
Parkinsonova nemoc s fenotypem progresivní supranukleární paralýzy – kazuistika
Parkinson’s Disease with a Phenotype of Progressive Supranuclear Palsy – a Case Report
Current clinical diagnostic criteria for Parkinson’s disease and progressive supranuclear palsy (PSP) are considered relatively safe tools for the clinical differentiation of the two neurodegenerative diseases; neither diagnosis includes the possibility that Parkinson’s disease might duplicate the clinical genotype of PSP. We document the case of a male patient who first manifested parkinsonian signs at the age of 56 years. Parkinsonian syndrome developed over the course of the following three years without tremor, but with typical subcortical dementia, gaze palsy and impairment of conjugated gaze upwards, eyelid opening apraxia and gait apraxia. The disease then progressed to sudden falls, typical Lhermitte’s frontal signs, utilizations and imitations. Response to L-DOPA therapy disappeared relatively quickly. The findings of paraclinical examinations (MRI, electrophysiology) also suggested a diagnosis of PSP. After approximately four years, the patient died of intercurrent disease. Pathological and histopathological examinations and evaluation of the results by two independent pathologists documented and confirmed changes typical of Parkinson’s disease. This case indubitably raises the question of overlap of phenotypes of different types of neurodegenerative diseases with parkinsonian syndrome dominant.
Key words:
Parkinson’s disease – progressive supranuclear palsy – Lewy bodies – synucleinopathy
Authors:
K. Farníková 1; J. Ehrmann 2; L. Tučková 2; P. Kaňovský 1
Authors‘ workplace:
LF UP a FN Olomouc
Centrum pro diagnostiku a léčbu neurodegenerativních onemocnění, Neurologická klinika
1; LF UP a FN Olomouc
Ústav patologie
2
Published in:
Cesk Slov Neurol N 2011; 74/107(6): 695-699
Category:
Case Report
Overview
Dosud platná diagnostická kritéria pro Parkinsonovu nemoc a progresivní supranukleární paralýzu (PSP) jsou považována za poměrně spolehlivý nástroj vzájemného klinického rozlišení obou neurodegenerativních onemocnění. Ani jedna kritéria nepřipouštějí možnost takové manifestace Parkinsonovy nemoci, která by kopírovala klinický genotyp PSP. Budeme dokumentovat případ pacienta, muže, u kterého se první příznaky parkinsonizmu objevily v 56 letech. Během následujících tří let se vyvinul klinický obraz parkinsonského syndromu bez třesu, s typickou demencí, okohybnou poruchou charakteru poruchy konjugovaného pohledu vzhůru, apraxií otevření víček a apraxií chůze. U pacienta se v průběhu progrese nemoci objevily také náhlé pády a typické Lhermittovy frontální příznaky, utilizace a imitace. Odpověď na léčbu L-DOPA poměrně rychle vymizela. Paraklinická vyšetření (MR mozku, elektrofyziologie) taktéž nasvědčovala pro diagnózu PSP. Po zhruba čtyřech letech průběhu nemoci pacient zemřel na interkurentní onemocnění. Patologické a histopatologické vyšetření a zhodnocení jeho výsledků dvěma nezávislými patology prokázalo a potvrdilo změny zcela typické pro Parkinsonovu nemoc. Případ nepochybně nastoluje otázku vzájemného průniku fenotypů různých forem neurodegenerativního onemocnění s parkinsonským syndromem jako dominantním příznakem.
Klíčová slova:
Parkinsonova nemoc – progresivní supranukleární paralýza – Lewyho tělíska – synukleinopatie
Úvod
V poslední době dochází k zásadním změnám v taxonomii parkinsonských syndromů působených neurodegenerativními onemocněními, zjevně v souvislosti se zlepšujícím se poznáním klinickopatologických korelací těchto chorob. Poměrně nedávná představa, že charakteristický klinický fenotyp je způsoben specifickým patologickým procesem a vice versa, bere postupně zasvé. Nedávno publikované studie přinesly důkazy, že neurodegenerativní onemocnění z okruhu atypických parkinsonských syndromů jsou klinicky natolik variabilní, že mohou úspěšně maskovat charakter patologického substrátu [1]. Ve většině případů šlo o „atypické“ manifestace atypického parkinsonského syndromu nebo Parkinsonovy nemoci (PN). Sami jsme recentně měli možnost pozorovat „typickou“ manifestaci progresivní supranukleární paralýzy (PSP), která se při autoptickém vyšetření ukázala být „typickou“ Parkinsonovou nemocí.
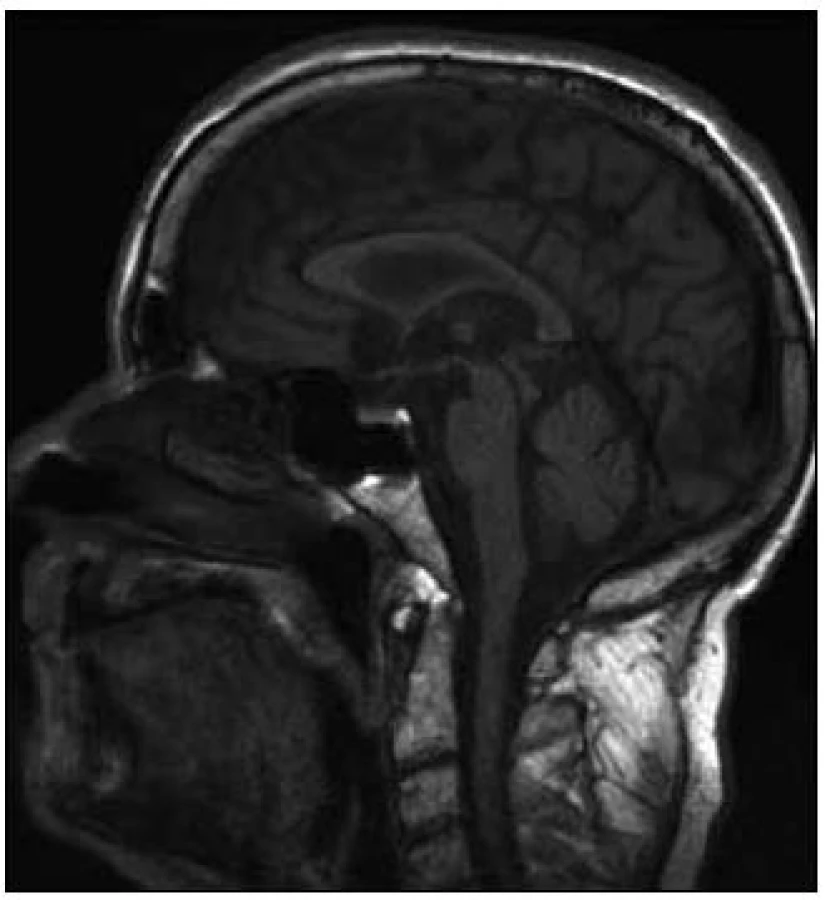
Progresivní supranukleární paralýza je jedním z atypických parkinsonských syndromů, charakterizovaný bradykinézou, rigiditou, absencí klidového třesu, náhlými pády, typickou poruchou konjugovaného pohybu (upward gaze palsy), apraxií otevření víček, progredujícím kognitivním deficitem a frontální symptomatologií zahrnující apraxii chůze, imitačním a utilizačním chováním. Korelátem při MR zobrazení mozku je ve většině případů atrofie tegmenta mesencefala vytvářející v sagitálních řezech obraz stojícího tučňáka tzv. standing penguin silhouette sign a v transverzálních řezech obraz svlačce tzv. morning glory sign [2]. Z patologického hlediska je PSP tzv. tauopatie. Poprvé byla nemoc klinicky popsána Steelem a Richardsonem et al v roce 1963 a patologicky Olszewskim et al v roce 1964 [3,4].
Kazuistika
Anamnéza a klinický průběh
Pacient byl muž ve věku 59 let, s negativní osobní i rodinnou anamnézou. První projevy parkinsonského syndromu se objevily v 56 letech. Jednalo se o celkové zpomalení hybnosti a zhoršující se neobratnost při provádění drobných pohybů. V průběhu dalšího půl roku se rozvinul klidový třes horních končetin, nejprve levé horní končetiny (LHK), velmi rychle se rozšířil i na pravou horní končetinu, přičemž stále byla zachována asymetrie postižení s těžším nálezem na LHK. Během následujících šesti měsíců tento třes však zcela vymizel. V dalším průběhu onemocnění postupně progredovalo, bradykinéza a rigidita se stávaly stále více zřetelnými. V průběhu dalšího roku se rozvinula typická paréza vertikálního pohledu směrem nahoru a současně se objevily náhlé, neočekávané pády.
V té době byl pacient hospitalizován na Neurologické klinice LF UP a FN Olomouc, kde podstoupil vyšetření podle protokolu vytvořeného za účelem podrobné diagnostiky pacientů se suspektním neurodegenerativním onemocněním. Základní laboratorní skríning a likvorologické vyšetření byly v normě. Na MR mozku byla popsána počínající atrofie mozku a drobné postischemické změny periventrikulárně oboustranně, spolu s oploštěním dorza mezencefala a napřímením kurvatury mozkového kmene (tzv. hummingbird sign nebo standing penguin silhouette, obr. 1). Ultrazvukové vyšetření mozkových tepen bylo s normálním nálezem, substantia nigra nebyla popsána. Podrobným neuropsychologickým vyšetřením byla zjištěna spíše lehká demence s dominujícím dysexekutivním syndromem, MMSE 24 bodů. Logopedické vyšetření odhalilo amnestickou afázii. Elektromyografické vyšetření (kondukční studie, jehlová EMG análního sfinkteru) a vyšetření multimodálních evokovaných potenciálů byly s normálními nálezy. Při apomorfinovém testu došlo pouze k nepatrnému zlepšení stavu, UPDRS III bylo 27/23 bodů. Jako součást protokolu byla zhotovena i podrobná videodokumentace.
Po zhruba čtyřech letech průběhu onemocnění klinický obraz zahrnoval bradykinezi, rigiditu, parézu konjugovaného pohledu směrem vzhůru, apraxii otevření očních víček, poruchu iniciace chůze, apraxii chůze, dysartrii, demenci s dominujícím dysexekutivním syndromem, posturální instabilitu, utilizační a imitační chování a typický „applauding sign“. Odpověď na terapii L-DOPA byla již minimální. Na základě tohoto zcela typického obrazu byla stanovena klinická diagnóza progresivní supranukleární paralýzy neboli Steele-Richardson-Olszewskiho nemoci.
V dalším – již krátkém – průběhu onemocnění docházelo k progresivnímu horšení všech symptomů, nejnápadnější byla progrese demence. Dysartrie se rozvinula prakticky v anartrii. Pády byly stále četnější, pacient postupně nebyl schopen samostatného stoje a chůze. Z autonomních potíží se rozvinula inkontinence, hypersalivace a paroxysmální tachykardie doprovázená dušností. Po pěti letech průběhu onemocnění pacient onemocněl závažnou bronchopneumonií, pro tuto diagnózu byl hospitalizován a i přes intenzivní antibiotickou a podpůrnou terapii během hospitalizace zemřel.
Patologický nález
Za účelem vyloučení tau patologie byla hodnocena bazální ganglia, dále substantia nigra, pontinní a mezencefalické tegmentum, frontotemporální a limbický kortex. Bylo provedeno makroskopické zhodnocení uvedených struktur, konvenční mikroskopie, impregnace solemi stříbra jako histochemická metoda k detekci specifických inkluzí, imunohistochemické vyšetření monoklonální protilátkou proti tau proteinu a barvení kongo červení k detekci amyloidu. Autoptické vyšetření mozku, kdy patologické i histopatologické nálezy byly zhodnoceny dvěma nezávislými patology, překvapivě prokázalo změny typické nikoli pro PSP, ale pro Parkinsonovu nemoc. Při makroskopickém zhodnocení mozkové tkáně byla přítomna dekolorizace pigmentových jader mozkového kmene.
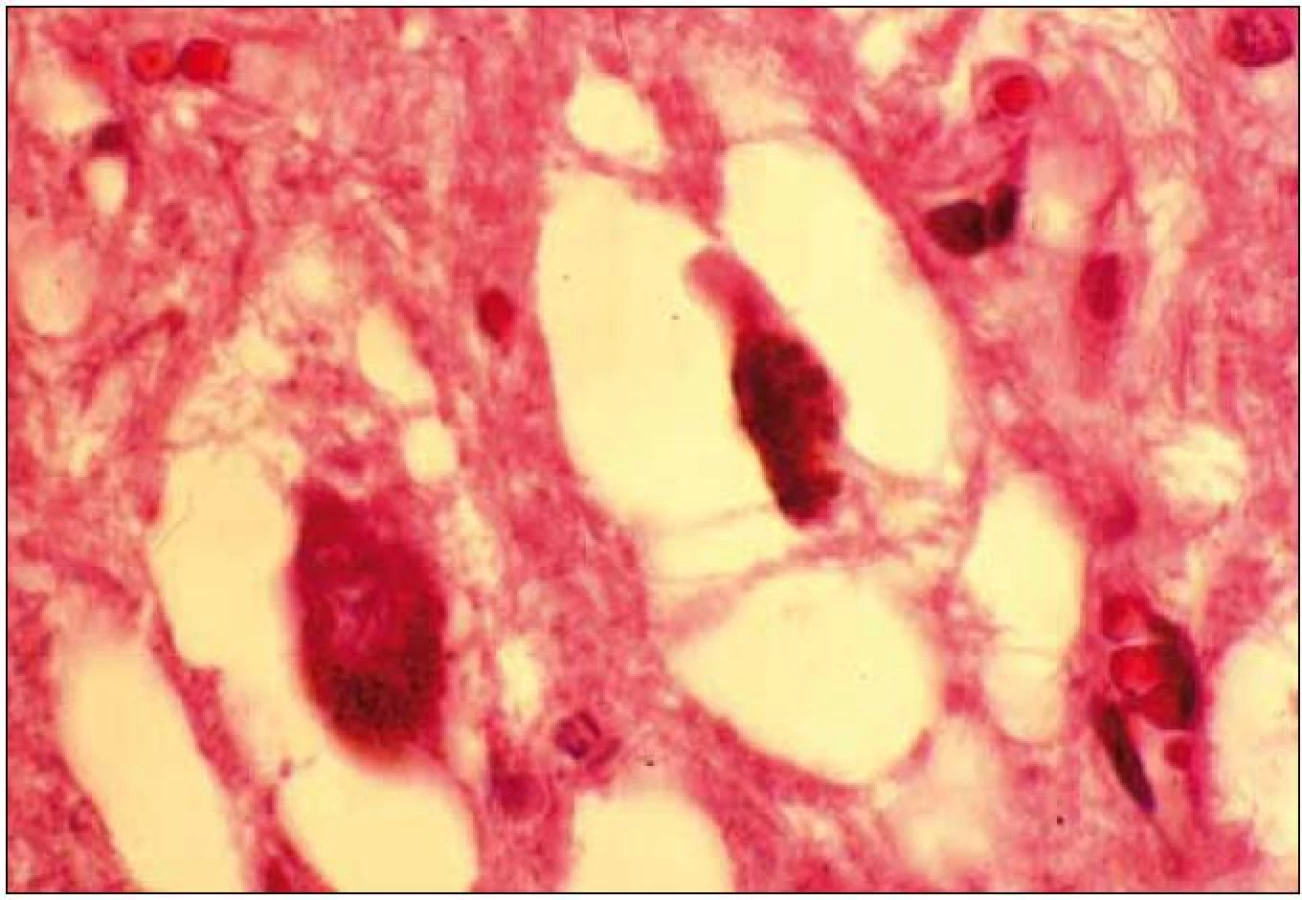
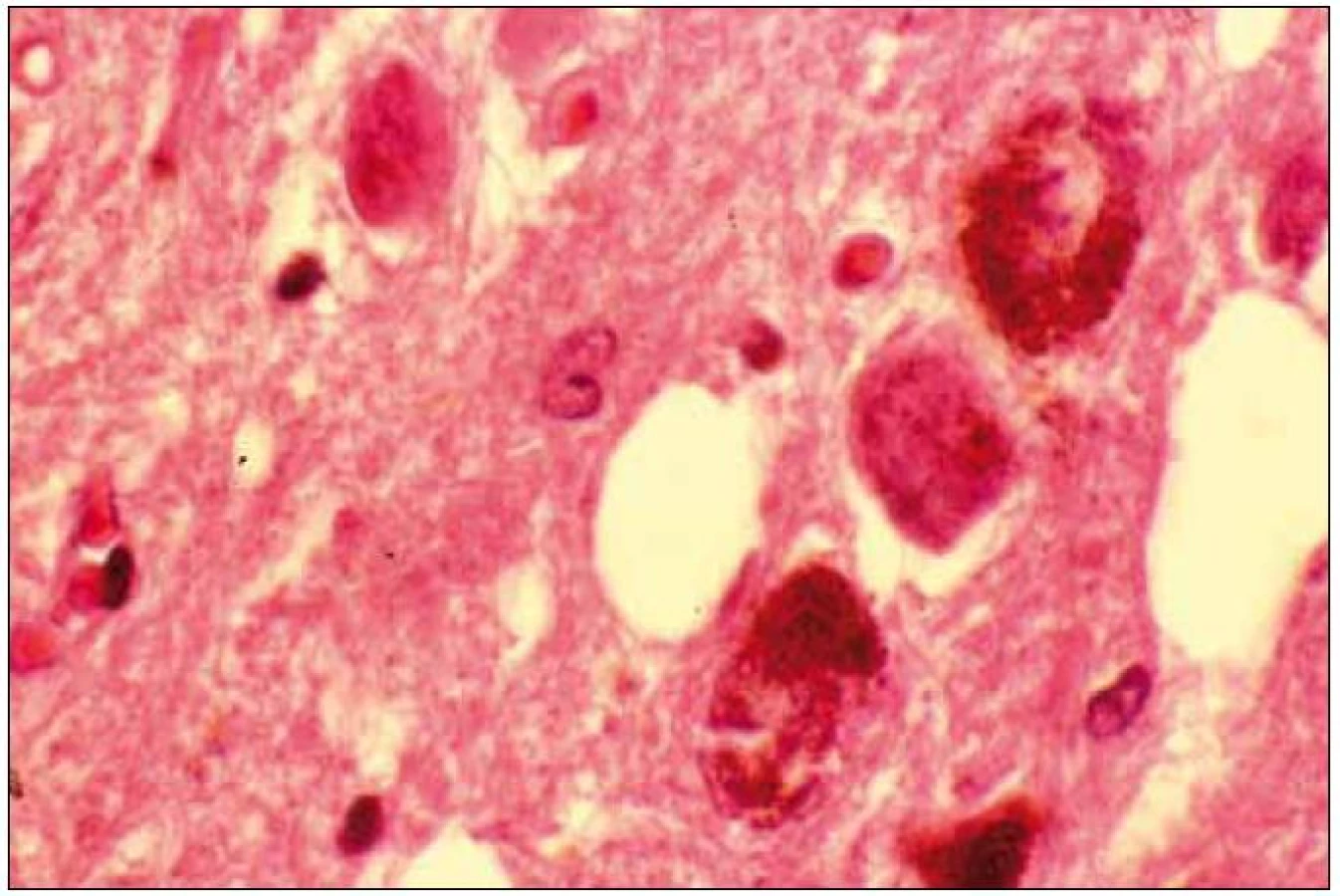
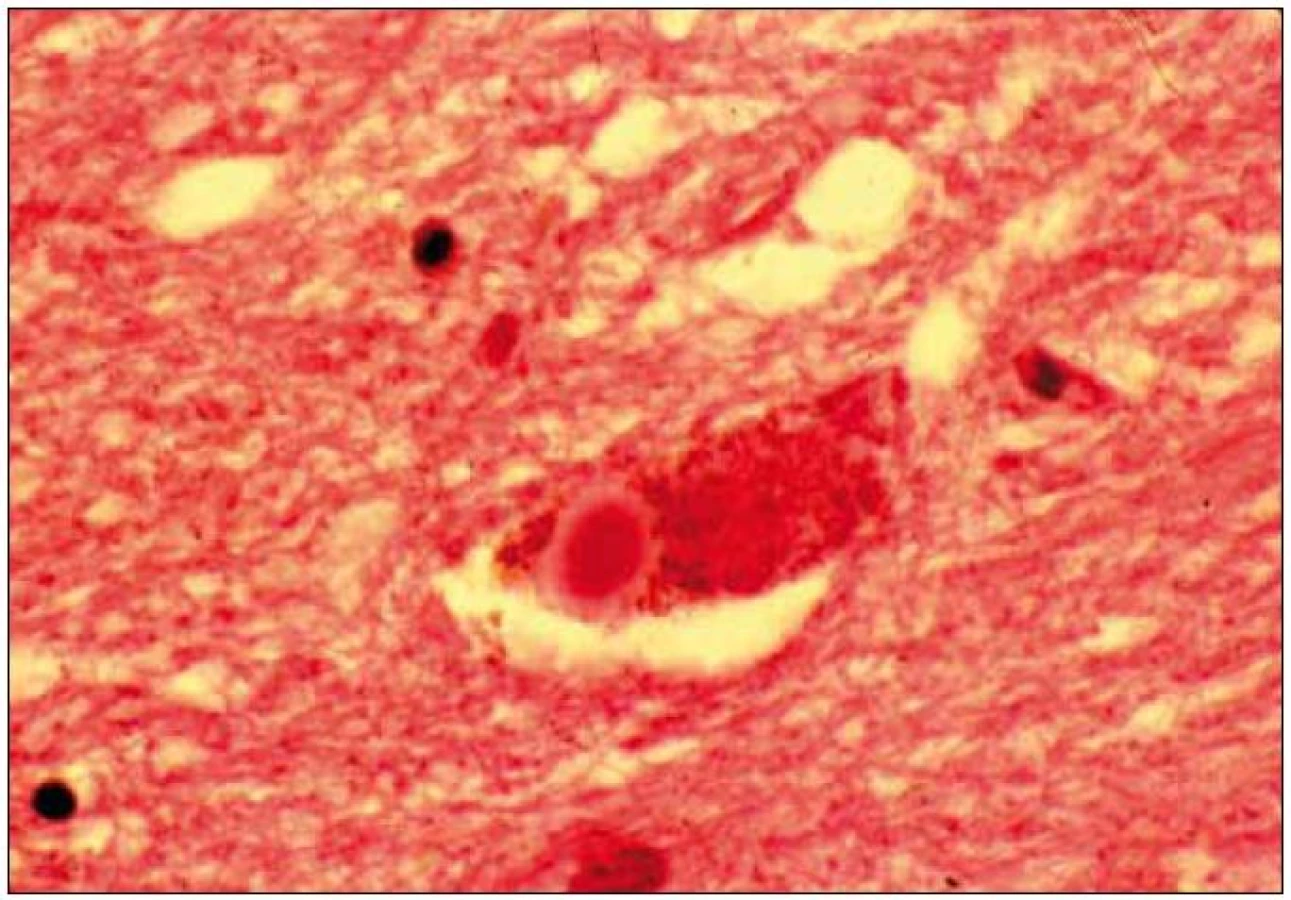
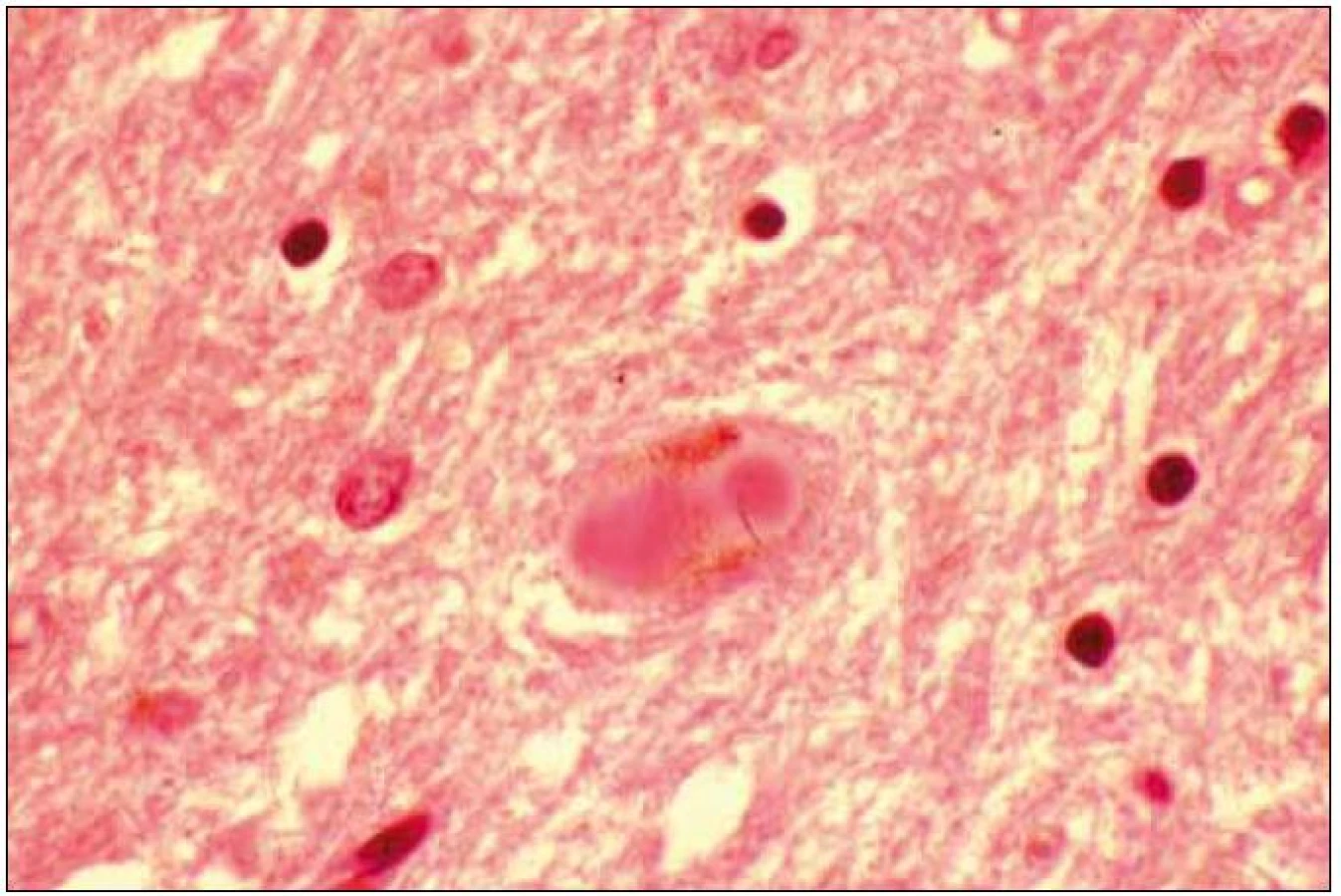
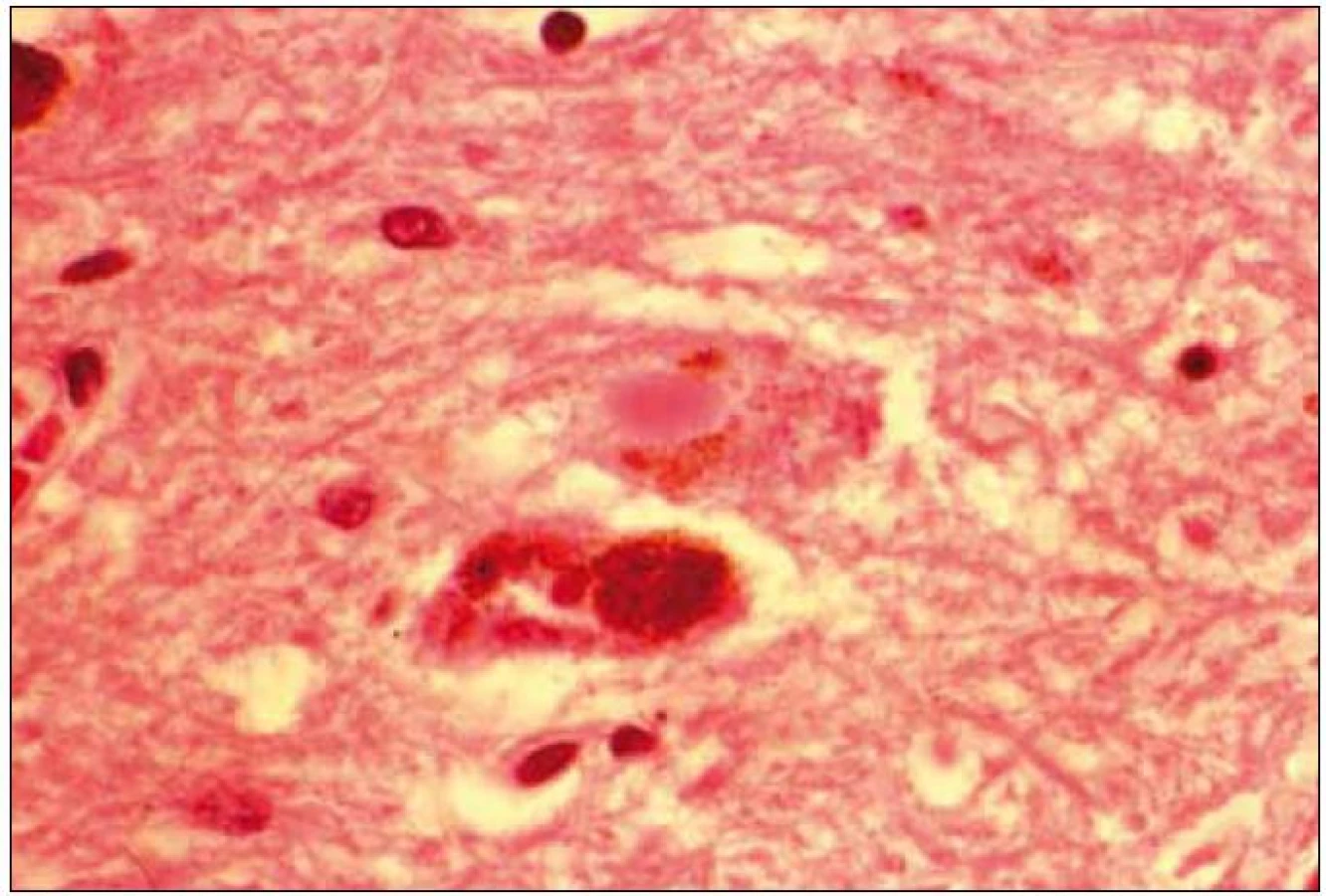
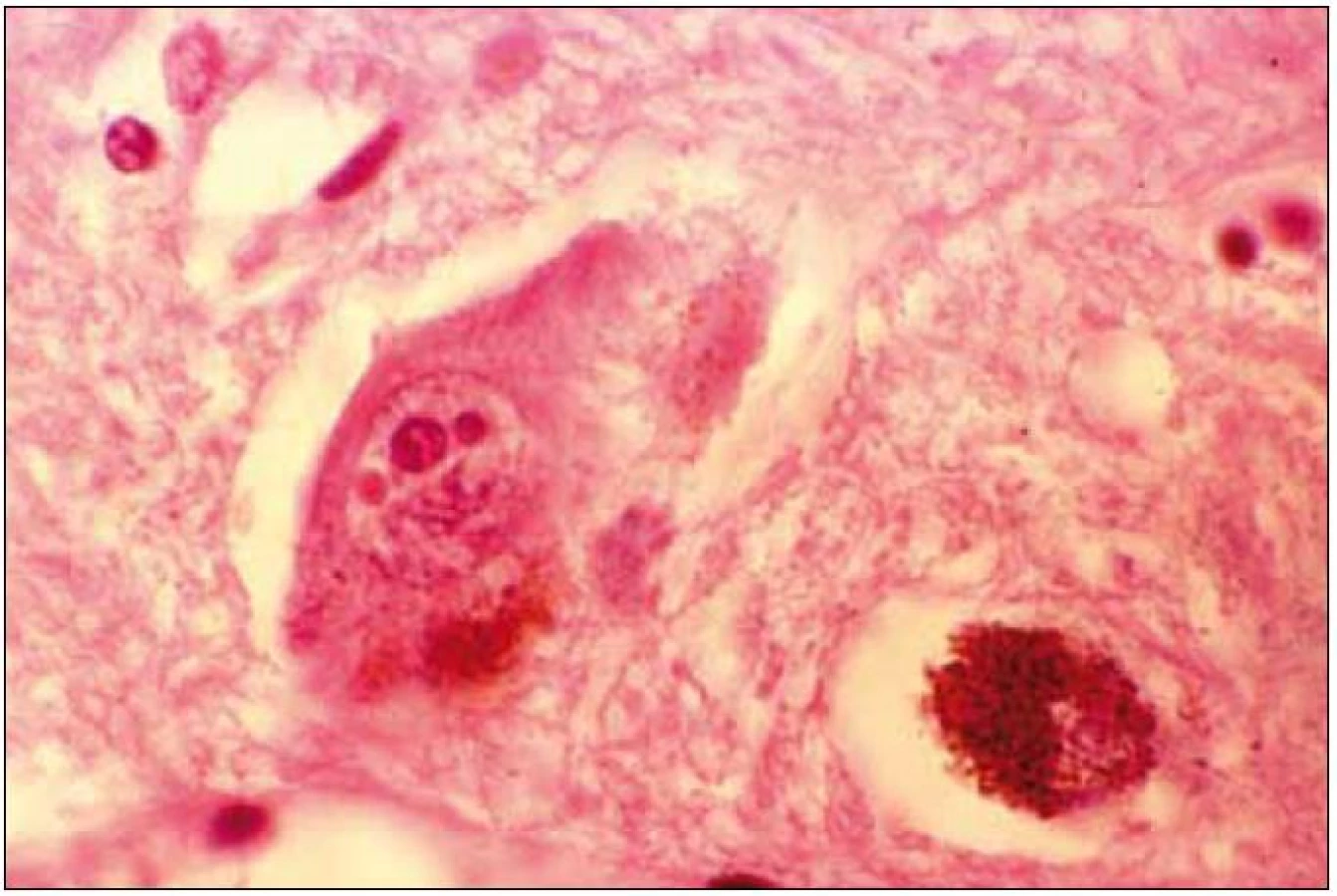
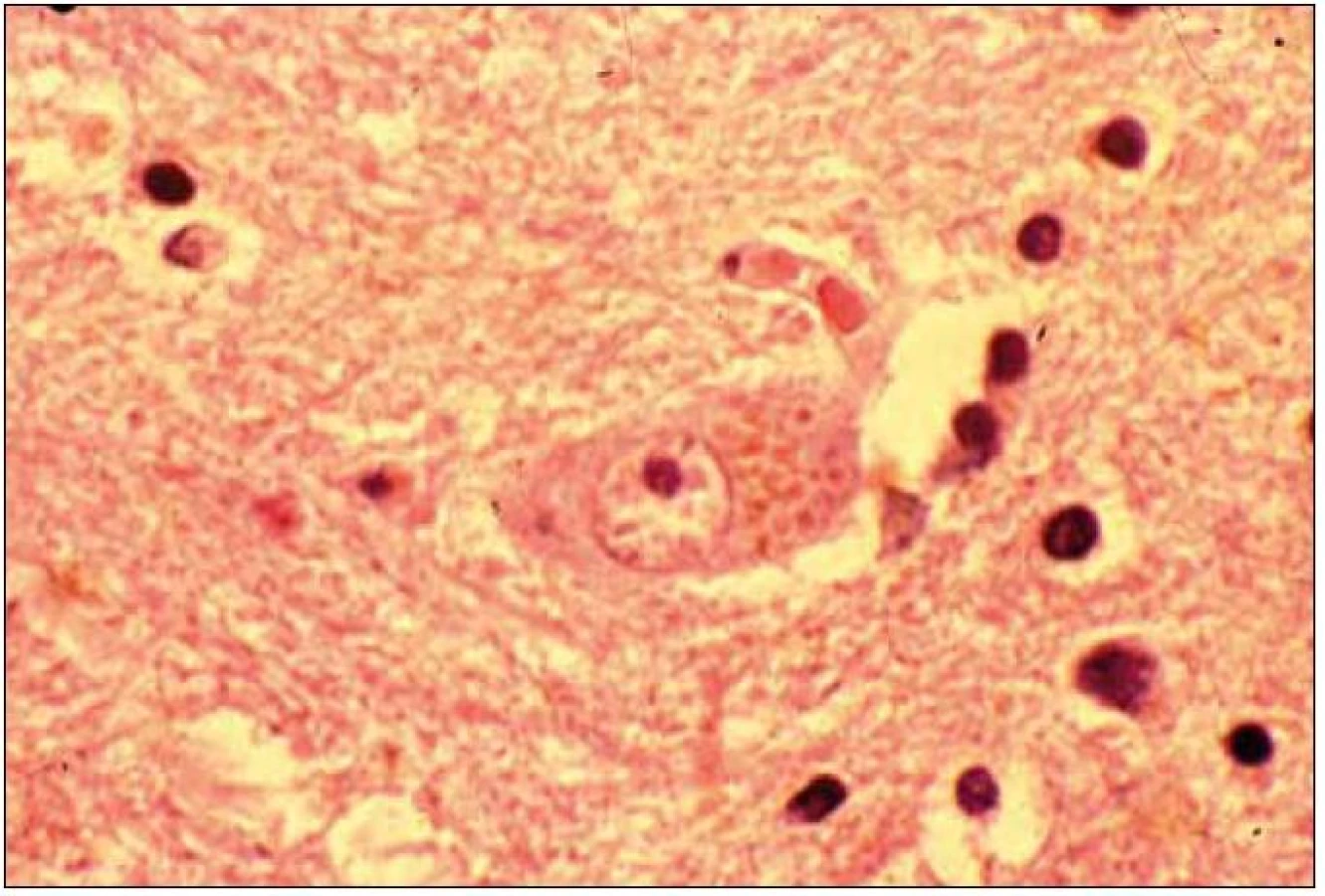
Při mikroskopickém vyšetření byla zjištěna přítomnost pigmentovaných a depigmentovaných neuronů v substantia nigra v různém stupni regresivních změn (obr. 2, 3), neuronů obsahujících typická Lewyho tělíska s maximem v substantia nigra a mesencefalu (obr. 4–6), v mozkovém kortexu Lewyho tělíska detekována nebyla, dále byly přítomny depigmentované neurony v mesencefalu obsahující Marinescova tělíska (obr. 7) a zanikající neurony v oblasti mozkového kmene s akumulovaným lipopigmentem (obr. 8). Zároveň nebyly nalezeny změny typické pro tau patologii.
Závěr
Uvedená kazuistika pacienta s klinickým obrazem typickým pro PSP a následně patologickým nálezem jednoznačně odpovídajícím Parkinsonově nemoci je dokladem toho, že Parkinsonovu nemoc již jednoduše nelze považovat za onemocnění, za které jsme ji doposud považovali. Tedy onemocnění manifestující se typickou dominantní tetrádou příznaků, k nimž se řadí klidový třes, hypokineze, bradykineze a posturální instabilita. Námi prezentovaný pacient je evidentním příkladem, kdy se tzv. Lewy body patologie, nebo alfa-synukleinopatie, dosud považovaná za typickou pro Parkinsonovu nemoc, manifestovala fenotypem odpovídajícím PSP. Rovněž časová souvislost mezi rozvojem poruch hybnosti a kognitivní poruchy, která byla v mírném stupni detekována v druhém roce onemocnění, poměrně rychle progredovala, u níž dominoval dysexekutivní syndrom doprovázený frontálními příznaky, utilizačním a imitačním chováním, svědčila spíše pro PSP než pro Parkinsonovu nemoc. Zcela jistě nejde o ojedinělý případ. V literatuře je dokumentována celá řada případů, kdy patologický nález odpovídal úplně jiné klinické entitě než té, na níž se podle charakteru fenotypické manifestace s vysokou mírou tzv. diagnostické jistoty usuzovalo [5–8].
Poslední dobou se stále častěji nabízí otázka, zda bychom neměli postupně upustit od striktní klasifikace parkinsonských syndromů, tj. dělení na idiopatickou Parkinsonovu nemoc a tzv. „atypické parkinsonské syndromy“, jež jsou dále rozděleny do dalších několika klinických jednotek s možnou další subdiferenciací. S přibývajícím množstvím klinickopatologických studií je stále jasnější, že z fenotypu onemocnění nelze jednoznačně a spolehlivě odhadnout charakter patologického procesu, tudíž již nelze tato onemocnění „škatulkovat“ do dnes známých skupin. Ve starší i poměrně recentní literatuře lze nalézt výsledky řady klinickopatologických studií a kazuistik, kdy neuropatologické nálezy pacientů s typickým klinickým obrazem PSP obsahovaly kromě PSP patologie i Lewyho tělíska [9], v jiném případě jenom difuzní Lewyho tělíska s neurofibrilárními klubky a neuritickými plaky [10]. Naopak u pacientů s typickým klinickým obrazem Alzheimerovy demence či frontotemporální demence byly zjištěny neuropatologické nálezy, jež odpovídaly PN a PSP [11,12]. Podobné průniky fenotypů i patologických nálezů byly popsány i mezi frontotemporální demencí, kortikobazální degenerací a PSP [13]. S takovýmito výsledky klinickopatologických korelačních studií by se patrně měl pozvolna měnit náš dnešní pohled na skupinu parkinsonských syndromů; nelze se ubránit dojmu, že jde možná o jeden či dva prototypy postižení nervové tkáně manifestující se nejrůznějšími fenotypy, jež jsou obrazem spíše distribuce než charakteru primárního patologického neurodegenerativního procesu [14].
Pro bližší porozumění zákonitostem tohoto onemocnění, pokud vůbec nějaké existují, bude nezbytné podrobné zkoumání neuropatologické a hledání určitých specifik toho kterého fenotypu ve vztahu ke konkrétním neuropatologickým změnám. Jinými slovy, dokud nebudeme u konkrétního případu znát přesnou povahu patologického procesu, neměli bychom asi příslušný konkrétní soubor příznaků nazývat onemocněním, ale spíše jenom syndromem [15].
Poděkování
Práce byla podpořena grantovým projektem IGA UP LF_2011_012.
MUDr. Kateřina Farníková
Neurologická klinika
LF UP a FN Olomouc
I. P. Pavlova 6
775 20 Olomouc
e-mail: katmen@centrum.cz
Přijato k recenzi: 4. 2. 2011
Přijato do tisku: 22. 4. 2011
Sources
1. Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002; 125(Pt 4): 861–870.
2. Grambalová Z, Hluštík P, Heřman M, Kaňovský P. Přítomnost tzv. typických MR nálezů u multisystémové atrofie a progresivní supranukleární paralýzy – retrospektivní pilotní studie. Cesk Slov Neurol N 2010; 73/106(5): 538–541.
3. Richardson JC, Steele J, Oslzewski J. Supranuclear ophtamoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of “heterogenous system degeneration“. Trans Am Neurol Assoc 1963; 88 : 25–29.
4. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: a heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 10 : 333–359.
5. Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LK, Williams DR et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010; 133(Pt 7): 2045–2057.
6. Abhinav K, Marsch L, Crain B, Reich SG, Biglan K. Co-existence of Parkinson’s disease and progressive supranuclear palsy: case report and a review of the literature. Neurol Sci 2011; 32(1): 159–163.
7. Wadia PM, Lang AE. The many faces of corticobasal degeneration. Parkinsonism Relat Disord 2007; 13 (Suppl 3): S336–S340.
8. Wakabayashi K, Takahashi H. Pathological heterogeneity in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 2004; 24(1): 79–86.
9. Mori H, Yoshimura M, Tomonaga M, Yamanouchi H. Progressive supranuclear palsy with Lewy bodies. Acta Neuropathol 1986; 71(3–4): 344–346.
10. Fearnley JM, Revesz T, Brooks DL, Frackowiak RS, Lees AJ. Diffuse Lewy body disease presenting with a supranuclear gaze palsy. J Neurol Neurosurg Psychiatry 1991; 54(2): 159–161.
11. Gearing M, Olson DA, Watts RL, Mirra SS. Progressive supranuclear palsy: neuropathologic and clinical heterogeneity. Neurology 1994; 44(6): 1015–1024.
12. Judkins AR, Forman MS, Uryu K, Hinkle DA, Asbury AK, Lee VM et al. Co-occurrence of Parkinson’s disease with progressive supranuclear palsy. Acta Neuropathol 2002; 103(5): 526–530.
13. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006; 66(1): 41–48.
14. Galpern WR, Lang AE. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol 2006; 59(3): 449–458.
15. Strupp M. Corticobasal syndrome: a field of uncertainty. J Neurol 2011; 258(1): 173–175.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2011 Issue 6
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Miller Fisherův syndrom – čtyři vlastní pozorování a přehled současných poznatků
- Současný pohled na patofyziologii migrény
- Novelizace české verze Addenbrookského kognitivního testu (ACE-CZ)
- Operační léčba poranění plexus brachialis