EEG-záznamu, případně z domácí video-nahrávky, popis záchvatu druhou osobou může být zavádějící. Vzhledem k tomu, že až 1/3 refrakterních epilepsií je ve skutečnosti neepileptickými záchvaty různého původu, je nutné všechny informace pozorně hodnotit (never accept secondhand description).
Krok 2. Určení typu epileptického záchvatu včetně případné lokalizace (asymetrický tonický záchvat vycházející z pravé suplementární motorické arey) a u reflexních záchvatů popis vyvolávajících faktorů (např. čtení textu u reading epilepsy).
Krok 3. Zařazení záchvatu do rámce některého z mnoha epileptických syndromů (myoklonicko-astatická epilepsie nebo Lennox-Gastautův syndrom), ne vždy však lze syndromickou diagnózu provést. Krom toho existuje řada nově popsaných syndromů, které v mezinárodní klasifikaci zatím zařazeny nejsou. Do rámce tohoto článku jsou zavzaty jen dobře definované homogenní syndromy a neuvádíme žádné z extrémně heterogenní podskupiny II.C.2.a, kde se skrývá několik desítek diagnostických jednotek a syndromů spojených s epileptickými záchvaty.
Krok 4. Etiologická diagnóza - zjištění patologického substrátu (nádor nebo dysplazie) u symptomatických epilepsií či genetického pozadí u idiopatických epilepsií. Zlatým standardem v etiopatogenetické diagnostice dětských epilepsií je provedení kvalitního zobrazení mozku pomocí magnetické rezonance (1.5 T), u refrakterních epilepsií s podezřením na kortikální dysplázií je nutný speciální protokol. U velmi diskrétních lézí lze využít citlivější zobrazení pomocí l H MR spektroskopie (CSI=chemical shift imaging spektrální mapy).
Krok 5. Zjištění míry postižení (podle Impairment classification WHO ICID). V České republice lze uplatnit škálu Stupně kompenzace pacientů s epilepsií navrženou Českou ligou proti epilepsii, která byla akceptována pro posudkové účely Ministerstva práce a sociálních věcí.
6. Léčba věkově vázaných epileptických syndromů
Výběr vhodného antiepileptika vyžaduje na jedné straně dostatečnou znalost specifik jednotlivých syndromů opírající se o dlouhodobé vlastní zkušenosti a na druhé straně by měl být podložen kvalitními kontrolovanými studiemi. Současná situace je paradoxní – i když bylo v posledních 20 letech rozpoznáno více než 30 věkově vázaných syndromů s relativně homogenním elektroklinickým obrazem, většina publikovaných studií je orientována na léčbu heterogenních souborů parciálních či generalizovaných epileptických záchvatů u dospělých a dětí starších 12 let. Výsledky se pak mnohdy mechanicky přenášejí do dětského věku - zaslepené kontrolované studie u většiny dětských syndromů k disposici nejsou. Lze však vycházet z dlouhodobého sledování větších souborů a u vzácnějších syndromů se opírat i o jednotlivé kazusitiky. U dětských idiopatických epileptických syndromů obvykle se zahájením léčby vyčkáváme mnohem déle, než je tomu u dospělých pacientů a v některých indikacích nenasazujeme medikaci z důvodů epileptických záchvatů, ale pro kognitivně behaviorální problémy spojené se subklinickými epileptiformními výboji na EEG. Opačnou situací je časné a agresivní léčení syndromů s potenciálně katastrofickým průběhem. To se týká zejména Westova syndromu, kdy zahájení terapie do 2 měsíců trvání hypsarytmie zlepšuje prognózu. Obdobně časné rozpoznání a léčba syndromů s kontinuálními výboji v pomalém spánku může příznivě ovlivnit finální kognitivně behaviorální profil dítěte. U ostatních syndromů by i při zahájení léčby mělo platit pravidlo citlivé titrace prvního zvoleného antiepileptika. Obykle začínáme co nejnižší denní dávkou, kterou velmi pomalu zvyšujeme (start low, go slow) - to platí zejména pro karbamazepin, lamotrigin, ale levetiracetam a topiramát. Po dosažení předpokládané optimální dávky je vhodné ověřit, jak pacient lék metabolizue a zda jej správně užívá pomocí stanovení plazmatické koncentrace. Pokud stále přetrvávají záchvaty a jde o syndrom s potenciálně nepříznivou prognózou, je třeba dosáhnout maximální možné hladiny, kterou pacient z hlediska vedlejších účinků toleruje. Je-li jisté, že monoterapie lékem první volby je zcela neúspěšná, lze postupně zaměnit první lék za jiný nebo v případě částečného úspěchu přidat druhý lék a léčit pacienta tzv. racionální kombinací (s vědomím rizik sčítání nežádoucích účinků aj). Ukončování dlouhodobé antiepileptické terapie se v dětském věku vyznačuje řadou odlišností od doporučených postupů u dospělých pacientů. Zásadou je, že u idiopatických syndromů, jako je rolandická epilepsie nebo dětské absence, je možné zvážit vysazování léčby už po jednom roce bez jakýchkoliv záchvatů. Velmi opatrné vysazení léčby je možné i u některých kryptogenních či symptomatických epilepsií. Na druhé straně u Lennox-Gastautova syndromu a podobných syndromů nelze terapii vysadit ani po delším relativně klidovém období. Značně diskutabilní je vysazování léků po úspěšně provedeném epileptochirurgickém výkonu. Chybou je jak alibistické opatrné dlouhodobé či dokonce trvalé ponechání předoperační medikace (obvykle jde o kombinaci dvou i více AE), tak předčasné či příliš rychlé vysazení léčby. Jinou známou zkušeností je i dlouhodobé léčení relativně příznivé idiopatické epilepsie, jako je juvenilní myoklonická epilepsie, kdy je dlouhodobá udržovací terapie nezbytná a pokusy o úplné vysazení nebývají úspěšné.
Specifika farmakologické léčby dětských epi syndromů
Léčba antiepileptiky má v dětském věku řadu specifik. Jednak je třeba při dávkování počítat s věkem se měnící farmakokinetikoui farmakodynamikou - např. dávky u novorozenců by měly být nižší vzhledem k ne zcela vyvinutým degradačním mechanizmům, naopak u kojenců a batolat podáváme dávky relativně vyšší než u dospívajících a dospělých. Velmi specifické je podávání léku jinak zcela opomíjeného v léčbě epilepsie dospělých, a sice sultiamu u rolandických epilepsií, LKS a CSWS.
Určitou atypií je léčení kortikoidy, resp. ACTH. Lékem první volby je u Westova syndromu, ale efektivní bývá i u LKS a CSWS, efekt lze čekat i u MAE, ABPE a LGS.
Nefarmakologickou léčbu epileptických syndromů u dětí lze rozdělit na kauzální epileptochirurgické resekční výkony, na výkony diskonekční rázu kalozotomie, a na výkony stimulační (implantace vagového stimulátoru - VNS - vagal nerv stimulation). Specifickou nefarmakologickou metodou léčby je ketogenní dieta.
7. Přehled jednotlivých epileptických syndromů podle věkové vazby
7.1 Epileptické syndromy v neonatálním období
Novorozenecké křeče postihují asi 3 děti z 1000 živě narozených, u nedonošených dětí je výskyt více než 20násobně vyšší. Semiologicky jsou velmi rozmanité - nejčastěji jsou pozorovány subtilní nevýrazné formy záchvatů, klonické a myoklonické jsou častější než tonické [2,4]. Ze syndromologického hlediska jsou v tomto období v mezinárodní klasifikaci uvedeny 2 epileptické encefalopatie a 2 idiopatické benigní syndromy. Diferenciálně diagnosticky jsou křeče u novorozenců velmi heterogenní skupinou - zmíněné 4 epileptické syndromy jsou poměrně vzácné a novorozenecké křeče se vyskytují mnohem častěji v souvislosti s různými formami dysbalance vnitřního prostředí (hypokalcemie, hypoglykemie aj), s hypoxicko ischemickým inzultem, neuroinfekcemi či se závažnou vrozenou metabolickou poruchou [7]. Tyto nesyndromické novorozenecké křeče léčíme buď cíleně úpravou iontogramu či glykemie, nebo symptomaticky barbituráty, fenytoinem a méně často benzodiazepiny.
7.1.1. Ohtaharův syndrom
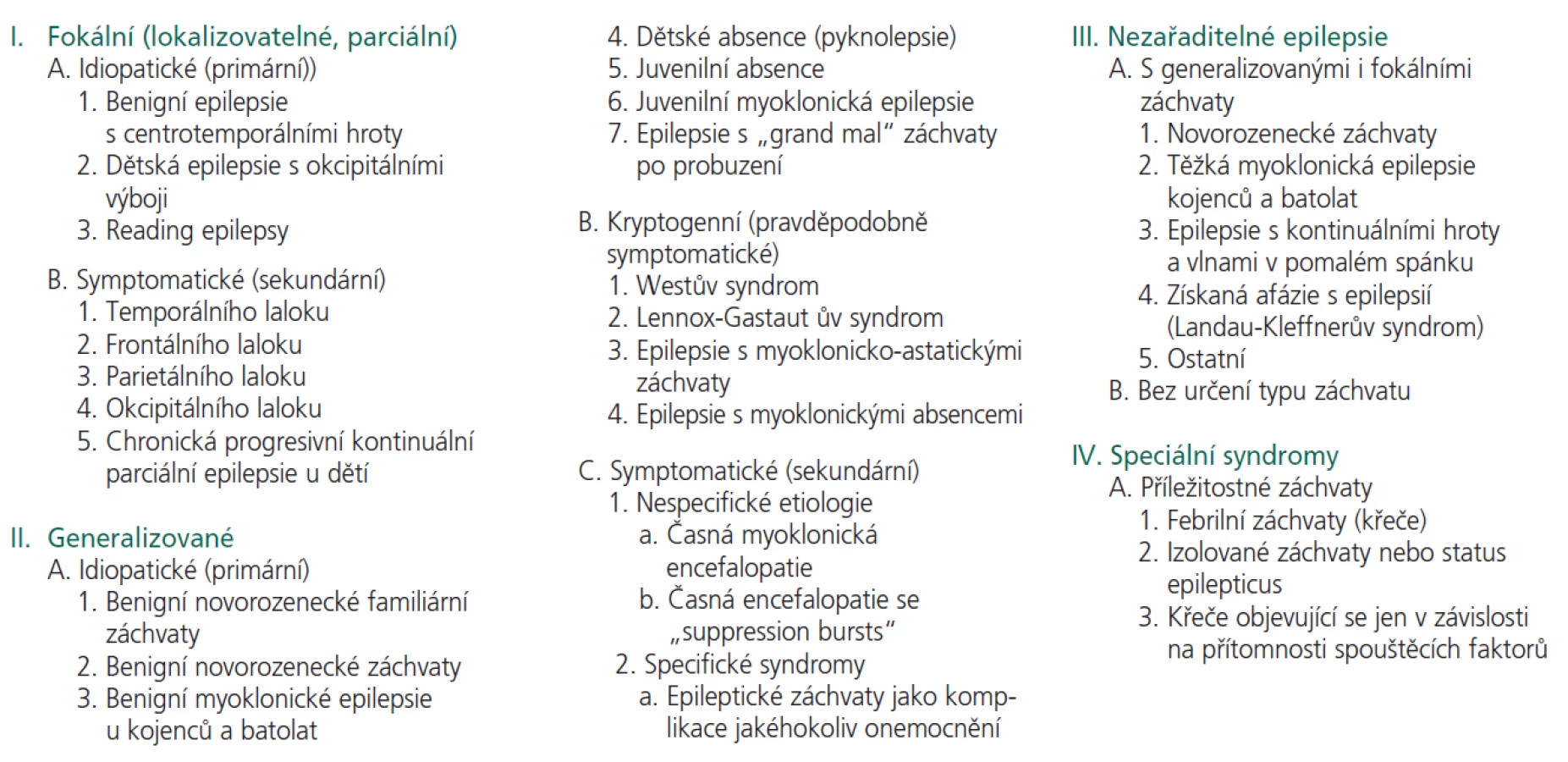
Ohtaharův syndrom (early infantile epileptic encephalopathy - EIEE) je relativně vzácnou jednotkou neonatálního období s nejčastějším začátkem v prvních 10 dnech po narození. Semiologicky jsou typické tonické flekční spazmy krátkého v trvání od 2 do 10 sekund, a to izolovaně i v sériích. Objevují se v bdění i ve spánku a mohou být jak symetrické, tak asymetrické. Asi u 1/3 třetiny mohou mít ráz eratických fokálních motorických záchvatů nebo hemikonvulzí. Obykle nebývají záchvaty myoklonické [11].
Charakteristický je nález tzv. suppression-bursts (SB) vzorců v bdělém EEG (obr.1a, 1b). Tento obraz krátkých úseků vysokovoltážních výbojů hrotů (burst) a následné utlumené aktivity (suppression) popisoval Lesný jako jev vyhasínání u závažných organických cerebrálních postižení. Jedná se o vzorec odlišný od dyskontinuální hypsarytmie ve spánku, u dětí s Westovým syndromem.


Asi u 2/3 pacientů se klinický i EEG-obraz Ohtaharova syndromu může dále vyvíjet směrem k Westovu a později Lennox-Gastautovu syndromu [8]. Diferenciálně diagnosticky je třeba myslet na Aicardiho syndrom, který zahrnuje agenezi corpus kalosum, chorioretinální abnormality, infantilní spazmy a bilaterálně nezávislé výboje na EEG vykazující rysy supression burst.
Etiologie Ohtaharova syndromu zahrnuje různé varianty vrozených poruch vývoje mozku včetně hemimegalencefalie, porencefalie či rozsáhlých kortikálních dysplazií. Prognóza je nepříznivá, s mortalitou až v polovině případů, přežívající děti mají různě závažné reziduální neurologické nálezy. Léčba klasického Ohtaharova syndromu nebývá téměř nikdy úspěšná. Časný epileptochirurgický výkon rázu hemisferektomie či multilobárních resekcí dává naději jen malému procentu postižených [9], v ojedinělých případech byla úspěšná léčba vigabatrinem či ACTH, v námi popsané kazuistice byl efektivní chloralhydrát [10].
7.1.2. Časná myoklonická encefalopatie
Časná myoklonická encefalopatie (EMEE - early myoclonic epileptic encephalopathy) je druhou rovněž vzácnou variantou novorozenecké encefalopatie, která má oproti EIIE odlišnou semiologii, podobný EEG obraz (SB vzorce) a stejně nepříznivou prognózu. Záchvaty se objevují již v prvních hodinách po narození a mají pro EME typický charakter eratických (bloudivých) myoklonů, které se nahodile stěhují z jednotlivých částí těla nebo méně často ráz generalizovaných (masivních) myoklonických záškubů, mohou se též objevit záchvaty s autonomní složkou (zrudnutí, apnoe) a ojedinělé asymetrické tonické záchvaty [12] .
Etiopatogeneticky jde o multifaktoriální onemocnění, kdy na první místě uvažujeme o non-ketotické hyperglycinemii a kromě dalších poruch metabolizmu aminokyselin přicházejí v diferenciální diagnostice v úvahu onemocnění mitochondriální či paroxizomální. Léčba není téměř nikdy úspěšná, nicméně není chybou otestovat efekt pyridoxinu v dávce 100 až 200 mg na 24 hodin a vyčkat klinické a EEG-odezvy. V případě vzácné pyridoxinové dependence je odezva na terapii jednoznačná a překvapivě úspěšná.
7.1.3 Benigní familiární neonatální záchvaty
Jedná se o relativně vzácnou jednotku, která byla dosud popsána u více než 50 rodin. Podkladem je autosomálně dominatní draslíková iontoforopatie s mutacemi genů na 20q13.3 (KCNQ2 - BFNC1 ) a na 8q24 (KCNQ3 – BFNC2), které mohou mít podobný fenotypický obraz. Křeče se manifestují obvykle druhý den po narození a prakticky u všech dětí se objeví do 8. týdne života. Semiologicky mívají ráz krátké tonické křeče s apnoe nebo s vokalizací, mohou se nakupit až do počtu 30 za den. V této situaci se u tohoto syndromu doporučuje klonazepam nebo midazolam. Úplnou remisi lze obvykle očekávat, a to i bez jakékoliv terapie, do několika měsíců po začátku onemocnění. Prognóza je sice trvale příznivá, ale asi 1/10 dětí má později febrilní záchvaty nebo jinou formu idiopatické, nejčastěji rolandické, epilepsie .
7.1.4. Benigní idiopatické (nefamiliární) novorozenecké křeče
Protože se vyskytují převážně mezi 4.-6. dnem, jsou označovány synonymem křeče pátého dne. Postihují častěji chlapce a mají klonický charakter spojený někdy s apnoickou pauzou. Záchvaty mohou někdy přejít do unilaterálního statu záškubů obličejových a končetinových svalů. Za těchto okolností se, tak jako u ostatních nesyndromických novorozeneckých křečí, doporučuje zahájit léčbu bolusem fenobarbitalu cca 20 mg/kg rychlostí 5 mg za minutu a případně pokračovat udržovací dávkou kolem 4mg/kg/den. Dalším lékem je fenytoin v iniciální dávce 20 mg/kg pomalu intravenózně 0,5 mg/kg/min (udržovací dávka je obvykle 5-10 mg/kg), ale je třeba počítat s extrémně dlouhým poločasem degradace phenytoinu, a tedy i rizikem předávkování.
7.2. Idiopatické epileptické syndromy u kojenců a batolat
7.2.1 Febrilní záchvaty
FZ jsou definovány jako příležitostné poruchy vědomí s křečemi v souvislosti s horečkou nad 38 ºC. Nezbytnou podmínkou pro stanovení diagnózy FZ je nepřítomnost intrakraniální infekce nebo jiných zjevných příčin záchvatu. Jedná se o nejčastější epileptický syndrom postihující až 4 % evropských dětí mladších 6 let . Febrilní záchvaty se objevují obvykle mezi 6. a 36. měsícem, horní hranicí výskytu je 5 až 6 let. Semiologicky převažují symetrické klonické záchvaty, někdy s úvodní tonickou složkou, v některých případech s ochabnutím, kdy diferenciálně diagnosticky zvažován tzv. febrilní kolaps. Nekomplikované FZ mají krátké trvání, křeče jsou klonické, symetrické. Komplikované febrilní záchvaty trvají 15 minut a déle, bývají lateralizované s případnou pozáchvatovou hemiparézou. V těchto případech je riziko pozdějšího rozvoje temporální epilepsie, jinak je prognóza u více než 80 % příznivá a křeče se v pozdějším věku neopakují. Výjimkou je familiární syndrom, v rámci kterého se vyskytují febrilní křeče i po 6. roce plus afebrilní generalizované záchvaty. Jedná se o syndrom GEFS+, jehož podkladem je nejčastěji mutace pro alfa podjednotku napěťově řízených sodíkových kanálů (SCN1A).
K patofyziologickým mechanizmům patří cytokinová reakce na noxu (nejčastěji virový infekt), jejíž součastí jsou téměř současně centrální aktivace hypotalamická (vzestup teploty) a talamokortikální (ospalost, hypersynchronizace a u predisponovaných jedinců i křeče).
Terapie při febrilních záchvatech je shodná s léčbou epileptických záchvatů, tj. podání Diazepamu nejlépe i.v. nebo per rectum v roztoku pomocí speciálního dávkovače. Dětem do 15 kg podáváme 5 mg, u dětí nad 15 kg podáváme 10 mg Diazepamu. V současné době je alternativou bukální podání midazolamu. Trvalé podávání antiepileptik po febrilním záchvatu není indikováno, a to ani u opakovaných záchvatech (FZ se opakují přibližně u 1/3 dětí) nebo komplikovaných (ty tvoří asi 5 % dětí s FZ). Ukázalo se, že zavedení preventivních opatření, spočívajících v intermitentním podávání Diazepamu (cca 0,7 mg/kg/24h) současně s antipyretiky (paracetamol, brufen), sníží výskyt opakovaných křečí z cca 30 % na 5-10 %
7.2.2. Benigní myoklonická epilepsie u kojenců
Tento syndrom tvoří asi 2 % ze všech epilepsií začínajících před 3. rokem, objevuje se nejčastěji mezi 3.-36. měsícem, a to častěji u chlapců. Semiologicky se manifestuje myoklonickými záškuby v první hodině po usnutí nebo po probuzení a diferenciálně diagnosticky je třeba odlišit benigní spánkové myoklony. Záchvaty dobře reagují na terapii valproátem a léčbu lze zpravidla po 3 letech vysadit
7.3. Epileptické encefalopatie
7.3.1. Westův syndrom
Westův syndrom patří mezi časné epileptické encefalopatie a svou incidencí je srovnatelný s dětskou mozkovou obrnou - tj. postihuje cca tři promile dětí. Devadesát sedm procent případů se objeví do l roku věku, maximum výskytu je mezi 3. až 7. měsícem. Pro diagnosu Westova syndromu jsou semiologicky charakteristické infantilní spasmy nazývané též bleskové křeče (flekční tonická křeč opakující se v serii několikrát denně) , na EEG bývá hypsarytmie (obr.2) a pokud se nejedná o idiopatickou a včas léčenou variantu WS, tak lze očekávat zástavu psychomotorického vývoj i jeho regres. Dlouhodobá prognóza závisí tedy nejen na příčině, ale též na včasné diagnostice a adekvátní terapii [14]. Častou příčinou Westova syndromu je tuberózní skleróza, případně různé vrozené vývojové vady mozku. Mnohdy je spouštěcím, nikoliv etiologickým, faktorem prvních infantilních spasmů vakcinace (zejména proti pertussi), případně jiné zatížení imunitního systému.
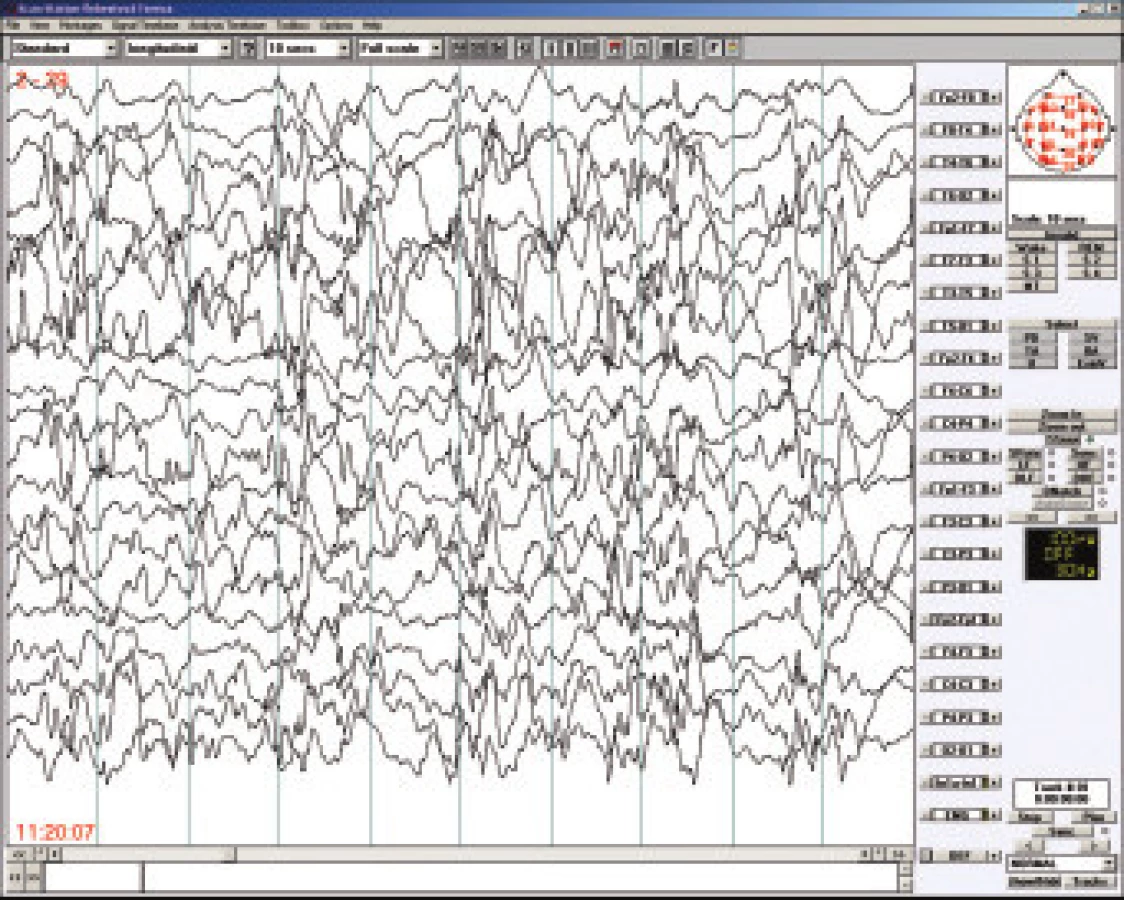
Mezi léky první volby patří ACTH a vigabatrin [15]. VGB se upřednostňuje u dětí s TS [16], u idiopatických forem může být efektivní valproát. U dětí do jednoho roku podáváme depotní formu ACTH v dávce 0,5mg (20 IU) až 1,0mg (40 IU) denně po dobu dvou týdnů, další dva týdny ob den a poslední dva týdny 2x týdně. U respondérů pozorujeme vymizení hypsarytmie i redukci záchvatů již během několika dnů po zahájení kůry. Existují práce doporučující podávání ACTH jen po dobu dvou týdnů. Léčba ACTH je provázena řadou nežádoucích účinků, ze závažných je to především arteriální hypertenze, riziko infekčních komplikací a sterodiní diabetes, z méně závažných kromě cushingoidního habitu a nárůstu tělesné hmotnosti, i změny chování, dráždivost a poruchy spánku. Steroidy jsou méně účinné než ACTH, podává se obvykle Prednizon v dávce 1-2mg/kg a den po dobu dvou týdnů a s následnou redukcí během několika týdnů. Dalším lékem volby je vigabatrin, který u dětí s Westovým syndromem dáváme v počáteční dávce 50 mg/kg/den a postupně po 5-7 dnech zvyšujeme až do dosažení optimální terapeutické odpovědi, kterou můžeme očekávat do dvou týdnů. Obvyklá udržovací dávka je 100 mg/kg/den, v případě potřeby může dosahovat až 150 mg/kg/den. Jak již bylo napsáno, VGB je velmi efektivní u dětí s TS, kdy mu dáváme přednost před ACTH [16]. K nežádoucím účinkům VGB u dětí patří excitace a hyperaktivita, je-li podáván v kombinaci tak je třeba mít na paměti, že snižuje plazmatické hladiny fenytoinu Z ostatních antiepileptík se podává valproát v dávce od 30 do 60 mg/kg (je třeba pečlivě hlídat jaterní testy a krevní obraz), dále existují studie dokazující efektivitu nitrazepamu i novějších léčiv lamotriginu, topiramátu a nejnověji levetiracetamu. V ojedinělých případech je indikována epileptochirurgická oprace [17].
7.3.2. Těžká myoklonická epilepsie u kojenců (Dravetové syndrom)
Výskyt se udává 1 na 30000 detí a vyskytuje se dvakrát častěji u chlapců. Z epilepsií v prvním roce života tvoří asi pět procent.
Obvykle začíná v prvním roce života u dětí s normálním psychomotorickým vývojem pod obrazem protrahované parciálního či generalizovaného záchvatu v průběhu febrilního onemocnění.. Může tak imitovat komplikovanou variantu febrilních záchvatů. Odlišuje se tím, že ve druhém roce života se přidávají myoklonické záchvaty postihující převážně horní končetiny a trupové svalstvo. Mohou se objevit i atypické absence nebo parciální záchvaty s autonomní komponentou. Charakteristická je pozitivní odpověď na fotostimulaci a časté je nakupení nekonvulsivních záchvatů. Později převládají klonické záchvaty po probuzení. V EEG jsou generalizované výboje sw či psw komplexů o o frekvencii 3-5 Hz. V batolecím období se objevuje regres vývoje a ataxie, takže dítě může připomínat klinický obraz neuronální ceroid-lipofuscinosy. Kromě mentální retardace se popisují i poruchy chování a autistické rysy.
Etiopatogeneticky jde geneticky podmíněné onemocnění s mutací sodíkového kanálu a někdy se považuje za nejtěžší fenotypickou variantu syndromu s mutací SCN1A genu.
Léčba je velmi svízelná, existuje randomizovaná studie dokladující efektivitu stiripentolu [18], který je t.č. již v ČR právě pro tuto diagnózu registrován. Efektivnost stiripentolu byla popsána v kombinaci s valproátem. Kontraindikováno je podání carbamazepinu, phenytoinu i lamotriginu, které mohou záchvaty u SMEI agravaovat. V ojedinělých případech je odezva na ketogenní dietu.
7.3.3. Lennox-Gastautův syndrom
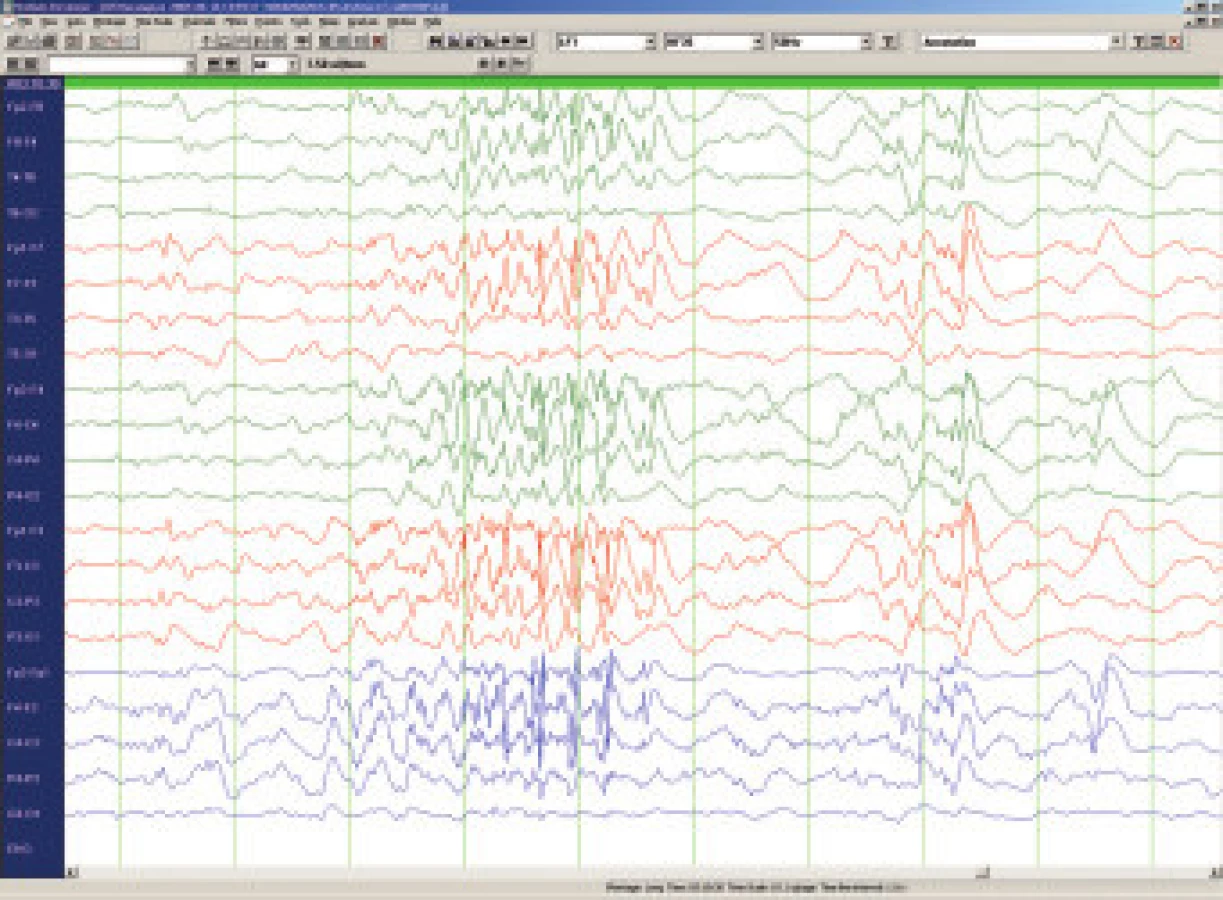
LGS postihuje 5-10% ze všech dětí s epileptickými záchvaty, incidence se udává 3 na 10 tisíc živě narozených dětí. Jde o jednu z nejobtížněji léčitelných epilepsií, a to především vzhledem k současnému výskytu rozličných typů epileptických záchvatů. Ze semiologického hlediska je pro diagnosu klíčová přítomnost tonických obvykle nočních záchvatů charakteru axiálních spasmů, časté jsou atypické absence s pomalými komplexy hrot vlna ( 2-2,5/s) na EEG, nebezpečné jsou astatické záchvaty s rizikem zranění při pádu a relativně méně časté jsou myoklonické záchvaty. Na EEG kromě zpomalené základní aktivity a zmíněných pomalých komplexů hrot vlna lze zachytit i pro LGS patognomické rychlé rytmické výboje ostrých vln (8 - 10/s) ve spánku (obr.3). Etiopatogeneticky se jedná téměř vždy o symptomatické či pravděpodobně symptomatické epilepsie v důsledku metabolického onemocnění , stacionární encefalopatie nebo difusních abnormalit CNS. LGS je téměř v 90 % provázen závažnou mentální retardací a dlouhodobá prognosa je prakticky vždy nepříznivá. Terapie se musí řídit vedoucím typem záchvatu. Tonické záchvaty lze ovlivnit felbamátem, který je pro tuto indikaci v ČR schválen. Doporučuje se topiramát [19], lamotrigin [20] a valproát - tyto léky jsou efektivní i u atypických absencí, myoklonií a někdy u astatických záchvatů. Benzodiazepiny a ethosuccinimidy se nedoporučují vzhledem k riziku aktivace tonických záchvatů, karbamazepin, vigabatrin a tiagabin mohou naopak aktivovat myoklonické záchvaty a absence. Phenobarbital i phenytoin mívají efekt jen výjimečně. U LGS je jednoznačně indikována ketogenní dieta. Nově je pro přídatnou léčbu záchvatů doporučován rufinamid. Resekční epileptochirurgické výkony jsou prováděny jen vzácně a to vzhledem k difusnímu charakteru postižení mozku, přechodný efekt může přinést u četných astatických záchvatů kalozotomie. Implantace vagového stimulátoru je považována za pravděpodobně efektivní a kromě snížení frekvence záchvatů přináší v některých případech i zlepšení chování a tím i kvality života dítěte i celé rodiny.
7.3.4. Landau - Kleffnerův syndrom
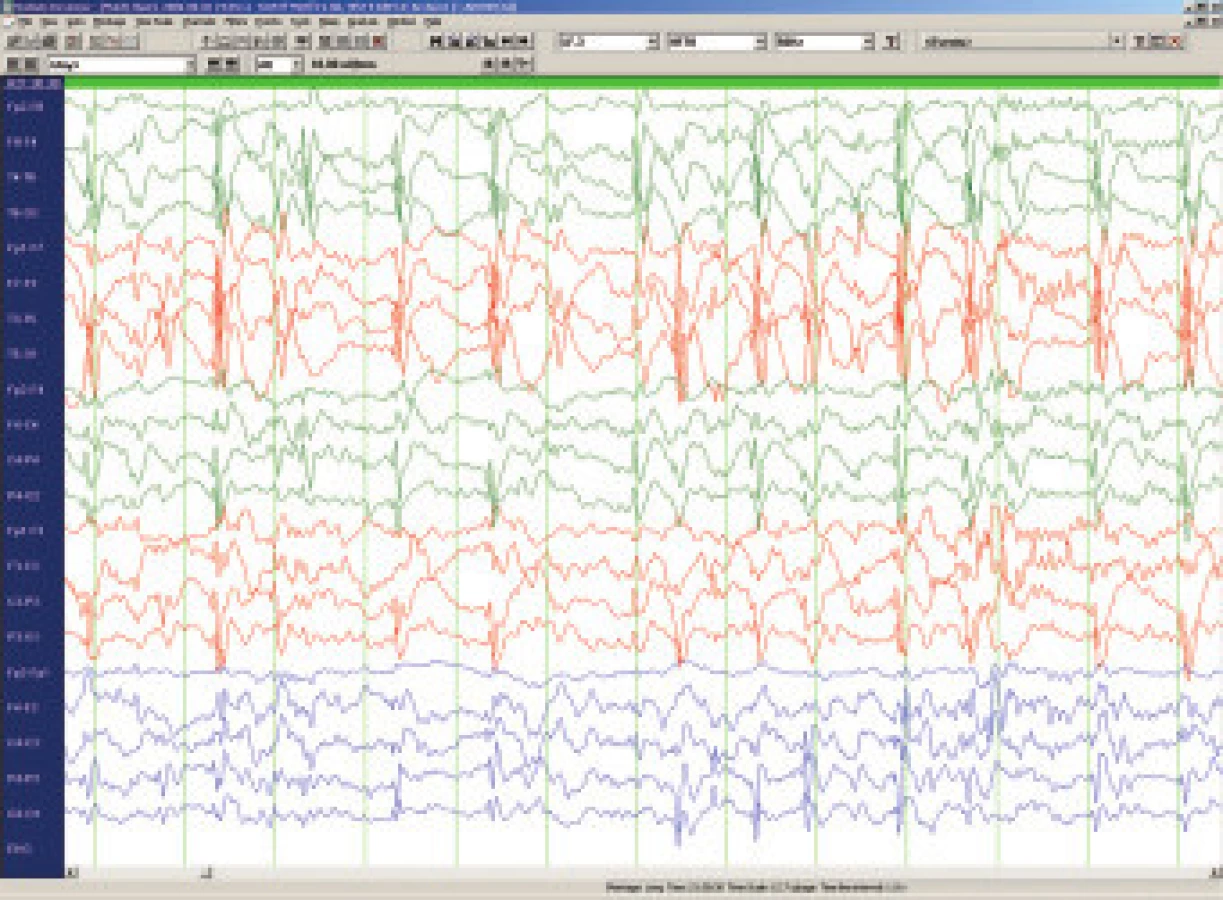
LKS je též nazýván syndromem získané afázie s epilepsií. Objevuje se obvykle kolem 4. až 8. roku a to o něco častěji u chlapců. Prvním příznakem jsou obtíže porozumět mluvenému slovu (akustická agnózie), postupně se rozpadá řeč postiženého dítěte, které se stává neklidné, hyperaktivní až agresivní. Epileptické záchvaty nejsou četné, často přicházejí až po delším období afázie a dominujícím problémem je porucha verbální komunikace. Pro diagnostiku LKS jsou charakteristické EEG hroty a pomalé vlny s nekonstantním centrotemporálním maximem s výraznou tendencí ke generalizované kontinuální sw aktivitě v pomalém synchronním spánku [21]. Při pečlivém vyšetření spánkového EEG nalezneme téměř u všech dětí s plně rozvinutým LKS obraz tzv. elektrického statu (electrical status epilepticus in slow sleep neboli ESES), kdy generalizované vysokovoltážní komplexy sw nahrazují v 85 a více procentech fysiologickou N-REM aktivitu (obr.4). Proto pokud možno vyšetřujeme u všech dětí s poruchou řeči spánkové EEG, je však otevřenou otázkou zda epileptiformní spánkové EEG - resp. tzv. subklinický elektrický status - kauzálně souvisí s vývojovými poruchami u dětí [22].
V léčbě se uplatňují benzodiazepiny, kortikoidy – ACTH , sultiam a valproát . Kontroversní jsou výsledky podávání vigabatrinu, objevily se nové informace dokladující efekt levetiracetamu. Efektivní je i podávání imunoglobulinů, otázkou je zda aplikaci má předcházet vyšetření likvoru za účelem zjištění vyšších titrů IGG jako indikačního faktoru. V liratuře nepanuje jednotný názor, osobně vyšetření likvoru doporučuji. Kontraindikován je carbamazepin, po lamotriginu jsou popsány jak případy se zlepšením tak i se zhoršením klinického a EEG nálezu. Efekt terapie lze posoudit na základě vymizení sw výbojů ve spánkovém EEG , nicméně toto zlepšení nemusí být provázeno klinickou úpravou řeči a v řadě případů zůstává trvalý deficit. Po 12 roce lze u většiny dětí očekávat spontánní zlepšení EEG a mírný ústup klinických obtíží.
7.3.5. Syndrom s kontinuálními hroty a vlnami v pomalém spánku
Mezi odborníky se stále vede diskuse zda CSWS je samostatným syndromem či variantou LKS (nebo naopak zda LKS je jednou z forem CSWS.) Jak LKS tak CSWS vykazují velmi podobné elektroencefalografické rysy (ESES), věkovou vazbu, odezvu na léky i prognosu. Klinický obraz kognitivně behaviorálních poruch začíná obvykle ve věku 4.-5. let a předpokládá se, že elektrický spánkový status nastupuje dříve, ale nebývá včas rozpoznán . Záznam bdělého EEG může před plným rozvinutím elektrického statu ozřejmit fokální či multifokální ostré vlny s pomalou složkou. První záchvaty bývají unilaterální, noční, někdy v nakupení, ve druhé fázi se připojují atypické či myoklonické absence, někdy atonické záchvaty. Tonické záchvaty pozorovány nejsou. Po několika letech obvykle dochází k remisi a zklidnění z hlediska záchvatů, nicméně kognitivně behaviorální problémy mohou přetrvávat a to zejména pokud ESES trval déle než tři roky. Léčba CSWS je prakticky totožná s doporučeními uvedenými u Landau-Kleffnerova syndromu [23], hlavní důraz se kromě valproátu klade jednak na kortikoidy [24] a pulsní terapii benzodiazepiny [25].
7.3.6. Rasmussenův syndrom
Rasmussenův syndrom též bývá definován jako " epilepsia partialis continua dětského věku" a měl by být odlišen od ostatních věkově nezávislých parciálních statů, které obvykle mívají dobře určitelnou etiologii (nádor, dysplasie, mitochondropatie aj). Jde o vzácně se vyskytující syndrom, jehož incidence není přesně definována. Postihuje děti mezi prvním až desátým rokem věku, maximum začátku se udává kolem 5-6 roku. Semiologicky se jedná v první fázi onemocnění o isolované jednoduché častěji motorické než sensorické záchvaty, které se postupně shlukují až vyústí v obraz kontinuální parciální epilepsie. Jsou přísně jednostranné - kontralaterálně k postižené hemisféře. Hemiparesa bývá v první fázi jen postiktální s mírným deficitem i interiktálně. Ve druhé klinické fázi se zhruba po čtvrt roce a delším trvání stav výrazně zhoršuje, narůstá četnost záchvatů a rozvíjí se hemiplegie, přidává se mentální deficit. Pokud nedojde včas k provedení epileptochirurgického zákroku , dospěje onemocnění do finálního stádia s katastrofickým scénářem nezvladatelného status epilepticus a mentální deteriorace [26]. Etiopatogeneze není známa, předpokládá se autoimunní proces s neurogliální a T-lymfocytární reakcí připomínající sclerosis multiplex a v.s. i se specifickými protilátkami proti glutamátovým receptorům jedné hemisféry. Není známo zda se v patogenezi uplatňují virové infekce či vakcinace jako spouštěcí faktor a jakou roli může hrát genetická disposice. Diferenciálně diagnosticky je velmi obtížné jen na základě kliniky a MRI nálezů odlišit nádorové či neuroinfekční procesy imitující obraz Rasmussenovy encefalitidy. Nicméně podezření z Rasmussenova syndromu by mělo být vysloveno vždy v případě refrakterních parciálních záchvatů a unilaterálního pomalu progredujícího procesu s postupnou destrukcí mozkové tkáně. Léčba antiepileptiky nebývá úspěšná a může v první fázi jen modulovat průběh onemocnění. Dočasný efekt lze očekávat od aplikace imunoglobulinů, kortikoidů či plasmaferesy, pozoruhodná je studie týkající se pozitivního efektu tacrolimu. Zásadní pro osud pacientů je optimální načasování epileptochirurgického zákroku (funkční případně anatomická hemisferektomie), který by měl být proveden ve fázi rozvíjející se hemiparesy.
7.3.6 Migrující fokální epilepsie u kojenců
Migrující fokální epilepsie u kojenců se objevuje mezi prvním týdnem života a 7 měsícem, zpočátku jsou parciální motorické křeče v průměru jedenkrát týdně, ale během několika týdnů až měsíců se stávají stále frekventnější a objevují se v nakupení o počtu 5-30 několikrát denně. Diagnostickým problémem mohou být autonomní záchvaty charakteru apnoe s cyanosou či ataky zrudnutí, které nemusí být správně rozpoznány. Terapie je svízelná, byl popsán efekt stiripentolu a clonazepamu, carbamazepin a vigabatrin mohou průběh onemocnění zhoršit.
7. 4. Idiopatické benigní fokální syndromy
Idiopatické benigní fokální záchvaty jsou po febrilních záchvatech druhou nejpočetnější "rodinou" a vyskytují se až u čtvrtiny všech "non-febrilních" věkově vázaných epileptických syndromů [27,28,29]. Z klasifikačního hlediska jsou velmi homogenní skupinou s velmi nadějnou prognosou. Osobně přiznávám, že po semiologickém a EEG rozboru idiopatických fokálních záchvatů, rád předávám rodičů dvě zprávy - špatnou (vaše dítě má epilepsii) a dobrou (po patnáctém roce bude bez záchvatů a bez terapie s šancí na prakticky normální život). Je však třeba doplnit i třetí ambivalentní informaci: je možné, že tato epilepsie nepříznivě ovlivní školní výkony dítěte.
7.4.1. Benigní epilepsie s rolandickými hroty
Benigní epilepsie s rolandickými hroty (BERS) je totožná se syndromem benigní epilepsie s centrotemporálními hroty (BECTS) a tvoří 15 až 20 % všech dětských epilepsií. Vyskytuje se zejména u chlapců mezi 3. až 9 rokem . Semiologicky se jedná o krátké klonické křeče jednotlivých svalových skupin v obličeji - obvykle ústního koutku ( orofaciální), někdy v kombinaci se záškuby na horní končetině (brachiofaciální). Časté je chrčení, polykání, dysarthrie, slinění, někdy předchází nebo následují parestesie v oblasti jazyka, poloviny obličeje, ruky. Záchvaty nejsou striktně jen na jedné straně, další záchvat může být druhostranný. Bývá tendence k nakupení, nebývá porucha vědomí a ve spánku (vzácněji ve dne) přechod do generalizovaného tonicko-klonického záchvatu. Charakteristický je EEG nález vysokovoltážních bi - až trifázických hrotů s následnou pomalou vlnou v centrotemporální oblasti na pozadí věku odpovídající základní aktivity (obr.5). V ospalosti a ve spánku jsou výboje výrazně četnější a mohou mít charakter kontinuálního fokálního statu.
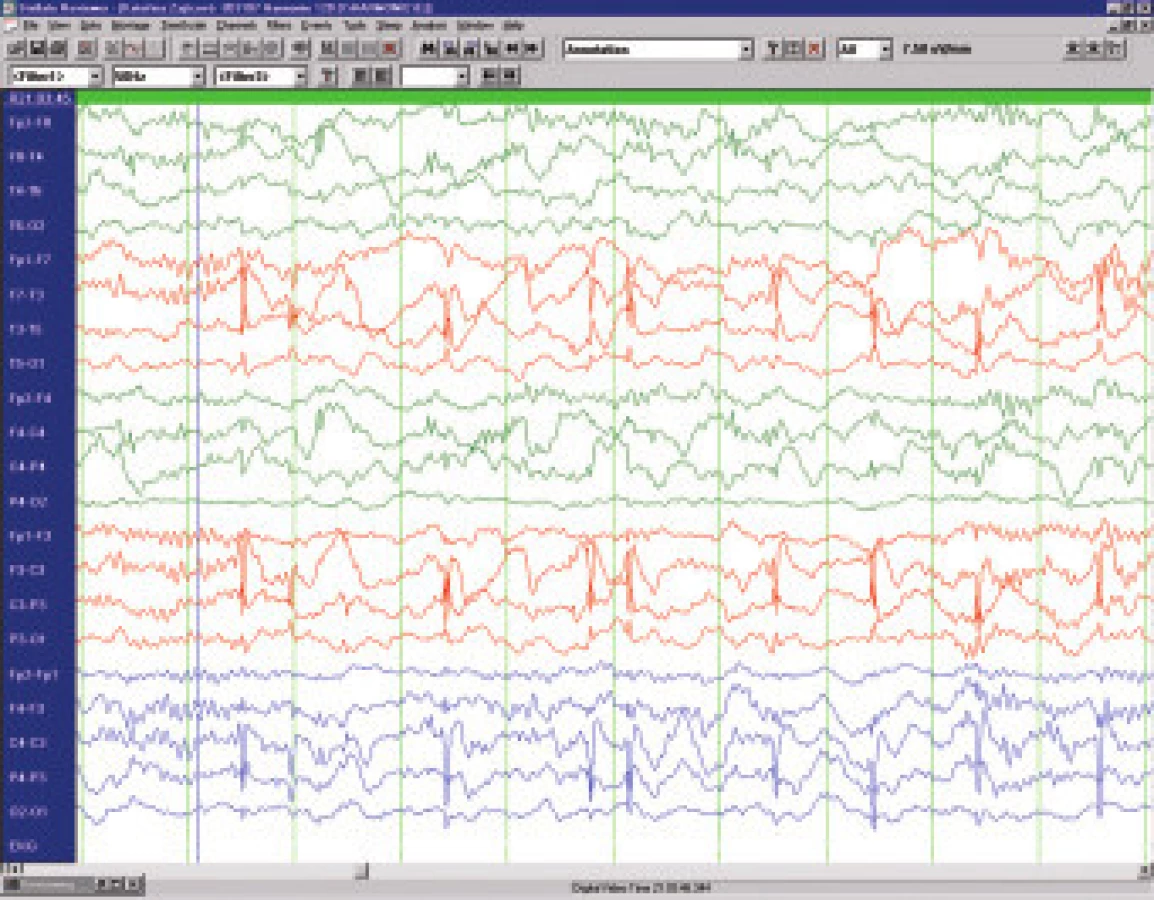
Léčba BERS bývá obvykle zdrženlivá - pokud záchvaty nejsou časté, na EEG není extrémní frekvence výbojů a spolupráce s rodinou je dobrá, tak se zahájením dlouhodobé terapie vyčkáváme. Věčně diskutovanou otázkou je zda "léčit" děti s frekventními výboji a školními problémy.
Za lék první volby osobně považuji sultiam , u mladších školních dětí v obvyklé dávce 150 až 300 mg na noc nebo rozděleně do několika denních dávek.. Sultiam jako lék první volby byl potvrzen v randomizované dvojitě slepé placebo kontrolované studii Ratinga [30] na souboru sledovaném po dobu 6 měsíců. Bez záchvatů bylo 81% (25/31) léčených sultiamem oproti 29% (10/35) užívajících placebo (p = 0.00002 .V současnosti je sultiam kromě Německa a Izraele lékem první volby i v řadě dalších zemí včetně České republiky a Slovenska.. Paradoxně nejen v rozvojových zemích, ale i v USA je jako lék první volby doporučován carbamazepin nebo fenytoin či lamotrigin , které mohou zhoršit průběh níže uvedené atypické varianty. V řadě doporučení vítězí jako universální lék valproát a nelze opomenout i studii, která potvrdila vyšší efektivnost gabapentinu v dávce 30 mg/kg v porovnání s placebem.
Atypická benigní parciální epilepsie (ABPE) nebo-li pseudo-Lennoxův syndrom (PLS) je svéraznou variantou rolandického syndromu, a jeho povaha se mnohdy ozřejmí až v souvislosti se zhoršením EEG i klinického stavu po nasazení carbamazepinu. U tohoto syndromu se sice u 56 % vyskytují téměř kontinuální výboje v pomalém spánku („near ESES“ vzorec), ale u 72 % dětí dojde k EEG normalizaci, a u 100 % vymizí po 15. roce záchvaty. Nicméně více než 50 % potřebuje pomoc pro dílčí oslabení školního výkonu a dle Hahnovy studie muselo docházet do speciálních škol [31]. Kromě rolandických záchvatů se u těchto dětí může objevit (zejména po CBZ) nakupení malých záchvatů se ztrátou kontaktu, sliněním a drobnými myokloniemi, ale žádné dítě s ABPE nemělo tonické záchvaty či výboje 10-12 Hz ve spánkovém EEG. V maximálně vyjádřené variantě se ABPE elektroencefalograficky podobá LKS a CSWS a klinicky se jen málo liší od Dooseho či Lennox Gastautova syndromu . Proto je zejména německými autory označován termínem pseudo Lennoxův syndrom. U refrakterních variant ABPE zkoušíme kromě sultiamu a valproátu i benzodiazepiny, kortikosteroidy či levetiracetam.
7.4.2 Benigní epilepsie s occipitálními hroty - Panayitopoulosův syndrom
Panayitopoulosova varianta - někdy nazývaná EBOS ( early onset benign childhood occipital seizures ) má maximu výskytu mezi 2. - 6. rokem , dominují noční záchvaty s deviací bulbů, zvracením a alterací vědomí, častá je iktální/postiktální cefalea , na EEG jsou occipitální hroty aktivující se zavřením očí. Diferenciálně diagnosticky je zejména nutné odlišit migrénosní záchvaty. Prognosa je příznivá a to navzdory pozorovaným zhoršením průběhu až rázu autonomního status epilepticus.
Z léčebného hlediska je pozoruhodné, že sám autor tohoto syndromu doporučuje pro rekurentní záchvaty carbamazepin, případně clobazam, levetiracetam a sultiam. Je pravda, že nebyly publikovány údaje o agravaci tohoto syndromu CBZ, ale osobně bych se přiklonil spíš k valproátu než carbamazepinu.
7.4.3. Benigní epilepsie s occipitálními hroty - Gastautův syndrom
Gastautova varianta BEOS má maximum výskytu mezi 3.-12 . rokem, semiologicky dominují visuální halucinace, poruchy visu a forsírované zavírání (mrkání) očí, na EEG jsou sw occipitálně nebo vzadu temporálně tlumící se otevřením očí, časté jsou i bilaterálně synchronní sw a polysw s maximem centrotemporálně. Asi deset procent pacientů má iktální i postiktální bolesti hlavy a opět je nutné odlišit migrenosní záchvaty. V terapii je rovněž doporučován carbamazepin.
7.5. Idiopatické generalizované epileptické syndromy
7.5.1 Myoklonicko-astatická epilepsie
Prevalence Dooseho syndromu je do 2 % ze všech dětských epilepsií. Objevuje se ve věku od 7 měsíců do 6ti let s peakem mezi 2. a 4. rokem a manifestuje se symetrickými myoklonickými záškuby s následnou axiální atonií resultující v pokles hlavy, podlomení kolen a případný prudký pád s rizikem zranění. Asi polovina pacientů může mít nakupení malých záchvatů rázu absencí a třetina může být postižena nonkonvulsivním statem [32,33].
Semiologie záchvatů v rámci MAE bývá někdy zaměňována za iniciální stadia LGS, ale u MAE nikdy nebývají tonické záchvaty a prognosa je mnohem příznivější.[34]. Na EEG jsou výboje nepravidelných vícečetných hrotů o vysoké amplitudě s následnou pomalou vlnou, které mohou být vystřídány vysokovoltážními synchronními vlnami (obr. 6.). Základní aktivita bává převážně z pásma theta, častá je fotoparoxysmální odpověď. V terapii se osvědčil valproát v monoterapii nebo lze otestovat kombinaci valproátu s malými dávkami clonazepamu nebo clobazamu či jiných benzodiazepinů, lze doporučit levetiracetam, topiramát (v průměrné dávce kolem 7 mg na kg váhy a den) i lamotrigin, případně ACTH, v úvahu přichází i sultiam, naopak zcela nevhodný je carbamazepin.
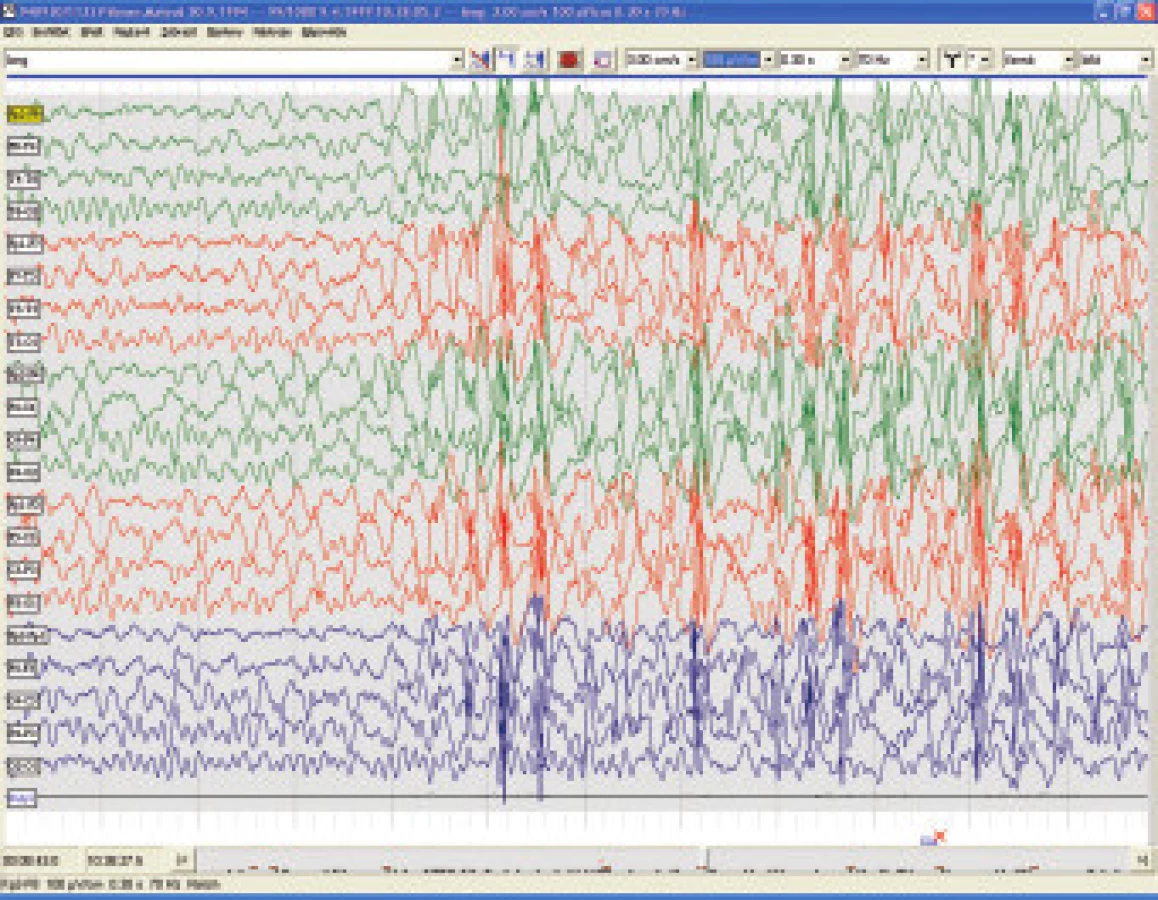
7.5.2. Dětské absence
Dětské absence (též CAE - childhood absence epilepsy) jsou též označovány synonymem pyknolepsie pro nahuštění záchvatů do několik stovek za den. Postihují až deset procent ze všech epilepsií dětského věku v rozmezí mezi 3. až 10 rokem - maximum je kolem 5-6. roku.
Semiologicky je charakteristický náhlý krátkodobý (5-15 sekund) výpadek (absence) kontaktu s okolím, obvykle bez výraznějších motorických projevů (simplexní - prostá absence). Lze rozlišit absence s klonickou komponentou (např. záškuby víček v rytmu 3/s), s atonickou komponentou (nepatrné poklesnutí hlavy někdy v rytmu 3/s), vzácněji s tonickou komponentou (tonické stočení očí vzhůru až záklon hlavy), případně i s autonomní komponentou (rozšíření zornic, zblednutí či zrudnutí, tachykardie). Dětské absence lze velmi dobře vyprovokovat pomalým hlubokým dýcháním a/nebo ospalostí. Patognomický je EEG nález se synchronním výskytem hrotů a vln 3/s, obvykle s centrálním a/nebo frontálním maximem. Etiopatogeneticky lze absence a ostatní idiopatické genralizované syndromy řadit mezi geneticky podmíněné iontoforopatie, kdy u absencí se jedná o poruchu funkce talamických neuronů s nízkoprahovými vápníkovými kanály T typu [35].
Diferenciálně diagnosticky je třeba odlišit samostatný syndrom myoklonických absencí, kde se absence kombinují s prudkými myoklony a až v polovině případů jsou refrakterní na terapii [36]. EEG nález je obdobný jako u syndromu CAE, ale v iktálním video-EEG je každý hrot doprovázen myoklonickým záškubem (viz obrázek 7).


V léčbě absencí je na prvním místě monotoreapie valproátem (u myklonických absencích ve vysokých dávkách) nebo etosuximidem ( s rizikem, že ETS nepokryje koincidentální GTKZ) nebo spíše lamotrigin, který podobně jako valproát potlačí i případné generalizované tonicko klonické záchvaty. Nověji je doporučován i levetiracetam, chybí však kontrolovaná studie[37]
7.5.3 Juvenilní absence
Syndrom juvenilních absencí tvoří 2-3 procenta ze všech epilepsií a až 10 procent ze všech IGE . Nejčastěji se objeví mezi 9-13 rokem. Juvenilní absence mají na rozdíl od CAE spíše než pyknoletickou spanio - resp. cykloleptickou distribuci záchvatů a rychlejší frekvenci sw komplexů na EEG. prognosa je rovněž méně příznivá – až u ¾ se objeví gneralizovaný tonicko klonický záchvat ( často rázu GMA) a asi jedna čtvrtina postižených má sporadické myoklony. Nicméně asi u 80 procent lze očekávat komepnsaci na monoterapii. Úplné vysazení AE je u pacientů s výskytem GTKZ a myoklonií obtížné. Léčba je méně úspěšná než u CAE, kromě valproátu je doporučován lamotrigin a levetiracetam.
7.5.4 Juvenilní myoklonická epilepsie
JME se vyskytuje až u 10 procent všech pacientů s epilepsií, typický peak výskytu je kolem 14-15. rokem. Semiologicky jsou typickým projevem jsou prudké (“impulzívní”) myoklony převážně extenzorů horních končetin, obvykle oboustranné, které nemusí být plně symetrické. Charakteristický je jejich výskyt po probuzení - u snídaně, v koupelně, aktivují se předchozí spánkovou deprivací a mohou být někdy “spuštěny” prudkou změnou osvětlení. Myoklonické záchvaty se mohou kombinovat s velkými tonicko - klonickými záchvaty po probuzení. Na EEG je iktálně typický nález krátkých výbojů charakteru mnohočetných hrotů s následnou pomalou vlnou (polyspike and wave komplex - PSW), které jsou někdy doprovázeny klinickým záškubem. Interiktálně jsou PSW kratšího trvání (maximálně dva až tři hroty před pomalou vlnou). Při podezření na JME je třeba vždy vyšetřovat fotostimulací, někdy i po předchozí spánkové deprivaci. Pozitivní odpověd na intermitentní fotostimulaci bývá u 30 % chlapců a 40 % dívek. Etiopatogeneticky jde o idiopatickou geneticky podmíněnou chorobu, část pacientů má mutaci lokalizovanou na 6. chromosomu, dynamická zobrazovací vyšetření naznačují možnost dysunkce mesiofrontálních struktur.
V terapii se obvykle uvádí na prvním místě valproát, ale u dospívajících dívek je třeba zvážit jak endokrinní rizika tak eventuelní možné riziko pro plod v případě gravidity. Je třeba poznamenat, že riziko aktivace syndromu polycystických ovarií valproátem bývá přeceňováno a lze předpokládat určité predispoziční faktory u postižených žen. Lékem první volby je rovněž lamotrigin, v současnosti se mezi léky první volby zařadil i levetiracetam Obsolentnější a nikoli na důkazech založená je terapie primidonem. Zcela nevhodný je carbamazepin, který mylně nasazený na fokální klonické záchvaty, může průběh onemocnění i zhoršit Prognosa je dobrá, ale trvalá terapie nízkými dávkami antiepileptik a dodržování životosprávy je obvykle nutná.
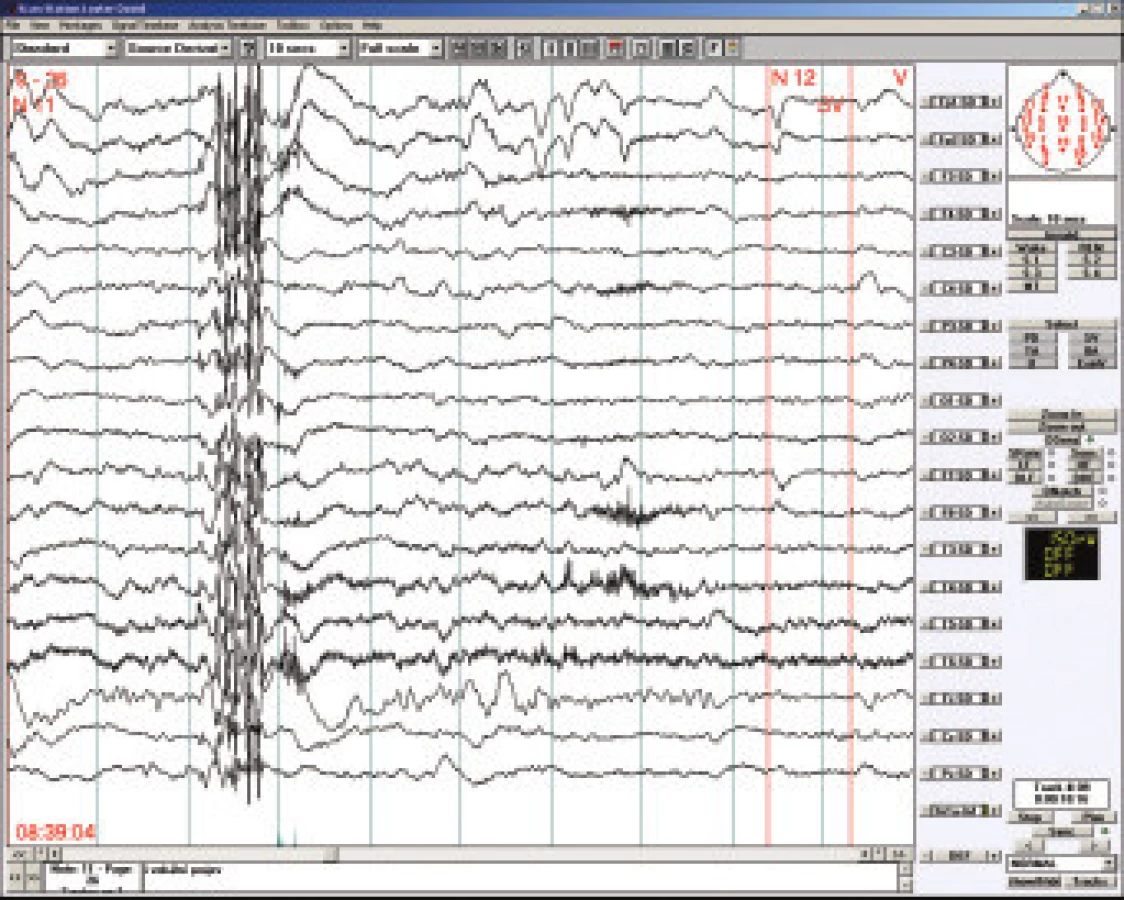
7.5.5. Grand mal vázaný na probuzení
Isolovaný výskyt čistých GTZK je relativně vzácný a tvoří asi 1% ze všech IGE. Nejlépe prostudovanou formou je tzv. „grand mal on awakening“ (GMA) se záchvaty po ranním probuzení ze spánku a to v rozmezí od několika minut až do dvou hodin. někdy předchází spánková deprivace. Ve dne se mohou záchvaty objevit při odpolední relaxaci a náhlém probrání z krátkého spánku. Prudké osvětlení může záchvat aktivovat. Vhodným lékem je večerní maximální dávka retardovaného valproátu, dobré zkušenosti jsme
měli s primidonem nebo lamotriginem, podávaným rovněž ve večerní maximální dávce. V současné době lze zvážit i podávání levetiracetamu. Důležitá je úprava životosprávy, vyvarovat se zkrácení spánku a nácvik správného stereotypu probouzení.
7.6. Reflexní epilepsie
Reflexní epilepsie bývá někdy definována jako "syndrom u kterého jsou veškeré epileptické záchvaty vyvolány sensorickým podnětem". V duchu této definice do tohoto syndromu nepatří agravace EEG výbojů či záchvatů fotostimulací. Nicméně někteří autoři navrhují do rámce tohoto zastřešujícího syndromu zavzít i epilepsie, u kterých je sice většiná záchvatů vyprovokována sensorickým (nejčastěji světelným) podnětem, ale sporadicky se objevují i spontánní záchvaty (např. JME či GMA). Charakter vyvolávajícího stimulu je značně různorodý: od obecně známého přerušovaného světla, přes televizní obrazovku či videohry až po velmi specifické podněty (čtení jen určitého druhu textu, poslech konkrétní písně apod). Za reflexní se nepovažují záchvaty vyvolané vysazením alkoholu či iniciální fází virového onemocnění . Semiologicky se obvykle jedná o generalizované záchvaty myoklonické, generalizované tonicko klonické a absence, méně často o fokální visuální, motorické či sensorické. Etiopatogeneticky se téměř výhradně jedná o idiopatické epilepsie, zajímavou variantou je okcipitální epilepsie vyvolaná přerušovaným světlem, příkladem jasně vyhraněné jednotky je Jeavonsův syndrom
7.6.1 Jeavonsův syndrom
Objevuje se asi u tří procent pacientů trpících epilepsií a tvoří asi 13% ze všech IGE Vyskytuje se dvakrát častěji u u ženského pohlaví , v dětském věku obvykle mezi 3.-12. rokem s maximem mezi 6.-8. rokem. Semiologicky se jedná o myoklonie víček, které mohou být spojeny s absencemi, typická je aktivace EEG výbojů i klinických záchvatů zavřením očí. Vzácně se objeví i myklonie na HK. Všichni postižení jedinci jsou fotosensitivní, asi u pětiny pacientů se může vyskytnout nakupení až status Na EEG jsou charakteristické 2-3 sekundové vysokovoltážní výboje sw či psw od 3 do 6hz.
V léčbě se uplatní valproát, clonazepam, clobazam, etosuximid a levetiracetam.
7.6.2. Idiopatická čtecí epilepsie (idiopatic reading epilepsy)
Poměrně vzácná forma postihující asi 2 z tisíce epilepsií trpících dětí, nejčastější je u dospívajíchc s téměř dvounásobnou převahou mužského pohlaví. Semiologicky jsou typické kratičké periorální myoklonie, někdy téměř neviditelné a pociťované pacientém jako záchvěvy. Jsou výhradně vázané na čtení textu a pokud pacient čtení nepřeruší, může se rozvinout generalizovaný tonicko klonický záchvat. V léčbě je na prvním místě valproát, efektivní je clonazepam a recentně i levetiracetam.
Závěr
Uvedený přehled vybraných 25 věkově vázaných epileptických syndromů není a nemůže být zcela úplný. Chybí desítky specifických jednotek (namátkou Rettův či Angelmannův syndrom, Laforova choroba, Neuronální ceroid lipofuscinosa aj), u kterých jsou sice dramatické epileptické záchvaty v popředí klinického obrazu, ale nelze je řadit mezi relativně homogenní a vyhraněné epileptické syndromy, na jejichž rozpoznání a klasifikaci měla zásluhu především Gastautova marseillská škola. Některé syndromy se sice částečně překrývají nebo do sebe přecházejí, nicméně lze odlišit jejch obrysy a rozhodnout se pro adekvátní léčebný postup.
Navržená antiepileptika pro jednotlivé syndromy je třeba vnímat jen jako orientační ukazatele, které sice vycházejí z mezinárodních (ILAE), evropských (EUREPA) a českých (EpiStop) doporučených postupů i z recentních publikací , ale jsou nepochybně kontaminovány vlastní zkušeností autora a „Dolanského epileptologickou školou" zdůrazňující individuální přístup. Vždy je třeba myslet na riziko paradoxního účinku některých antiepileptik, která mohou aktivovat epileptické záchvaty [38] a být tak nejen dobrým sluhou, ale i zlým pánem.
Recenzenti:
doc. MUDr. Pavol Sýkora, CSc.
MUDr. Hana Ošlejšková
MUDr. Dana Šišková
Přijato k recenzi: 25. 6. 2007
Přijato do tisku: 30. 8. 2007
doc. MUDr. Vladimír Komárek, CSc.
Klinika dětské neurologie a Centrum pro epilepsie 2. LF UK a FN v Motole
V úvalu 84, 150 06 Praha 5
e-mail: vladimir.komarek@lfmotol.cuni.cz
Doc. MUDr. Vladimír Komárek, CSc.

Po promoci v roce 1973 pracoval jako dětský lékař v Teplicích a Duchcově, od roku 1979 působil jako dětský neurolog v Praze 4, od roku 1984 pracuje na Klinice dětské neurologie UK 2. LF ve FN Motol, přednostou kliniky je od roku 1991. Absolvoval studijní stáž v Centru pro epilepsie v Heemstede a byl na kratších pracovních pobytech v epileptologických centrech v Clevelandu, Sandvice a Bielefeldu, v současnosti spolupracuje s univerzitními pracovišti v Bonnu a Kiehlu. Je dlouhodobě aktivním členem výboru České ligy proti epilepsii a České společnosti dětské neurologie, v roce 1999 organizoval jako generální sekretář 23. světový epileptologický kongres, v roce 2003 obdržel ocenění ILAE „Ambassador for Epilepsy“ a v roce 2002 získal certifikát „EUREPA-Trainer“. Je členem ILAE Commisssion on Pediatrics, členem redakční rady „Epileptic Disorders“ a členem širší redakční rady "Neurologie pro praxi". Je zakladatelem a předsedou správní rady Nadace dětský mozek a členem Akademického senátu fakulty. Byl hlavním řešitelem jednoho mezinárodního grantového projektu podporovaného US-Czech S&T Fund a sedmi grantů IGA MZČR. Napsal monografii Epileptické záchvaty a syndromy, je spoluatorem monografie Dětský autizmus a spolueditorem učebnice Dětské neurologie. Dále je autorem řady kapitol v našich i zahraničních monografiích a 140 časopiseckých publikací.
Vědomostní test
1. AE první volby u novorozeneckých záchvatů jsou:
- a) VPA a CBZ
- b) benzodiazepiny
- c) PB a PHE
- d) ACTH
2. AE první volby u Westova syndromu jsou:
- a) VPA
- b) nitrazepam
- c) ACTH nebo VGB
- d) TPM
3. Nejčastějším věkově vázaným syndromem je:
- a) WS
- b) BERS
- c) febrilní záchvaty
- d) JME
4. Nasazení CBZ může vyvolat:
- a) tonické záchavaty
- b) zvýšení frekvence sw výbojů ve spánkovém EEG
- c) myoklonie
- d) zmnožení parciálních komplexních záchvatů
5. AE první volby u JME jsou:
- a) VPA a LTG
- b) CBZ
- c) PHE
- d) LEV
6. U dětí s hmotností nižší než 15 kg podáváme při záchvatu rektální diazepam v dávce:
- a) 10 mg
- b) 5 mg
- c) 15 mg
- d) 2 mg
7. Synonymem pro syndrom získané afázie s epilepsií je:
- a) Lennox-Gastautův syndrom
- b) Landau-Gastautův syndrom
- c) Landau-Kleffnerův syndrom
- d) Lennox-Kleffnerův syndrom
8. Kortikosteroidy podáváme v léčbě epilepsie u:
- a) JME
- b) LKS
- c) WS
- d) LGS
9. Pro Panayitopoulusův syndrom je charakteristické:
- a) noční výskyt záchvatů
- b) periiktální bolest hlavy
- c) myoklonie
- d) poruchy vizu
10. EEG vykazující rysy „supression burst“ je u:
- a) Westova syndromu
- b) Ohtaharova syndromu
- c) Lennox-Gastautova syndromu
- d) Aicardiho syndromu
11. Porucha funkce sodíkových kanálů byla nalezena u:
- a) benigních familiárních novorozeneckých křečí
- b) GEFS+
- c) JME
- d) SMEI
12. Mezi léky první volby u syndromu JME nepatří:
- a) VPA
- b) CBZ
- c) LTG
- d) LEV
13. Kontinuální výboje ve spánku jsou časté u:
- a) Lennox Gastautova syndromu
- b) Landau Kleffnerova syndromu
- c) atypické benigní parciální epilepsie
- d) JME
14. Mezi léky indikované u BERS nepatří:
- a) sulthiam
- b) fenytoin
- c) valproát
- d) karbamazepin
15. U Jeavonsova syndromu se vyskytují:
- a) tonické křeče
- b) myoklonie víček
- c) absence
- d) automatizmy
16. U Gastautovy varianty BEOS nebývají:
- a) poruchy vizu
- b) absence
- c) mrkání víčky
- d) myoklonie
17. Fotosenzitivní odpověď se vyskytuje u:
- a) JME
- b) LGS
- c) LKS
- d) GMA
18. V rámci GMA syndromu se vyskytují:
- a) tonické záchvaty před probuzením
- b) tonicko-klonické záchvaty po probuzení
- c) myoklonie vázané na probuzení
- d) atatické záchvaty po probuzení
19. Felbamát je indikován u:
- a) myoklonií v rámci MAE
- b) Westova syndromu
- c) tonických záchvatů v rámci LGS
- d) absencí
20. U syndromu myoklonicko-astatické epilepsie nejsou:
- a) absence
- b) tonické záchvaty
- c) astatické záchvaty
- d) myoklonie
správná je jedna nebo více odpovědí
Správné odpovědi: WWW.CSNN.EU
Sources
1. Menkes JH, Sarnat HB, Maria BL. Child Neurology 7th ed. Philadelphia: Lippincott Williams& Wilkins 2006 : 857-942.
2. Roger J, Bureau M, Dravet Ch,Genton P, Tassinari CA, Wolf P. Epileptic syndromes in infancy, chidlhood and adolescence 3rd ed. Eastkeight UK: John Libbey&Co Ltd. 2002.
3. Komárek V. Epileptické záchvaty a syndromy. Praha: Galén 1997.
4. Klein P, van Passel-Clark LMA, Pezzullo JC. Onset of epilepsy at the time of menarche. Neurology 2003; 60 : 495-497.
5. Panayitopoulos CP. The Epilepsies: Seizures, Syndromes and Management. Bladon Medical Publishing 2004.
6. Dolanský J. Současná epileptologie. Praha: Triton 2000.
7. Rennie JM. Neonatal seizures and their treatment. Curr Op Neurol 2003 : 16; 177-181.
8 Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression burst. J Clin Neurophysiol 2003; 20(6): 398-407.
9. Kršek P, Tichý M, Zámečník J, Paulas L, Faladová L, Jiruška P et al. Life.saving epilepsy surgery for status epilepticus caused by cortical dysplasia. Epileptic Disorders 2002, 4(3): 203-208.
10. Kršek P, Sebroňová V, Procházka T, Maulisová A, Komárek V. Successful treatment of Ohtahara syndrome with chloral hydrate. Pediatric Neurology 2002; 27(5): 388-391.
11. Sýkora P. Liečba epileptických syndromov v detskom veku s nepriaznivou prognózou. Neurol pro praxi 2007; 2 : 87-90.
12. Dalla Bernardina B, Dulac O, Fejerman N, Dravet C, Capovilla G, Bondavalli S et al. Early myoclonic epileptic encephalopathy (EMEE). Eur J Pediatr 1983; 140 : 248-252.
13. Offringa M. Seizures associated with fever in childhood. Rotterdam: Mosby 1995.
14. Riikonen R. A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics 1982; 13 : 14-23.
15. Mackey MT, Weiss SK, Adams-Webber MLS, Ashwal S, Stephens D, Ballaban-Gill K et al. Practice parameter: medical treatment of infantile spasms. Report of the American Academy of Neurology and the Child Neurology Society. Neurology 2004; 62 : 1668-1681.
16. Hancock E, Osborne JP. Vigabatrin in the treatment of infantile spasms in tuberous sclerosis: literature review. J Child Neurol 1999; 14 : 71-74.
17. Uthman BM, Reid SA, Wilder BJ, Andriola MR, Beydoun AA. Outcome for West syndrome following surgical treatment. Epilepsia 1991; 32 : 668-671.
18. Chiron C, Marchand MC, Tran A, Rey E, d´Athis P, Vincent J et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet 2000; 356(9242): 1638-1642.
19. Glauser TA, Levisohn PM, Ritter F, Sachdeo RC. Topiramate in Lennox-Gastaut syndrome:open-label treatment of patients completing a randomized controlled trial. Epilepsia 2000; 41(Suppl 1): S86-90.
20. Trevathan E, Motte J, Arvidsson J, Manasco P, Mullens L. Safety and tolerability of adjunctive Lamictal for the treatment of the Lennox-Gastaut syndrome: results of multinational, double-blind, placebo-controlled trial. Epilepsia 1996; 37 : 202(Abstract).
21. Pearl PL, Carrazana EJ, Holmes GL. The Landau-Kleffner syndrome. Epilepsy Curr 2001; 1(2): 39-45.
22. Patry G, Laygoubi S, Tassinari CA. Subclinical electrical status epilepticus induced by sleep in children. Arch Neurol 1971; 24 : 242-252.
23. Inutsuka M, Kobayashi K, Oka M, Hattori J, Ohtsuka Y. Treatment of epilepsy with electrical status epilepticus during slow sleep and its related disorders. Brain Dev 2005; 24: l-6.
24. Kramer U, Watemberg N, Zelnik N, Shahar E, Lerman-Sagie T, Goldberg-Stern H et al. Efficacy of corticosteroid therapy in treating epileptic encephalopathies and refractory epilepsies other than West syndrome. J Pediatr Neurol 2006; 4(3): 147-153.
25. De Negri M, Baglietto MG, Battaglia FM, Gaggero R, Pessagno A, Recanati L. Treatment of electrical status epilepticus by short diazepam (DZP) cycles after DZP rectal bolus test. Brain Dev 1995; 17 : 330-333.
26. Pardo CA, Vining EPG, Guo L, Skolasky RL, Carson BS, Freeman JM. The Pathology of Rasmussen Syndrome: Stages of Cortical Involvement and Neuropathological Studies in 45 Hemispherectomies. Epilepsia 2004; 45 : 516–526.
27. Holmes GL. Clinical Spectrum of Benign Focal Epilepsies of Childhood. Epilepsia 2000; 41(8): 1051-1052.
28. Chahine LM, Mikati MA. Benign partial localization-related epilepsies Part II. Syndromes in childhood. Epileptic Disorders 2006; 8 : 243 - 258.
29. Komárek V. Léčba věkově vázaných epileptických syndromů s příznivější prognózou. Neurol pro praxi 2007; 2 : 87-90.
30. Rating D, Wolf Ch, Bast T. Sulthiame as monotherapy in children with benign childhood epilepsy with centrotemporal spikes: a 6-month randomized, double-blind, placebo-controlled study. Epilepsia 2000; 41: 1284-1288.
31. Hahn A. Atypical benign partial epilepsy/pseudo-Lennox syndrome. Epileptic Disord 2000; 2 (Suppl 1): S11-17.
32. Neubauer BA, Halm A, Doose H, Tuxhorn I. Myoclonic-astatic epilepsy of early childhood-definition, course, nosography, and genetics. Adv Neurol 2005; 95 : 147-155.
33. Weber P. Tillmann B. Myoclonic-astatic epilepsy in early childhood: review of clinical signs, EEG features, etiology, and therapy. Klin Paediatr 2002; 214(5): 279-284.
34. Oguni H, Tanaka T. Treatment and Long-Term Prognosis of Myoclonie – Astatic Epilepsy of Early Childhood. Neuropediatrics 2002; 33(3): 122-132.
35. Khosravani H, Zamponi GW. Voltage-Gated Calcium Channels and Idiopathic Generalized Epilepsies. Physiol Rev 2006; 86(3): 941–966.
36. Perucca E. The Management of Refractory Idiopathic Epilepsies. Epilepsia 2001; 42 : 31–35.
37. Labate A. Levetiracetam in patients with generalised epilepsy and myoclonic seizures: an open label study. Seizure 2006; 15 : 214-218.
38. Sazgar B. Aggravation of Epilepsy By Antiepileptic Drug. Pediatric Neurology 2005; 33(4): 227-234.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2007 Issue 5
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Léčba epileptických syndromů u dětí
- Hodnocení edému terče zrakového nervu
- Jsou některé kontraindikace lumbální punkce dnes již obsoletní? Kazuistika
- Transforaminální lumbo-sakrální mezitělová fúze (TLIF) s instrumentací: prospektivní studie s minimálně 20měsíčním sledováním