
Tkáňové kultury pro studium prionových chorob
Cell Cultures for the Study of Prion Diseases
Transmissible spongiform encephalopathies, or prion diseases, comprise a group of fatal neurodegenerative disorders. Common to all these diseases is the accumulation of pathogenic prion protein isoforms in the brain. Much of the pathogenesis of prion diseases is studied in animal models. These experiments are time-consuming, expensive and ethically disturbing. In addition, animal models provide only limited scope for the study of biochemical aspects of these disorders in detail. Considerable efforts have been made to establish cell culture models supporting prion agent replication, but only a few permissive cell cultures have been found in which the stable propagation of prions can take place. Another restriction is associated with their ability to model only certain aspects of these diseases. The main advantage of cell culture is that it allows the rapid detection of prion infectivity and detailed study of processes linked to the propagation and spread of prions. Despite their limitations, cell cultures remain a powerful tool for the study of the mechanisms of prion propagation and the testing of anti-prion compounds that have much relevance to treatment of these diseases.
Key words:
transmission of spongiform encephalopathy – prions – cellular prion protein – cell cultures – experimental model
Authors:
K. Hobzová; O. Janoušková
Authors‘ workplace:
Ústav imunologie a mikrobiologie 1. LF UK v Praze
Published in:
Cesk Slov Neurol N 2010; 73/106(4): 379-386
Category:
Review Article
Overview
Transmisivní spongiformní encefalopatie neboli prionová onemocnění jsou smrtelné neurodegenerativní choroby, pro něž je charakteristická akumulace patologické izoformy prionového proteinu v mozku postižených jedinců. Běžným modelovým systémem pro studium jejich patogeneze jsou laboratorní zvířata. Experimenty na pokusných zvířatech jsou však zdlouhavé, drahé a eticky sporné. Navíc možnost detailního studia biochemických procesů spojených s těmito chorobami je velmi omezena. Řadu nevýhod odstraňuje použití tkáňových kultur. Buněčné linie schopné propagace patologické formy prionového proteinu umožňují rychle detekovat prionovou infektivitu a na molekulární úrovni studovat procesy spojené s přenosem a propagací prionů. Použití tkáňových kultur s sebou nese i omezení spojená s malým počtem linií schopných dlouhodobě propagovat priony a také se schopností modelovat pouze některé aspekty prionové infekce. Přesto tkáňové kultury významně posouvají možnosti studia prionových chorob, a to od výzkumu základních mechanizmů propagace prionů až po testování antiprionových látek, které by mohly najít uplatnění při jejich léčbě.
Klíčová slova:
transmisivní spongiformní encefalopatie – priony – buněčný prionový protein – tkáňové kultury – experimentální model
Zdroje podpory:
Grantová
agentura České republiky – projekty číslo GP310/09/P260 a GA310/08/0878,
Ministerstvo školství České republiky – projekt číslo MSM0021620806
Autoři děkují Ing. Karlu Holadovi, Ph.D., a MUDr. Robertu Rusinovi, Ph.D., za vstřícnost, ochotu a odborné rady.
Úvod
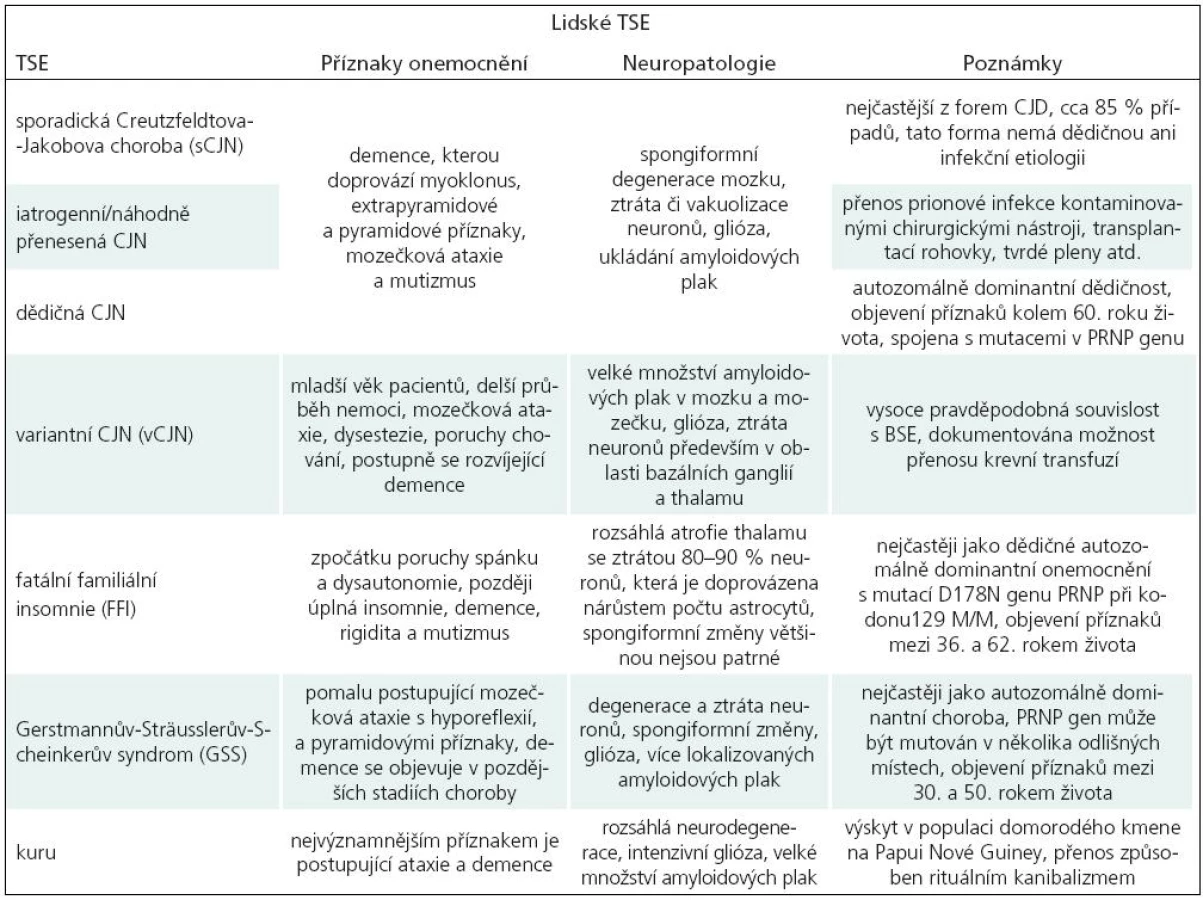
Transmisivní spongiformní encefalopatie (TSE) postihují jak člověka, tak zvířata. Lidské TSE mohou mít formu sporadickou (Creutzfeldtova-Jakobova choroba, sCJN), genetickou (GSS, FFI, fCJN) a infekční (iCJN, vCJN, kuru), viz tab. 1, více viz [1].
Zájem o TSE prudce stoupl během 90. let 20. století v důsledku rozsáhlé epidemie bovinní spongiformní encefalopatie (BSE) ve Velké Británii a následného přenosu BSE na člověka ve formě nové varianty CJN (vCJN). Pro tento přenos existuje řada nepřímých důkazů [2–4]. Zavedením bezpečnostních opatření, jako je zákaz zkrmování masokostní moučky a rozsáhlé testování dobytka s nucenou porážkou postižených stád, se podařilo epidemii BSE potlačit. Naštěstí se také nenaplnily katastrofické scénáře možného rozsahu epidemie vCJN. Od roku 2000 dochází k pozvolnému poklesu ročně zaznamenaných případů. Nicméně nadále zůstává nejasné, jaké množství lidí inkubuje vCJN priony bez klinických příznaků a zda mohou takto postižení jedinci být sekundárním zdrojem vCJN šířeného nosokomiální cestou. V současnosti se navíc v Severní Americe lavinovitě šíří chronické chřadnutí jelenovité zvěře (chronic wasting diseases, CWD), existují tedy obavy z možného přenosu CWD na hospodářská zvířata či přímo na člověka. Tato situace společně s dokumentovanou možností přenosu TSE transplantací rohovky [5,6] nebo krevní transfuzí [7,8] podtrhuje potřebu vývoje preklinického testu pro TSE a nalezení účinné léčby.
Hlavním diferenciálním znakem TSE je přítomnost konformačně odlišné patologické formy (PrPTSE) běžně se vyskytujícího buněčného prionového proteinu (PrPC), který je nejvíce exprimován v neuronální tkáni. PrPC je povrchový glykoprotein s doposud nepřesně specifikovanou funkcí, který se vyskytuje u všech savců, ale také u ptáků, plazů nebo ryb. Předpokládaný infekční mechanizmus stojící za prionovými onemocněními je schopnost abnormálního prionového proteinu PrPTSE změnit konformaci PrPC na PrPTSE bez účasti kódujících nukleových kyselin [9]. Přestože souvislost PrPTSE s prionovými onemocněními byla popsána před více než 25 lety, stále nejsou zodpovězeny základní otázky spojené s konverzí PrPC na PrPTSE, mechanizmem propagace PrPTSE v buňkách, podmínkami přenosu PrPTSE a fyziologickou funkcí PrPC. Hledání odpovědí na ně komplikuje řada faktorů, k nimž patří zejména velká podobnost molekul PrPTSE a PrPC, které se odlišují prakticky pouze rozdílnou konformací. PrPTSE obsahuje více než 40 % struktury β skládaného listu [10], zatímco PrPC obsahuje přibližně 3 % této struktury [11]. Dalším faktorem, který komplikuje studium PrP i prionových chorob obecně, jsou specifické fyzikálněchemické vlastnosti PrPTSE, především jeho sklon k tvorbě agregátů [12,13], a dále pak dlouhá inkubační doba, která je u člověka 5–30 let, u krávy 3–6 let a u myši asi 150 dní. Právě inkubační doba je jednou z největších nevýhod využití zvířecích modelů při studiu TSE. Dalšími nevýhodami jsou odlišnosti v expresi PrPC u různých živočišných druhů [10,14], finanční náročnost experimentů a obtížnost studia detailních biochemických procesů v živém organizmu.
Tkáňové kultury (TK) neboli buněčné linie schopné propagovat priony nabízejí možnosti, jak tyto limity překonat. TK jsou buňky, které jsou odvozeny od mnohobuněčných eukaryot, lze kultivovat in vitro v přesně kontrolovaných podmínkách (vhodná teplota, kultivační média, nasycení CO2 apod.). Primárně izolované buněčné linie, např. pokusných zvířat mají omezenou životnost, podléhají stárnutí a po určité době se buňky přestanou dělit. Široce využívané jsou proto tkáňové kultury, které jsou „nesmrtelné“. Zpravidla jde o buňky získané z nádorů nebo uměle imortalizované vnesením vhodných genů. V tomto případě se hovoří o kontinuálních buněčných liniích. I v nich si buňky obvykle zachovávají částečnou schopnost regulace buněčného dělení. Pokud ji ztratí a dělí se nekontrolovaně, mluvíme o transformované buněčné linii.
Většina buněčných linií vyžaduje k růstu vhodný povrch – adherentní kultury. Některé typy buněk dokáží přežívat a dělit se v suspenzi; například buněčné linie odvozené od krevních buněk.
Při dlouhodobé práci s buněčnou linií se zpravidla krátce před dosažením konfluence buňky od povrchu kultivační nádoby uvolní nejčastěji působením trypsinu nebo chelátorů dvojmocných iontů – EDTA. Vzniklá suspenze se naředí a nasadí do nových kultivačních nádob. Celý postup se označuje jako pasáž.
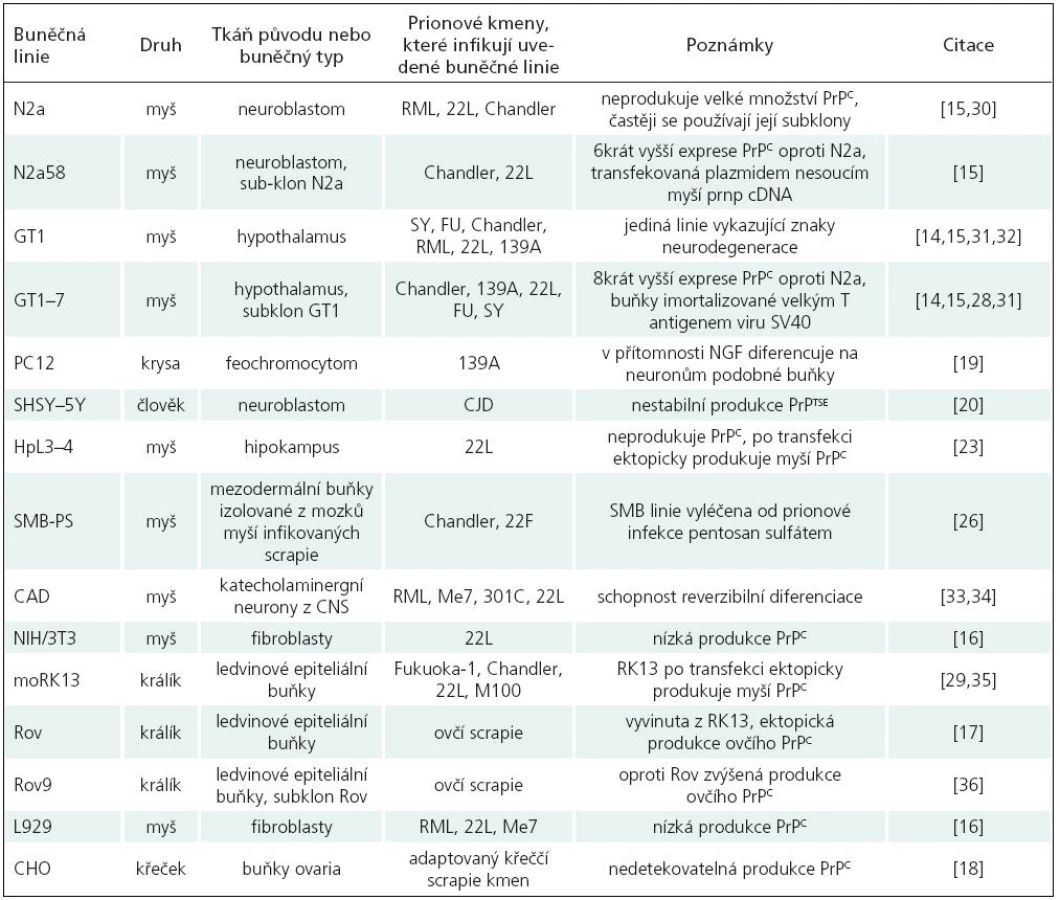
Pro studium prionových chorob je v současné době využíváno více než 15 adherentních buněčných linií, které jsou schopné dlouhodobé propagace PrPTSE.
Tkáňové kultury
Mezi nejpoužívanější buněčné linie patří především myší [15–17], několik králičích [18], křeččí [19] a krysí buněčné linie [20]; více viz tab. 2. Podařilo se sice vytvořit i několik lidských linií [21,22], bohužel žádná z nich zatím není spolehlivým modelem pro studium PrPTSE. Používané buněčné linie jsou jak neuronálního, tak i jiného původu (fibroblasty, ledvinné buňky, buňky ovaria).
TK nabízejí řadu výhod oproti experimentům využívajícím zvířecí modely. Je to snadná tvorba buněčných linií s ovlivnitelnou expresí PrPC a tvorba různých PrPC mutant, které pomáhají především při studiu vlastností prionových kmenů (různé PrPTSE se shodnou aminokyselinovou sekvencí, ale odlišnými biologickými a chemicko-fyzikálními vlastnostmi [23]), podstaty mezidruhové bariéry (minimalizace nebo neúčinnost přenosu infekce mezi jednotlivými živočišnými druhy) a mechanizmu konformační změny PrPC na PrPTSE nebo šíření PrPTSE z buňky na buňku. Dalším významným přínosem je využití TK pro rychlou detekci prionové infektivity v krátké době po infekci a snadné testování látek s anti-prionovým účinkem.
Použití tkáňových kultur má své limity, které je nutné brát při jejich aplikaci v úvahu. Především je obtížné získat buněčné linie senzitivní k prionové infekci, přičemž tyto buňky jsou schopné propagovat jen omezený počet prionových kmenů. Klíčovým kritériem pro dlouhodobou nebo trvalou senzitivitu buněk k prionové infekci je exprese PrPC. To bylo potvrzeno pokusy in vitro na HpL3–4 buněčné linii odvozené z buněk hipokampu myší s deletovaným PRNP genem [24], které byly zcela rezistentní k prionové infekci a shodovaly se s předchozími výsledky in vivo experimentů [25]. Exprese PrPC je klíčovým, ale pravděpodobně nikoli jediným nezbytným faktorem, který hraje roli v propagaci PrPTSE (více viz podkapitola Přenos prionové infekce v tkáňových kulturách).
Nejpoužívanější buněčné linie využívané pro studium prionového proteinu a aspektů prionových onemocnění
SMB
Linie odvozená z buněk myšího mozku infikovaného prionovým kmenem Chandler [26]. Neinfekční analog byl získán vyléčením buněk pomocí pentosan sulfátu, tj. polyaniontový sulfátový glykan [27].
PC12
Linie odvozená z buněk krysího feochromocytomu [20]. V přítomnosti nervového růstového faktoru (NGF) v nízké koncentraci dochází k diferenciaci, která vede k rozšíření neuritů, vývoji vzrušivé membrány a syntéze neurotransmiterů. Tato diferenciace umožnila studování funkce PrPC v buňkách s vlastnostmi neuronů.
N2a
Linie odvozená z myších neuroblastomových buněk je jednou z nejpoužívanějších buněčných linií. Sama o sobě neprodukuje velké množství PrPC, proto byly selektovány její subklony, které produkují jeho větší množství. V současné době patří mezi nejpoužívanější subklony PK1 linie [28].
GT1
Linie odvozená z myších hypothalamických buněk; oproti N2a stabilně produkuje až 8krát větší množství PrPC [16]. Patří k malému počtu buněčných linií, u kterých byla během prionové infekce pozorována neurodegenerace [15,29]. Slouží tedy jako jedinečný model pro studium neurodegenerace a apoptózy způsobené prionovou infekcí.
CAD
Linie je subklonem buněk odvozených z myších katecholaminergních neuronů CNS, vykazuje vlastnosti neuronů, ale postrádá jejich morfologii. Při kultivaci bez přídavku séra prochází reverzibilní morfologickou diferenciací do podoby neuronálních progenitorů. Slouží tedy jako jedinečný model pro studium neuronální diferenciace.
RK13
Linie odvozená z králičích epiteliálních buněk, produkuje minimální nebo žádné množství endogenního králičího PrPC. Buňky jsou však schopny ektopicky exprimovat ovčí (Rov linie) nebo myší (moRK13 linie) PrPC, a proto je možné je infikovat ovčí scrapie [18] nebo adaptovanými myšími prionovými kmeny [30].
HpL3–4
Tato linie má obdobné vlastnosti jako RK13; je odvozena z buněk hipokampu myší s deletovaným PRNP genem. Při ektopické expresi myšího PrPC byly buňky senzitivní k infekci adaptovanými myšími prionovými kmeny [24]. Spolu s RK13 buněčnou linií přináší možnost vytvoření nových modelů propagujících a přenášejících PrPTSE z druhů, ke kterým zatím neexistují buněčné linie.
SH-SY5Y
Tato lidská neuroblastomová linie po infekci mozkovým homogenátem pacientů trpících sporadickou formou CJN produkuje PrPTSE. Jeho přítomnost byla detekována i po 30 pasážích. Tato buněčná linie však není pro studium CJN spolehlivým modelem, protože produkce PrPTSE není stabilně detekovatelná [21].
Výše uvedený výčet buněčných linií není vyčerpávající, avšak postihuje nejdůležitější zástupce TK, které jsou pro studium PrP a aspektů prionových chorob používány nejčastěji. Z uvedených kultur jsou v naší laboratoři využívány myší linie neurálního původu CAD5, PK1, N2a, SMB-PS a fibroblastoidní linie L929 a NIH3T3. Linie CAD5 a PK1 patří v současné době k liniím, které jsou k infekci adaptovanými myšími kmeny scrapie nejcitlivější. Buněčné linie nám slouží ke sledování působení antiprionových látek, vlivu růstových podmínek na infikovatelnost buněk a studiu mechanizmu prionové infekce. Velkým přínosem bylo zavedení a v současné době již rutinní využívání in vitro metod detekce prionové infektivity.
Detekce PrPTSE
Spolehlivá detekce prionové infekce má význam především pro zabránění přenosu TSE, např. krevní transfuzí [7], kontaminovanými neurochirurgickými nástroji [38], kontaminovaným krmivem, nebo pro testování antiprionových látek.
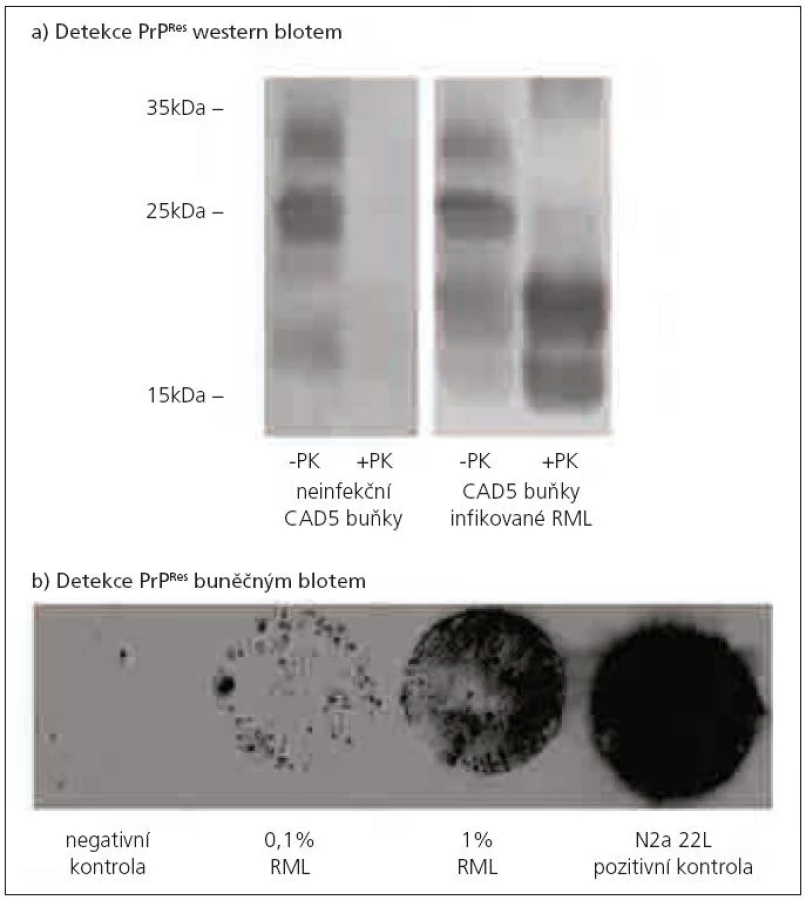
Potvrzení diagnózy TSE je obvykle založeno na stanovení přítomnosti PrPTSE ve vzorku mozkové tkáně, kde je koncentrace PrPTSE největší. Při jeho detekci se využívá rozdílná citlivost PrPC a PrPTSE k proteolytickému štěpení proteinázou K. PrPC je beze zbytku štěpen jako většina proteinů. Z PrPTSE je odštěpeno přibližně prvních 90 AK a zůstává neštěpitelný zbytek o velikosti asi 142 AK označovaný jako PrPRes, který lze detekovat řadou metod, jakými jsou např. imunohistochemie, PET blot, ELISA nebo western blot (obr. 1a pořízený v naší laboratoři). Průkaz PrPRes je považován za znak TSE infekce. V některých případech hladina PrPRes neodpovídá stadiu onemocnění [39]. A ve vzácných případech nemusí být prionové onemocnění spojeno s přítomností detekovatelného množství PrPRes [40]. Proto nepřítomnost PrPRes není vždy spolehlivým ukazatelem vylučujícím prionovou infekci.
Přímou detekci prionové infektivity umožňuje myší bioassay, která je jednou z nejcitlivějších metod. Principem je intracerebrální očkování indikátorových zvířat homogenátem studované tkáně. Pokud byl vzorek infekční, po několika měsících začne myš projevovat příznaky TSE a následně umírá [41]. Myší bioassay má kromě etického hlediska dvě zásadní nevýhody, které brání jejímu širokému použití, a to dlouhou dobu trvání (cca 20–40 týdnů) a velkou finanční náročnost.
Na bázi TK byly v posledních letech vyvinuty in vitro testy pro rychlou detekci prionové infektivity – metoda buněčného blotu [14] (obr. 1b z naší laboratoře) a scrapie buněčného testu [15], který se svou citlivostí vyrovná myší bioassay. Prionová infekce je detekovatelná po inkubaci indikátorových buněk s vyšetřovaným mozkovým homogenátem při jeho ředění 10-7 až 10-8. Metoda je však 10krát rychlejší a zhruba 100krát levnější než myší bioassay [42].
Právě výše zmíněné in vitro testy by mohly být do budoucna slibnou metodou, jak detekovat infekční vzorky zvířecích či lidských tkání v krátkém časovém úseku s dostatečnou citlivostí. Již dnes lze pomocí různé míry infekce indikátorových buněčných linií, nejčastěji CAD5 nebo PK1, charakterizovat odlišné prionové kmeny [35], podobně jako jsou např. charakterizovány odlišnými histopatologickými změnami v mozku postižených jedinců.
Využití tkáňových kultur při studiu prionového proteinu
TK se využívají jak k objasnění fyziologické funkce PrPC, jeho lokalizace v buňce, tak ke studiu lokalizace a průběhu konformační změny PrPC v PrPTSE. Dále slouží pro studium podmínek propagace a přenosu prionové infekce, charakterizaci prionových kmenů, mezidruhové bariéry, identifikaci buněčných vazebných partnerů PrPTSE a v neposlední řadě i pro testování látek s možným anti-PrPTSE účinkem.
TK na jedné straně umožňují detailně studovat nezodpovězené otázky, na straně druhé před nás kladou úkol dobře porozumět procesům probíhajícím in vitro tak, aby bylo možné poznatky aplikovat in vivo a do humánní medicíny.
Přenos prionové infekce v tkáňových kulturách
Většina TK neprojevuje po infekci priony žádné změny. Šíření prionů in vitro je ovlivněno typem buněk. V některých případech je přenos PrPTSE závislý na přímém buněčném kontaktu, v jiných případech však nikoli [24,36,43]. Jednotlivé kmeny prionů jsou selektivně propagovány pouze některými buněčnými liniemi. Kritériem pro odlišení prionových kmenů in vitro je především rozdílná elektroforetická pohyblivost PrPRes po štěpení proteinázou K a odlišné zastoupení jednotlivých glykoforem PrPTSE, které závisí kromě kmenu prionů i na typu buněk [16]. Studium podstaty těchto charakteristik by mohlo výrazně pomoci při hledání důvodu selektivní akumulace PrPTSE v odlišných oblastech mozku [44], objasnění rozdílné délky inkubační doby nebo vysvětlení otázky, proč dochází k rozdílným histopatologickým změnám v mozku infikovaných jedinců při infekci různými kmeny prionů.
Problematika druhové bariéry se objevuje i při experimentech in vitro. Podobně jako u experimentálních zvířat je velmi obtížné infikovat buněčné kultury priony pocházejícími z jiného živočišného druhu. Tento problém lze někdy obejít modifikací buněk tak, aby exprimovaly buněčný prionový protein odpovídající živočišnému druhu, jehož priony používáme pro infekci. S využitím TK bylo také ukázáno, že mezidruhový přenos není pravděpodobně ovlivněn jen rozdílnou aminokyselinovou sekvencí PrP, ale také konformací PrPTSE.
Typ buněk a rychlost jejich dělení rovněž ovlivňuje akumulaci prionů v buněčné kultuře [45,46]. Studie využívající myší neuroblastomové N2a buňky ukázaly, že požadavky pro dlouhodobou infekci buněčné kultury mají specifika doposud nepozorovaná v podmínkách in vivo. Weismann [46] popisuje, že při rychlosti buněčné proliferace, jež překračuje rychlost šíření prionů, nemůže docházet k perzistentní infekci, a naopak dochází k jejich ředění a možnému vymizení infekce. Oproti tomu Ghaemmaghami et al [45] došli při studiu N2a buněk k výsledkům naznačujícím, že buněčné dělení sice ovlivňuje hladinu exprese PrPTSE, avšak nedochází ani ke kompletnímu vyředění infekce, ani k exponenciálnímu nárůstu hladiny PrPTSE. Vorberg et al [47] navíc při pokusech s N2a a fibroblastoidní linií ukázali, že během prvních 24 hod po infekci docházelo ke vzniku nového PrPTSE. Tvorba nového PrPTSE ale automaticky neznamenala dlouhodobou nebo trvalou propagaci PrPTSE u obou linií. K dlouhodobé infekci došlo pouze u fibroblastoidní buněčné linie.
Výsledky řady studií ukazují významné rozdíly v přenosu a šíření infekce při použití rozdílného typu buněčných linií, prionových kmenů a také v uspořádání experimentů. Vedle esenciální úlohy PrPC tyto výsledky naznačují důležitost dalších faktorů, které mohou modulovat procesy spojené s průběhem a projevy onemocnění. Specifické faktory buňky, jako proteiny, glykosaminoglykany [48], endogenní retroviry [49] nebo mikro RNA [50], se mohou spolupodílet na těchto procesech jako předpovězený, ale dosud nenalezený „protein/faktor X“ [51,52].
Příznaky prionových onemocnění v tkáňových kulturách
Projevy neurodegenerace in vitro nejsou u většiny buněčných linií patrné a buňky nenesou žádné známky onemocnění. Jedním z důvodů může být imortalizace a transformace TK, u nichž se, na rozdíl od diferencované mozkové tkáně, neurodegenerace neprojevuje. Znaky neurodegenerace a apoptózy byly pozorovány u GT1 linie dlouhodobě propagující prionovou infekci [15,29]. Sedmiměsíční exprese PrPTSE se u GT1 buněk projevila změnami v životaschopnosti, době zdvojení, morfologii, cytopatologii a apoptóze. U prionem infikovaných GT1 buněk poklesla životaschopnost o 11 %, prodloužila se doba zdvojení z 1,5 na 2–2,5 dny. Buňky přestaly růst v jedné vrstvě a začaly vytvářet shluky, zvýšil se počet autofagických vakuol a v buňkách byly detekovány fragmenty DNA. Všechny tyto příznaky byly reverzibilně zmírnitelné přidáním nervových růstových faktorů [15]. Sledování cytopatologických změn in vitro může zprostředkovat bližší porozumění mechanizmům spongiformní dystrofie a tvorby amyloidových plak a následně napomoci hledání cest k eliminaci těchto patologických projevů.
Hledání látek pro léčbu prionových onemocnění
Možnost detekce malého či minimálního množství PrPTSE i během časných fází infekce nabízí využití TK jako vhodného nástroje pro hledání látek s anti-PrPTSE účinkem. Při studiích in vitro se během posledních let podařilo objevit několik látek s anti-PrPTSE účinkem, které je možné využít také při léčbě in vivo. Mezi nejvýznamnější patří cyklické tetrapyroly [37], větvené polyaminy [53], protilátky proti PrP [28,45–59] a sulfátové glykany [27].
Mezi cyklické tetrapyroly náleží porfyriny a ftalocyaniny. Tyto látky in vitro inhibují akumulaci PrPTSE v buněčné kultuře a in vivo prodlužují až 4krát inkubační dobu u infikovaných laboratorních zvířat. Mechanizmus účinku je pravděpodobně založen na inhibici vazby PrPTSE a PrPC [37].
Větvené polyamidy, mezi něž patří P-dendrimery, in vitro interagují s PrP. Inhibiční účinek byl pozorován u buněk již exprimujících PrPTSE. In vivo byla pozorována inhibice akumulace PrPTSE ve slezině myší intraperitoneálně infikovaných priony [53].
Monoklonální protilátky proti PrP inhibují in vitro konverzi PrPC na PrPTSE, dochází tak k potlačení vzniku PrPTSE. V některých případech vedla tato terapie k úplnému vyléčení buněk [55]. In vivo byla testována jak aktivní, tak i pasivní imunizace, které v řadě případů vedly k prodloužení inkubační doby a zmírnění projevů onemocnění [57,59]. Úspěch terapie je však závislý na použití určitých monoklonálních protilátek proti PrP [54,56,58].
Příkladem sulfátového glykanu je polyaniontový pentosan polysulfát (PPS), který byl použit k odléčení prionové infekce ve stabilně infikované buněčné kultuře [27]. PPS byl injektován pokusným myším přímo do mozku [60]. Tato terapie výrazně prodloužila inkubační dobu onemocnění. PPS byl také použit pro terapii v humánní medicíně. Na univerzitě Fukuoka v Japonsku se podrobilo intraventrikulárnímu podávání PPS 11 pacientů (3 s dědičným typem CJN, 2 s iatrogenní CJN, 6 se sporadickou CJN a 1 s GSS). Pacientům byl PPS podáván kontinuálně ventrikulárním katétrem zavedeným přímo do mozku a připojeným na subkutánní infuzní pumpu. Dávky PPS postupně stoupaly od 1 do 120 µg/kg/den. Terapie byla dobře snášena; pouze u některých pacientů se po intraventrikulárním podání PPS objevil subdurální hydrom. Průměrná doba života při podání PPS byla 24,2 měsíců; sedm osob zemřelo na sepsi a pneumonii. Terapie neovlivnila klinické příznaky, pouze prodloužila život pacientů [61]. Od roku 2003 absolvovalo terapii intraventrikulárním podáváním PPS více než 25 pacientů; výrazného benefitu však nebylo dosaženo.
Další látkou, jejíž antiprionové účinky byly testovány in vitro a také in vivo při experimentech na zvířatech, jakož i pro terapii v humánní medicíně, je quinacrine. Quinacrine je původně antimalarikum. Při použití in vitro byla tato látka schopna vyléčit infikované buněčné kultury [62,63]. V klinických experimentech však nepřinesla očekávaný výsledek. U některých pacientů přechodně zmírnila příznaky onemocnění, u většiny však neměla žádný efekt [64]. Quinacrine špatně prochází hematoencefalickou bariérou. U experimentálních zvířat byla penetrace zvýšena modifikací transportérů odpovědných za přenos látek do mozku.
Látky účinné při odléčení buněk od prionové infekce in vitro nemusí být vhodné pro použití in vivo. Důvodem může být především jejich toxicita, neschopnost prostupovat hematoencefalickou bariérou, ale také odlišné podmínky propagace prionů in vivo a in vitro. V kontinuálně se dělící buněčné kultuře může být účinnost antiprionových látek potencována ředěním prionů v průběhu dělení buněk, které in vivo v diferencované mozkové tkáni nepřispívá k procesu akumulace, sekrece a degradace PrPTSE.
Závěr
Tkáňové kultury již dnes představují rychlý a efektivní nástroj pro studium mechanizmu buněčné propagace a šíření prionů. Jejich využití by mělo přinést odpovědi na otázky týkající se fyziologické úlohy PrPC a mechanizmu stojícího za odumíráním priony infikovaných neuronů. Navíc jsou využitelné i pro rychlou detekci prionové infektivity a prvotní testování širokého spektra látek s antiprionovým účinkem, nezbytného pro nalezení účinného prostředku pro léčbu TSE. Důležitým krokem pro další využití tkáňových kultur bude příprava kultur schopných stabilně propagovat lidské priony a vytvoření buněčných modelů prionové infekce na neproliferujících buňkách lépe napodobujících situaci v mozkové tkáni. Přes svoje limity mají buněčné modely prionové infekce potenciál významně přispět k objasnění četných neznámých ve výzkumu prionových chorob, a pomoci tak jejich zařazení mezi léčitelné nemoci.
Mgr. Olga Janoušková, Ph.D.
Ústav imunologie a mikrobiologie 1. LF UK v Praze
Studničkova 7
128 00 Praha 2
e-mail: Olga.Janouskova@lf1.cuni.cz
Přijato k recenzi: 15. 12. 2009
Přijato do tisku: 14. 4. 2010
Sources
1. Matěj R, Rusina R, Koukolík F. 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury. Cesk Slov Neurol N 2007; 70/103(6): 637–642.
2. Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J et al. The same prion strain causes vCJD and BSE. Nature 1997; 389(6650): 448–450.
3. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A et al. Transmissions to mice indicate that ‚new variant‘ CJD is caused by the BSE agent. Nature 1997; 389(6650): 498–501.
4. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‚new variant‘ CJD. Nature 1996; 383(6602): 685–690.
5. Allan B, Tuft S. Transmission of Creutzfeldt-Jakob disease in corneal grafts. BMJ 1997; 315(7122): 1553–1554.
6. Hammersmith KM, Cohen EJ, Rapuano CJ, Laibson PR. Creutzfeldt-Jakob disease following corneal transplantation. Cornea 2004; 23(4): 406–408.
7. Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004; 363(9407): 417–421.
8. Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 364(9433): 527–529.
9. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216(4542): 136–144.
10. Holada K, Simák J, Vostal JG. Transmission of BSE by blood transfusion. Lancet 2000; 356(9243): 1772.
11. Safar J, Roller PP, Gajdusek DC, Gibbs CJ jr. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem 1993; 268(27): 20276–20284.
12. Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J Mol Biol 2005; 346(2): 645–659.
13. Ironside JW, McCardle L, Horsburgh A, Lim Z, Head MW. Pathological diagnosis of variant Creutzfeldt-Jakob disease. APMIS 2002; 110(1): 79–87.
14. Holada K, Simak J, Brown P, Vostal JG. Divergent expression of cellular prion protein on blood cells of human and nonhuman primates. Transfusion 2007; 47(12): 2223–2232.
15. Schätzl HM, Laszlo L, Holtzman DM, Tatzelt J, DeArmond SJ, Weiner RI et al. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J Virol 1997; 71(11): 8821–8831.
16. Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J et al. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J Virol 2000; 74(1): 320–325.
17. Vorberg I, Raines A, Story B, Priola SA. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J Infect Dis 2004; 189(3): 431–439.
18. Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, Lehmann S et al. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci U S A 2001; 98(7): 4055–4059.
19. Hijazi N, Kariv-Inbal Z, Gasset M, Gabizon R. PrPSc incorporation to cells requires endogenous glycosaminoglycan expression. J Biol Chem 2005; 280(17): 17057–17061.
20. Rubenstein R, Carp RI, Callahan SM. In vitro replication of scrapie agent in a neuronal model: infection of PC12 cells. J Gen Virol 1984; 65 (12): 2191–2198.
21. Ladogana A, Liu Q, Xi YG, Pocchiari M. Proteinase-resistant protein in human neuroblastoma cells infected with brain material from Creutzfeldt-Jakob patient. Lancet 1995; 345(8949): 594–595.
22. Kikuchi Y, Kakeya T, Sakai A, Takatori K, Nakamura N, Matsuda H et al. Propagation of a protease-resistant form of prion protein in long-term cultured human glioblastoma cell line T98G. J Gen Virol 2004; 85 (Pt 11): 3449–3457.
23. Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318(5852): 930–936.
24. Maas E, Geissen M, Groschup MH, Rost R, Onodera T, Schätzl H et al. Scrapie infection of prion protein-deficient cell line upon ectopic expression of mutant prion proteins. J Biol Chem 2007; 282(26): 18702–18710.
25. Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73(7): 1339–1347.
26. Clarke MC, Haig DA. Evidence for the multiplication of scrapie agent in cell culture. Nature 1970; 225(5227): 100–101.
27. Birkett CR, Hennion RM, Bembridge DA, Clarke MC, Chree A, Bruce ME et al. Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. EMBO J 2001; 20(13): 3351–3358.
28. Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A 2001; 98(16): 9295–9299.
29. Milhavet O, McMahon HE, Rachidi W, Nishida N, Katamine S, Mangé A et al. Prion infection impairs the cellular response to oxidative stress. Proc Natl Acad Sci U S A 2000; 97(25): 13937–13942.
30. Courageot MP, Daude N, Nonno R, Paquet S, Di Bari MA, Le Dur A et al. A cell line infectible by prion strains from different species. J Gen Virol 2008; 89 (Pt 1): 341–347.
31. Bosque PJ, Prusiner SB. Cultured cell sublines highly susceptible to prion infection. J Virol 2000; 74(9): 4377–4386.
32. Arjona A, Simarro L, Islinger F, Nishida N, Manuelidis L. Two Creutzfeldt-Jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc Natl Acad Sci U S A 2004; 101(23): 8768–8773.
33. Arima K, Nishida N, Sakaguchi S, Shigematsu K, Atarashi R, Yamaguchi N et al. Biological and biochemical characteristics of prion strains conserved in persistently infected cell cultures. J Virol 2005; 79: 7104-7112.
34. Qi Y, Wang JK, McMillian M, Chikaraishi DM. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J Neurosci 1997; 17(4): 1217–1225.
35. Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci U S A 2007; 104(52): 20908–20913.
36. Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol 2007; 211(5): 582–590.
37. Caughey WS, Priola SA, Kocisko DA, Raymond LD, Ward A, Caughey B. Cyclic tetrapyrrole sulfonation, metals, and oligomerization in antiprion activity. Antimicrob Agents Chemother 2007; 51(11): 3887–3894.
38. Flechsig E, Hegyi I, Enari M, Schwarz P, Collinge J, Weissmann C. Transmission of scrapie by steel-surface-bound prions. Mol Med 2001; 7(10): 679–684.
39. Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1994; 1(1): 19–30.
40. Lasmézas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM et al. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997; 275(5298): 402–405.
41. Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull 2003; 66 : 43–60.
42. Klöhn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci U S A 2003; 100(20): 11666–11671.
43. Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ et al. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol 2002; 12(7): 523–530.
44. Beringue V, Mallinson G, Kaisar M, Tayebi M, Sattar Z, Jackson G et al. Regional heterogeneity of cellular prion protein isoforms in the mouse brain. Brain 2003; 126 (Pt 9): 2065–2073.
45. Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE et al. Cell division modulates prion accumulation in cultured cells. Proc Natl Acad Sci U S A 2007; 104(46): 17971–17976.
46. Weissmann C. The state of the prion. Nat Rev Microbiol 2004; 2(11): 861–871.
47. Vorberg I, Raines A, Priola SA. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J Biol Chem 2004; 279(28): 29218–29225.
48. McBride PA, Eikelenboom P, Kraal G, Fraser H, Bruce ME. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J Pathol 1992; 168(4): 413–418.
49. Stengel A, Bach C, Vorberg I, Frank O, Gilch S, Lutzny G et al. Prion infection influences murine endogenous retrovirus expression in neuronal cells. Biochem Biophys Res Commun 2006; 343(3): 825–831.
50. Weissmann C. A ‚unified theory‘ of prion propagation. Nature 1991; 352(6337): 679–683.
51. Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A 1997; 94(19): 10069–10074.
52. Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83(1): 79–90.
53. Solassol J, Crozet C, Perrier V, Leclaire J, Béranger F, Caminade AM et al. Cationic phosphorus-containing dendrimers reduce prion replication both in cell culture and in mice infected with scrapie. J Gen Virol 2004; 85 (Pt 6): 1791–1799.
54. Féraudet C, Morel N, Simon S, Volland H, Frobert Y, Créminon C et al. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem 2005; 280(12): 11247–11258.
55. Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001; 412(6848): 739–743.
56. Perrier V, Solassol J, Crozet C, Frobert Y, Mourton-Gilles C, Grassi J et al. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J Neurochem 2004; 89(2): 454–463.
57. Sigurdsson EM, Brown DR, Daniels M, Kascsak RJ, Kascsak R, Carp R et al. Immunization delays the onset of prion disease in mice. Am J Pathol 2002; 161(1): 13–17.
58. Sigurdsson EM, Sy MS, Li R, Scholtzova H, Kascsak RJ, Kascsak R et al Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci Lett 2003; 336(3): 185–187.
59. White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003; 422(6927): 80–83.
60. Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R et al. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol 2004; 78(10): 4999–5006.
61. Tsuboi Y, Doh-Ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology 2009; 29(5): 632–636.
62. Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 2000; 74(10): 4894–4897.
63. Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A 2001; 98(17): 9836–9841.
64. Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol 2009; 8(4): 334–344.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2010 Issue 4
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine Eases Daily Life for Patients and Caregivers
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Dynamické vyšetření bederní páteře pomocí magnetické rezonance – kazuistika
- Farmakologická léčba epilepsie
- Funkční význam pólu temporálního laloku
- Léčba juxtafacetární cysty bederní páteře dynamickou interspinózní stabilizací – kazuistika