
Papilární nádor pineální oblasti u dítěte – kazuistika
Papillary Tumor of the Pineal Region in a Child – a Case Report
We describe a case of a five years old girl with a large tumor of the pineal region, the 3rd and 4th ventricles, presented with the increased intracranial pressure syndrome. Endoscopic third ventriculostomy with tumor biopsy was performed, and the tumor was classified as papillary tumor of the pineal region. Local relapse occurred fourteen months after complete resection and it was treated with stereotactic external fractionated radiotherapy. Papillary tumor of the pineal region is a rare tumor with a difficult diagnosis, uncertain prognosis, and a high risk of local recurrence. The only defined prognostic factor is radicality of the resection. Most adult patients are treated with a combination of surgery and adjuvant local radiotherapy. A standard of care for children has not been clearly defined yet.
Key words:
papillary tumor – pineal region – radiotherapy – chemotherapy
Authors:
Z. Pavelka 1; M. Smrčka 2; L. Křen 3; M. Keřkovský 4; J. Skotáková 5; P. Šlampa 6
![]() ; K. Zitterbart 1; J. Štěrba 1
; K. Zitterbart 1; J. Štěrba 1
Authors‘ workplace:
Klinika dětské onkologie LF MU a FN Brno
1; Neurochirurgická klinika LF MU a FN Brno
2; Ústav patologie LF MU a FN Brno
3; Radiologická klinika LF MU a FN Brno
4; Klinika dětské radiologie LF MU a FN Brno
5; Klinika radiační onkologie LF MU a MOÚ v Brně
6
Published in:
Cesk Slov Neurol N 2012; 75/108(6): 754-756
Category:
Case Report
Overview
Popisujeme případ pětileté dívky s rozsáhlým nádorem pineální oblasti, třetí i čtvrté mozkové komory, který se prezentoval příznaky syndromu intrakraniální hypertenze. Byla provedena endoskopická ventrikulostomie s biopsií tumoru, jenž byl odečten jako papilární nádor pineální oblasti. Po radikální resekci došlo s odstupem 14 měsíců k lokální recidivě, která byla ošetřena stereotaktickou zevní frakcionovanou radioterapií. Papilární nádor pineální oblasti je vzácný nádor s obtížnou diagnostikou, nejistou prognózou a vysokým rizikem lokální recidivy. Jediný známý prognostický faktor je radikalita resekce. Většina dospělých pacientů je léčena kombinací chirurgického výkonu a následné lokální radioterapie. Standardní léčba pro děti není jasně definována.
Klíčová slova:
papilární nádaor – pineální oblast – radioterapie – chemoterapie
Úvod
Nádory pineální oblasti jsou u dětí vzácné, tvoří pouze 3–4 % všech primárních nádorů centrálního nervového systému (CNS) dětí [1]. Anatomické struktury pineální oblasti dávají vznik různým histologickým typům nádorů s rozdílnou prognózou: parenchymální tumory (pineocytom), nádory ze zárodečných buněk, nádory z glie, primitivní neuroektodermální tumory (zde pineoblastom), meningeomy a cysty [1].
V roce 2003 byl poprvé jako nový typ tumoru popsán Jouvetem et al [2] papilární nádor pineální oblasti (PNPO). Jedná se o vzácný nádor nejisté biologické povahy, který sdílí morfologické rysy papilární varianty ependymomu a papilomu choroidálního plexu [2]. U dětí byly publikovány pouze jednotlivé případy, dosud není jednoznačně definována standardní léčba.
Kazuistika
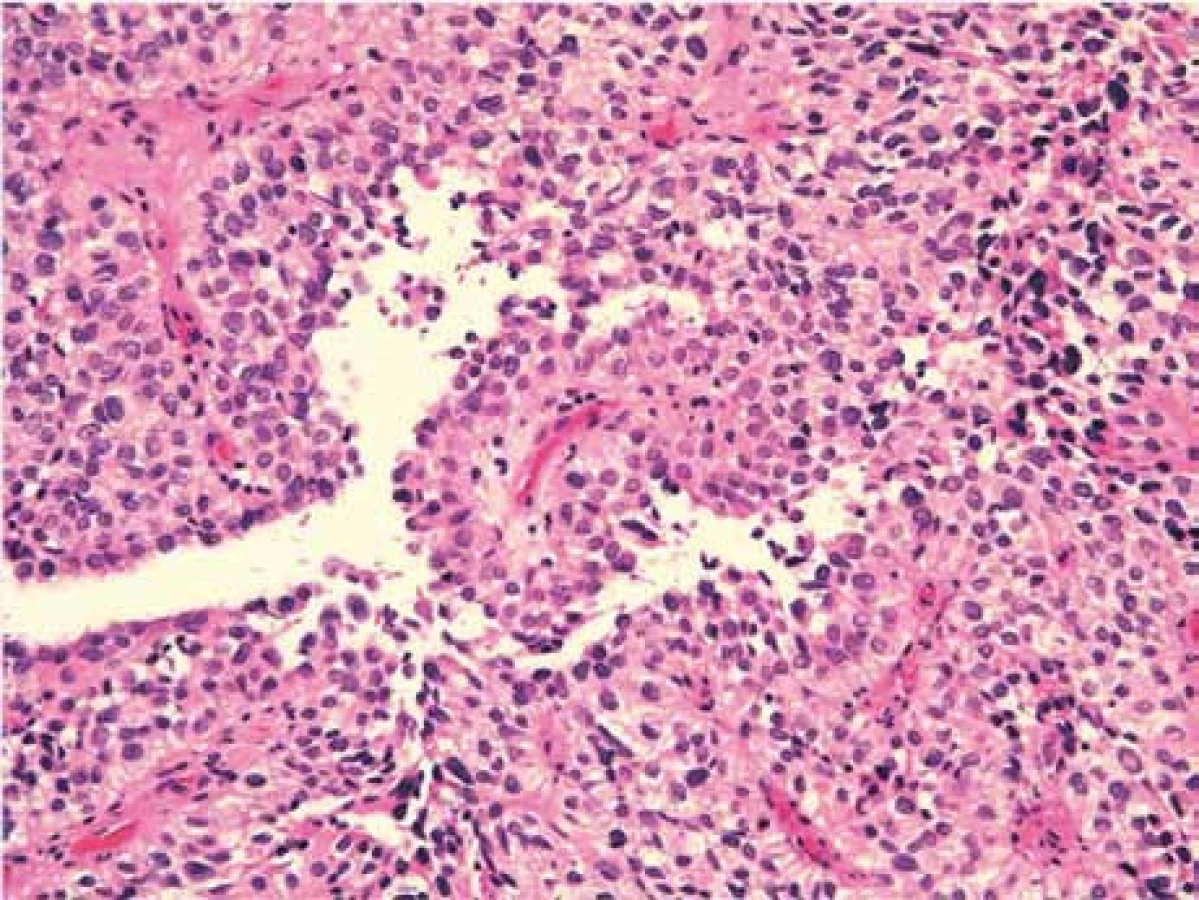
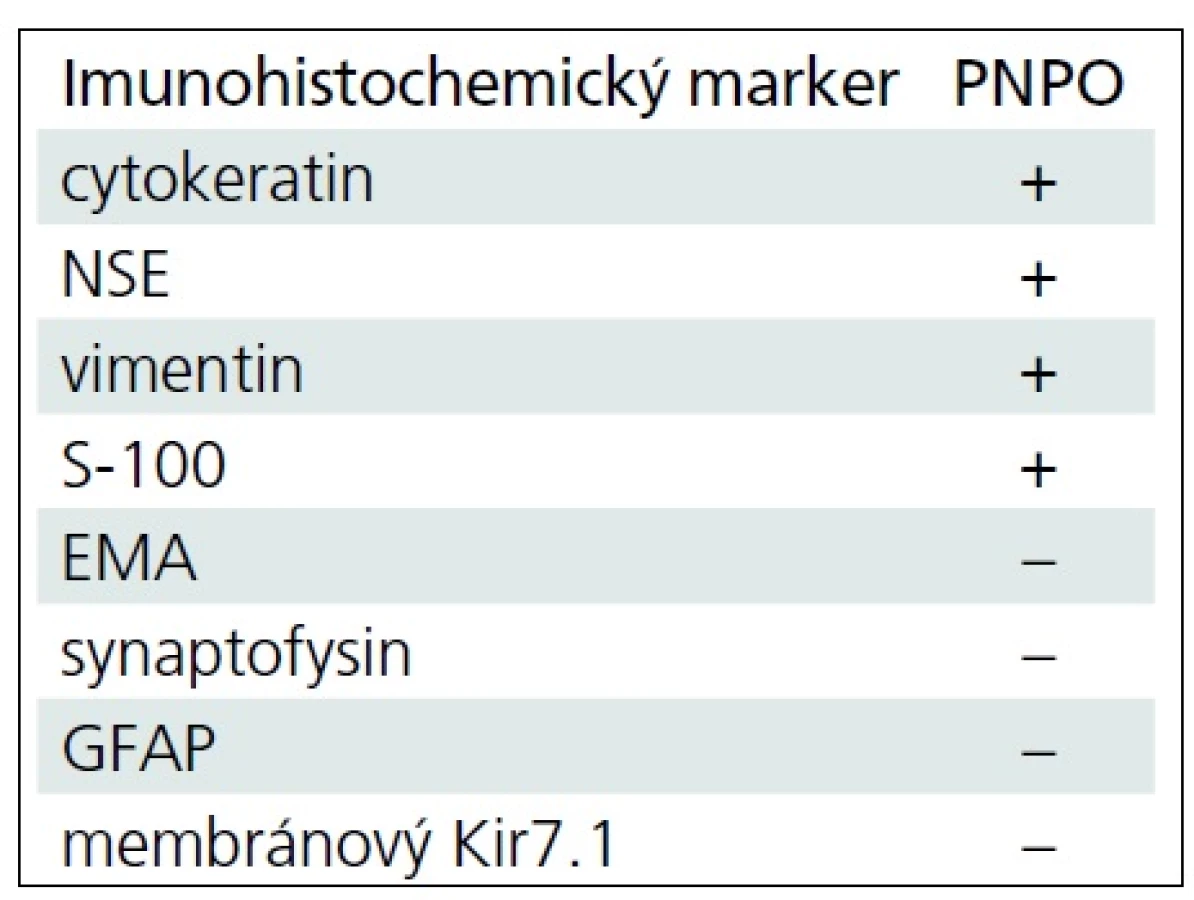
Pětiletá dívenka s negativní rodinnou anamnézou se dostavila s příznaky syndromu intrakraniální hypertenze. Vyšetření mozku magnetickou rezonancí (MR) prokázalo tumor pineální oblasti s postižením třetí komory a s propagací skrze Sylviův akvedukt do čtvrté mozkové komory. Tumor o celkových rozměrech 27 × 24 × 44 mm působil obstrukční hydrocefalus. Byl v MR obraze nehomogenní, obsahoval úseky cystické a solidní (obr. 1). Nejprve byla provedena endoskopická ventrikulostomie s biopsií tumoru i odběrem mozkomíšního moku. Hodnoty alfa-fetoproteinu a choriogonadotropinu v moku i séru byly negativní. V mikroskopickém obraze bioptického vzorku dominovalo papilární uspořádání s jemným fibrovaskulárním stromatem krytým nasedajícími nádorovými buňkami ve více řadách (obr. 2). Buňky byly cylindrické, vyšší, se světlou eozinofilní cytoplazmou. Solidní buněčné partie vykazovaly jadernou pleomorfii. Nádorové nekrózy nebyly zastiženy. Chyběly typické ependymální rosety, mitotická aktivita byla střední s Ki67 mezi 5 a 10 %. Imunohistochemický profil tumoru byl následující: pozitivní vimentin, cytokeratin, NSE, S100-P, negativní EMA, GFAP, synaptofyzin, membránový Kir 7.1 (tab. 1). Tumor byl klasifikován jako papilární nádor pineální oblasti s hraničními biologickými vlastnostmi (stupeň malignity 2 až 3 dle WHO 2007).
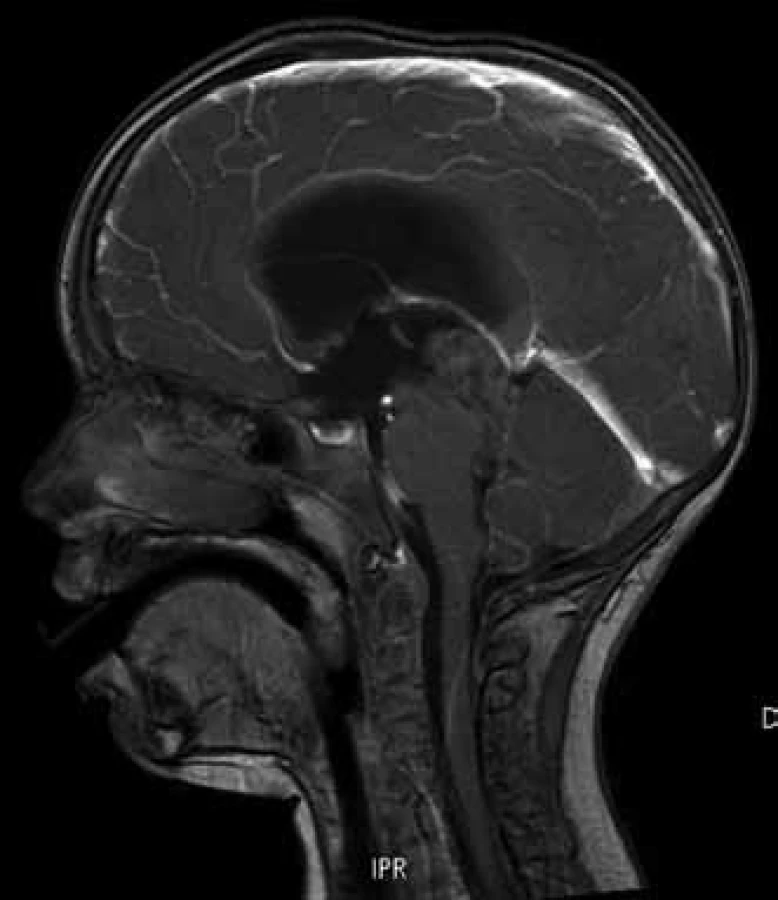
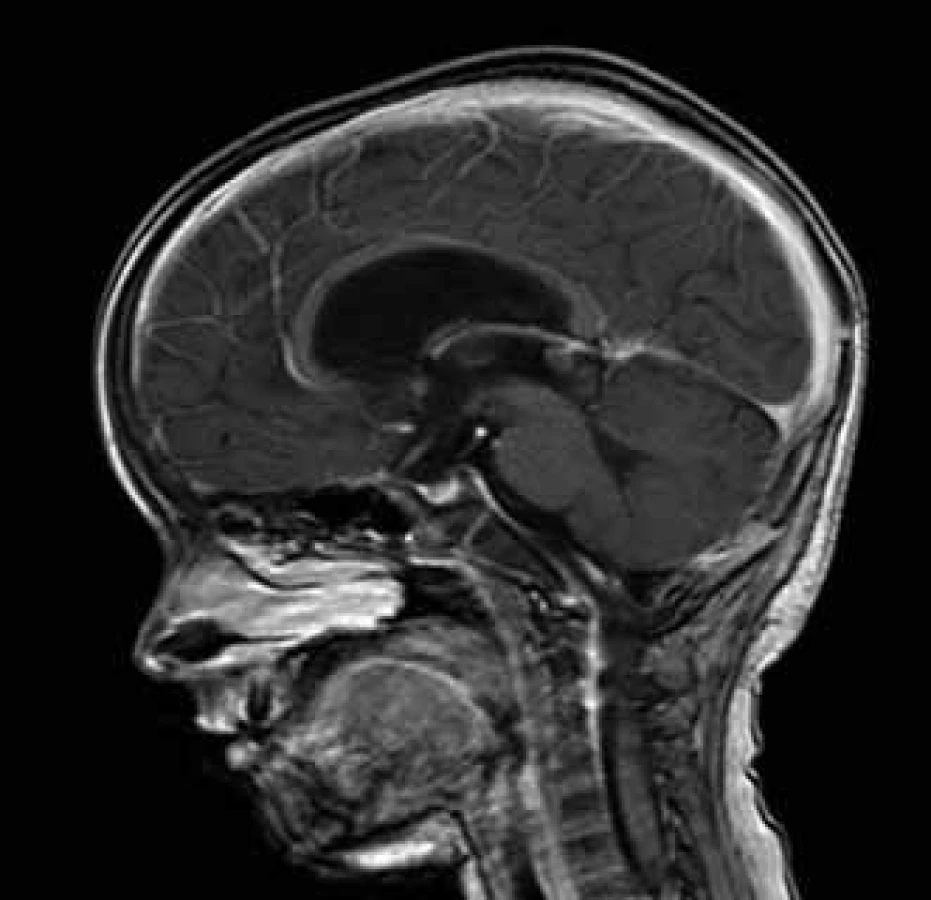
Diseminace nádoru v rámci CNS byla vyloučena negativním MR míchy a cytologickým vyšetřením moku. Vzhledem k rozsahu nádoru se současným postižením 3. i 4. komory byl operatérem zvolen dvoudobý operační výkon. Nejprve byla z přístupu supracerebelárního infratentoriálního odstraněna část tumoru ve třetí mozkové komoře až do akveduktu. Nádor se jevil šedavý, cystický, místy hlenovitý. Tumor se podařilo z této oblasti vyjmout s ponecháním možného mikroskopického rezidua v oblasti tekta, které bylo devitalizováno koagulací. V odstupu 14 dní následovala subokcipitální kraniotomie s přístupem do čtvrté komory s vermotomií. Z tohoto přístupu byla odstraněna porce tumoru ve čtvrté komoře a dolní části akveduktu. Časná pooperační MR 24 hod po výkonu nepopsala jasné měřitelné reziduum. Pooperačně bylo dítě bez významného neurologického deficitu vyjma okohybné poruchy. V této fázi jsme projednávali otázku následné zevní radioterapie, která však nakonec indikována nebyla a zůstala vyhrazena pro léčbu eventuální recidivy. Dítě bylo periodicky kontrolováno vyšetřením MR. Při rutinní MR kontrole 14 měsíců od operace byla zjištěna lokální recidiva v dorzální části stropu třetí mozkové komory o velikosti 13 × 10 × 8 mm (obr. 3), přežití bez události (Event Free Survival, EFS) 14 měsíců. Tato lokální recidiva byla ošetřena zevní stereotaktickou radioterapií lineárním urychlovačem o energii 6 MeV dávkou 48 Gy (16 frakcí á 3 Gy). Kontrolní MR vyšetření v odstupu šesti měsíců pro provedené stereotaktické radioterapii popsalo velikostní i strukturální regresi ložiska (velikost 10 × 9 × 6 mm). V současnosti je dítě plně mobilní, bez končetinových paréz, bez poruch taxe, s okohybnou poruchou ve smyslu omezené hybnosti očních bulbů ve vertikále. Druhý EFS činí 12 měsíců a celkové přežití (Overall Survival, OS) je v době prezentace sdělení 36 měsíců od doby prvotní diagnózy.
Diskuze
Nádory pineální oblasti představují pouze 3–4 % všech nádorů mozku u dětí a tvoří heterogenní skupinu [1,3]. V této lokalizaci se mimo PNPO dále vyskytují parenchymální tumory (pineocytom), nádory ze zárodečných buněk, nádory z glie, primitivní neuroektodermální tumory (pineoblastom), meningeomy a cysty [3]. Papilární tumor pineální oblasti je relativně nová jednotka prvně popsaná v literatuře v roce 2003 [2]. Dosud byly publikovány pouze malé série dospělých pacientů, u dětí pak jen jednotlivé kazuistiky [4–6]. MR obraz je nespecifický, diagnózu PNPO z něj nelze přesně určit a její stanovení je v rukou patologa.
Diferenciální diagnostika PNPO je obtížná a zahrnuje především papilární formu ependymomu a papilom choroidálního plexu, se kterými PNPO sdílí základní morfologické rysy. PNPO je neuroepiteliální nádor vycházející ze specializovaného ependymu subkomisurálního orgánu [2]. Biologická povaha PNPO je nejistá, stupeň malignity bývá označován jako 2 až 3. Nádorové buňky PNPO formují pseudorozety, imunohistochemický profil je dosti podobný nádorům plexu. Nádorové buňky PNPO konstantně exprimují NSE, S100-P, vimentin a cytokeratin KL1 a cytokeratin 18 (zvláště v papilárních zónách). Barvení pro GFAP a synaptofyzin bývá negativní [2,4]. Od papilomu plexu odlišuje PNPO spolehlivě absence barvení membránového kanálu pro draslík Kir7.1, negativita barvení pro EMA pak vylučuje papilární variantu ependymomu, který je GFAP a EMA pozitivní [4].
Pineocytom vycházející z pineálního parenchymu formuje podobné papilární struktury a pseudorozety jako PNPO, avšak odlišuje ho rozdílná imunoreaktivita: masivní pozitivita synaptofyzinu, zatímco cytokeratiny a vimentin nejsou exprimovány [2,4]. Cytogenetické změny nejčastěji prokazované u PNPO zahrnují delece chromozomů 10 a 22q a dále inzerce v oblasti chromozomů 4, 8, 9, 12. Prognostický význam těchto aberací není dosud znám [7].
Střední věk pacientů s PNPO v publikovaných souborech činí 30 let [4–6], převažující je ženské pohlaví. Vzhledem k raritě nádoru je málo známo o prognostických faktorech a není jednoznačně definován standard léčby, zejména u dětí. Metastatická manifestace či forma recidivy jsou raritní, naopak lokální recidivy jsou časté. V největším hodnoceném souboru autorů Fèvre-Montange et al bylo publikováno u 31 pacientů s PNPO celkové přežití 73 % v pěti letech a 58 % v deseti letech od diagnózy [4]. Za jediný dosud definovaný prognostický faktor je považována radikalita resekce. Přítomnost mitotické aktivity v tumoru (> 1 mitóza/10 zorných polí o vysokém rozlišení) neovlivňovala celkové přežití ani přežití bez progrese [4]. V případech lokální recidivy pak její radikální resekce a/nebo radioterapie vedla ke stabilizaci či zpomalení progrese onemocnění [4,8]. Většina autorů doporučuje proto po operaci adjuvantní radioterapii různého rozsahu od stereotaktického ozáření rezidua až po konformní radioterapii na lůžko po resekci [4–6,8]. Role radioterapie by měla být evaluována v prospektivní studii. Ještě méně je známo o efektivitě chemoterapeutických režimů, chemoterapie byla dosud podávána jenom v ojedinělých specifických případech (recidiva po předchozí radioterapii v ozařovaném poli, vzácné případy spinální diseminace), režimy byly postaveny na platinových derivátech nebo nověji temozolomidu [9]. Opět bude nezbytné efektivitu chemoterapie ověřit formou standardních prospektivních studií.
Závěr
Papilární nádor pineální oblasti je vzácný tumor centrálního nervového systému s obtížnou diagnostikou, nejistou prognózou a vysokým rizikem lokální recidivy. Standardní léčba pro děti není jasně definována. Tato kazuistika dokumentuje obtíže při stanovení diagnózy a volbě léčebné strategie u malého dítěte s PNPO, jehož dlouhodobá prognóza je nejistá.
MUDr. Zdeněk Pavelka
Klinika dětské onkologie
LF MU a FN Brno
Černopolní 9
613 00 Brno
e-mail: zpavelka@fnbrno.cz
Přijato k recenzi: 20. 2. 2012
Přijato do tisku: 29. 3. 2012
Sources
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114(2): 97–109.
2. Jouvet A, Fauchon F, Liberski P, Saint-Pierre G, Didier-Bazes M, Heitzmann A et al. Papillary tumor of the pineal region. Am J Surg Pathol 2003; 27(4): 505–512.
3. Gupta N, Banerjee A, Haas-Kogan D. Rare tumors. In: Gupta N, Banerjee A, Haas-Kogan D (eds). Pediatric CNS tumors. 2nd ed. Berlin, Heidelberg: Springer--Verlag 2010 : 205–219.
4. Fèvre-Montange M, Hasselblatt M, Figarella-Branger D, Chauveinc L, Champier J, Saint-Pierre G et al. Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol 2006; 65(10): 1004–1011.
5. Buffenoir K, Rigoard P, Wager M, Ferrand S, Coulon A, Blanc JL et al. Papillary tumor of the pineal region in a child: case report and review of the literature. Childs Nerv Syst 2008; 24(3): 379–384.
6. Sato T, Kirby PA, Buatti JM, Moritani T. Papillary tumor of the pineal region: report of a rapidly progressive tumor with possible multicentric origin. Pediatr Radiol 2009; 39(2): 188–190.
7. Gutenberg A, Brandis A, Hong B, Gunawan B, Enders C, Schaefer IM et al. Common molecular cytogenetic pathway in papillary tumors of the pineal region (PTPR). Brain Pathol 2011; 21(6): 672–677.
8. Reyns N, Hayashi M, Chinot O, Manera L, Péragut JC, Blond S et al. The role of Gamma Knife radiosurgery in the treatment of pineal parenchymal tumours. Acta Neurochir 2006; 148(1): 5–11.
9. Lorenzetti M, Motta F, Campanella R, Bauer D, Assi A, Arienta C et al. Adjuvant temozolomide chemotherapy for treatment of papillary tumor of the pineal region. World Neurosurg 2011; 76(1–2): 160–163.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2012 Issue 6
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Epidemie roztroušené sklerózy ve světě?
- Kortikální patologie u roztroušené sklerózy – morfologické, imunopatologické a klinické souvislosti
- Fázový model neurorehabilitace
- Endovaskulární léčba ischemické cévní mozkové příhody