
Molecular Genetic Analysis of Fetal Tissues from a Family Affected by Myotonic Dystrophy
Molekulárně genetická analýza tkáně plodu rodiny postižené myotonickou dystrofií
Vývoj patologického fenotypu u pacientů s myotonickou dystrofií závisí značnou měrou na toxickém účinku transkriptu expandované repetice genu DMPK a jeho interakci s vazebnými proteiny. Vztah mezi CUGexp tohoto genu a vazebnými proteiny MBNL1 a MBNL2 byl dosud studován na tkáňových kulturách myoblastů a na transgenních zvířatech. V tomto sdělení podáváme první molekulárně genetickou analýzu fetálních tkání plodu z rodiny postižené myotonickou dystrofií prvního typu (MD1). Intrauterinní vývoj, zrání a diferenciace kosterního svalstva plodu (360 g, 22. týden) byly jen mírně opožděny. Paternálně zděděná expanze repetice CTG v genu DMPK (350 CTG) byla potvrzena molekulárně genetickou analýzou a následné histopatologické vyšetření plodu prokázalo znaky myotonické dystrofie. Tkáně získané autopsií (kosterní sval, jícen, žaludek a střevo) byly vyšetřeny histologickými i molekulárně genetickými (imunohistochemickou analýzou proteinů MBNL1 a MBNL2, a in situ hybridizací DMPK CUGexp) metodami. Intranukleární ložiska (foci) s CUG transkripty byla zjištěna v kosterním svalu, ve svalovině (muscularis externa) zažívací trubice, v cévní medii, endotelu a intramurálních nervových pleteních i v epitelu. Protein MBNL1 přitom s ložisky expandovaných transkriptů extenzivně kolokalizoval ve všech tkáních. Naproti tomu byl protein MBNL2 nalezen též v cytoplazmě. Přítomnost transkriptu DMPK s expandovanou repeticí CUG a proteinu MBNL1 v intranukleárních fokusech fetálních tkání MD1 pacientů může teoreticky znamenat sekvestraci proteinu MBNL1, a tak přispět ke generaci patologického fenotypu MD1.
Klíčová slova:
myotonická dystrofie – DMPK mutace – fetální tkáň – proteiny MBNL
Authors:
Z. Lukas? 1; M. Falk 2; I. Falková 1,2; J. Feit 1; L. Fajkusová 3,4; J. Zítková 3,4; I. Valaskova 5
Authors‘ workplace:
Institute of Pathology, Faculty of Medicine, Masaryk University and University Hospital Brno
1; Institute of Biophysics, Academy of Sciences of the Czech Republic, Brno
2; IHOK, Centre of Molecular Biology and Gene Therapy, University Hospital Brno
3; CEITEC, Masaryk University Brno
4; Department of Clinical Genetics, University Hospital Brno
5
Published in:
Cesk Slov Neurol N 2012; 75/108(6): 730-736
Category:
Short Communication
Overview
Development of the pathological phenotype in patients with myotonic dystrophy (MD1) largely depends on toxic effects of expanded DMPK gene repeats (CUGexp) transcription and their interaction with binding proteins. The relationship between DMPK CUGexp, MBNL1 and MBNL2 has so far been studied on tissue cultures (myoblast cell lines) and transgenic animals. In this report, the first in situ molecular genetic analysis of fetal tissue from a family affected by MD1 is presented. Intrauterine development, maturation and differentiation of skeletal muscles of the fetus (360 g, 22nd week of pregnancy) were only slightly delayed. In the fetus, paternally inherited expansion of the CTG repeat in the DMPK gene (350 CTG) was confirmed by molecular analysis. Subsequent histopathological examination revealed signs of myotonic dystrophy. Fetal tissue obtained at autopsy (skeletal muscles, esophagus, stomach and intestines), were studied by histopathological, immunofluorescence (expression of MBNL1/MBNL2 proteins) and in situ hybridization (DMPK CUGexp) methods. Intranuclear CUGexp-containing foci were present in skeletal muscle fibers, muscularis externa of the esophagus, stomach and intestines, vascular smooth muscle, neurons and Schwann cells of intrinsic ganglionic plexuses, and epithelial cells. MBNL1 protein was largely co-localized with the CUGexp foci in all tissues examined. Contrarily, MBNL2 protein was also detected in the tissue cytoplasm. The presence of DMPK transcript with expanded CUG repeat and MBNL1 protein in the intranuclear foci of MD1 fetal tissues studied may theoretically result in sequestration of the protein and thus contribute to generation of the MD1 phenotype.
Key words:
myotonic dystrophy – DMPK mutation – fetal tissue – MBNL proteins
Introduction
Myotonic dystrophy type 1 (MD1, OMIM#160900) is an autosomal dominant genetic multi-systemic disorder with anticipation – the increase of the mutated allele expansion length and increased disease severity seen through successive generations [1]. Genetic cause of MD1 was identified as an expansion of the CTG triplet repeat in the 3´-untranslated region of the Dystrophia Myotonica Protein Kinase gene (DMPK, 19q13.3) [2,3]. Mutant DMPK alleles could contain up to several thousand triplets, whereas normal alleles have between 5 and 35 CTGs. Both normal and expanded alleles are transcribed but intracellular transport of the expanded mRNA is inhibited. This results in retention of the expanded transcripts in the cell nucleus and formation of ribonuclear CUGexp foci [4,5]. Molecular pathogenesis of MD1 includes sequestration of muscleblind proteins in the CUGexp foci causing a loss of their function in the nucleoplasm. Recruitment of human muscleblind (MBNL) proteins into CUGexp foci in MD1 was firstly suggested by Miller et al [6] and illustrated in vivo by Fardaei et al [7,8]. In addition to MBNL loss of function, there is also evidence that elevation of another family of CUG-binding proteins – CUG-BP(1) may play a role in MD1 pathogenesis [9–11].
MBNL proteins have been implicated in an alternative splicing of several specific pre-mRNA targets [12]. In addition to regulating terminal muscle differentiation through an alternative splicing control, MBNL proteins also participate in differentiation of photoreceptors, neurons, adipocytes, and blood cell types [13].
CUGexp mRNA of the DMPK gene and sequestration of MBNL1 protein were detected in skeletal muscles (both human and murine), heart, smooth muscles, brain, and some other differentiated tissues [14,15]. Retention of mutant RNA ribonuclear foci during fetal stages of development was only studied in cultivated myoblasts derived from the quadriceps muscles of three CDM fetuses with various clinical severity [16]; the relationship between CUGexp expression and recruitment of muscleblind proteins in fetal CDM tissues in situ has not been reported yet.
In this study, we present the first molecular genetic and histopathological in situ analysis of fetal tissues from a family affected by myotonic dystrophy (MD1). We found CUGexp retained in ribonuclear foci of the fetal tissue and co-localized with muscleblind proteins, and suppose that sequestration of the proteins may contribute to the generation of the MD1 phenotype.
Material and methods
Confirmation of MD1 diagnosis by TP-PCR
The family history of MD1 indicated a pregnancy with an increased risk of MD1. The father suffered from a mild form of adult DM with the disease onset at around 25 years, with myotonia and slight increase of creatine kinase (450 U/L) but with no muscle weakness. The mother was free of MD1 symptoms and the fetus did not express signs of congenital DM (decreased fetal movements, polyhydramnion or relevant ultrasound findings) during pregnancy. An analysis of the CTG repeat size in the DMPK gene was performed on genomic DNA isolated from peripheral blood leukocytes and from fetal cells (obtained by amniocentesis using standard procedures) by standard flourescence PCR (with primers P1 and P2) in combination with TP-PCR [17, as described in 18]. The family members (father, mother, and father’s parents) were examined before prenatal testing of the fetus. Since DNA test confirmed expansion in the fetal DMPK gene, the parents were given an option to terminate pregnancy. The termination of pregnancy was performed in accordance with the Czech legislation and ethical rules.
Determination of the CTG-repeat expansion size by Southern blotting
The size of CTG-repeat expansions was determined by Southern blot analysis as described in Guida et al [19].
Fetal tissue samples
Fetal tissue was extracted for diagnostic purposes at the 22nd week of pregnancy. Samples from cross-striated muscles (vastus lateralis and gastrocnemius), esophagus, stomach, and intestines were obtained at autopsy performed 9 hours after the abortion. The samples were deep-frozen in a propane-butane mixture cooled with liquid nitrogen. Cryostat sections were processed using a set of conventional histological and histochemical methods [20] supplemented by immunohistochemical detection of (1) fetal (developmental) myosin heavy chain (d-MHC) in cross-striated muscle fibers, and (2) neurofilaments in intramural fetal plexuses of esophagus, stomach, and intestine.
Immunofluorescence visualization (IFL) of MBNL1 and MBNL2 proteins
IFL was performed according to Holt et al [21]. Fresh frozen sections (6 µm thick) were placed on Superfrost Plus microscope slides (Menzel-Gläser), dried and fixed with acetone-methanol (1 : 1) at room temperature (RT). Alternatively, the sections were fixed in 4% paraformaldehyde. After washing in PBS, the slides were incubated for 1 hour at 37 °C with the MB1a (mouse, dilution 1 : 3), MB2a (mouse, 1 : 3) [22], MBNL1 (mouse, 1 : 200, Sigma) MBNL2 (rabbit, 1 : 200, Sigma) and NFh (Neurofilament-h, Novocastra) primary antibodies, respectively. Subsequently, the sections were washed in PBS at RT and incubated with a secondary antibody labeled with Alexa Fluor 488 (Alexa Fluor 488 Goat Anti-Mouse, SFX Kit, or Goat Anti-Rabbit Molecular Probes™, Invitrogen) for 1 hour at 37 °C. Following incubation, the slides were washed in PBS and mounted in Vectashield Mounting Medium (Vector Laboratories, USA). Nuclear chromatin was counterstained with 0.075 µg/ml diamino-2-phenylindole (DAPI). The image acquisition and evaluation of the results were performed on a Leica DMRXA2 fluorescence microscope; Leica 100x PlApo oil-immersion objective, NA 1.4.
Visualization of CUGexp ribonuclear foci by fluorescence in situ hybridization (FISH)
Visualization of CUGexp ribonuclear foci was performed by FISH on frozen muscle sections, as described in Holt et al [21]. Frozen sections (6 µm) were placed onto SuperFrost Plus slides (Menzel-Gläser), air dried at RT for 30 minutes, fixed in modified Carnoy’s fixative [73% ethanol, 25% acetic acid, 2% formalin (Sigma)] for 30 minutes at 4 °C, and washed in PBS (RT). Then, the sections were incubated in prehybridization buffer [30% formamide (Sigma) in 2× SSC (Eppendorf)] for 5 minutes at RT and thereafter hybridized for 16 hours with 200 nM of the probe (see below) at 37 °C in hybridization buffer [consisting of 30% formamide (Sigma), 2× SSC, 0.02% BSA (Invitrogen), 66 ng/µl of yeast tRNA (Invitrogen), and 2 mM vanadyl complex (Sigma)]. The probe for CUGexp (in the DMPK gene) was 5’-CAG CAG CAG CAG CAG CAG CAG-3’2’–O-Me-RNA5’Fl (Generi Biotech, Czech Rep.). The hybridized sections were washed in a post-hybridization solution [30% formamide (Sigma) in 2× SSC] for 30 minutes at 45 °C, followed by 1× SSC wash for 30 min/RT. Finally, the sections were mounted with DAPI (Vectashield Mounting Medium with DAPI, Vector Laboratories, USA). The slides were examined in a Leica DMRXA2 fluorescence microscope; Leica 100x PlApo oil-immersion objective, NA 1.4.
Combined immunofluorescence and fluorescence in situ hybridization (IFL-FISH)
For IFL-FISH, the sections were dried, fixed with acetone-methanol (1 : 1) at RT, and then incubated with the primary and secondary antibodies as described above. Following the incubation, the slides were washed in 2× SSC, fixed in 4% paraformaldehyde in PBS for 5 minutes at RT, and prehybridized in 30% formamide in 2× SSC for 5 minutes (RT). Next, the tissue sections were hybridized for 16 hours at 37 °C with 200 nM of 5’-end cy3-labeled RNA probe, containing CUG repeats and mounted as described above.
Results
Clinical and histopathological features of the fetus
The fetus was aborted in the 22nd week of pregnancy in a family with MD1 history (Tab. 1). Its body weight (360 g) corresponded to the 19th –20th week rather than the 22nd week of the normal development. Many skeletal muscle fibers were in fact myotubes with central nuclei, and only a minority of muscle fibers with peripheral sarcolemmal nuclei were identified (Fig. 1a). Muscle fibers were arranged in loose fascicles and interfascicular connective tissue was focally increased. The majority of muscle fibers/myotubes was small, measuring 3–10 µm only. However, the mean size of muscle fibers was 5.7 ±1.1 µm in the vastus lateralis and 7.2 ±1.3 µm in the gastrocnemius; these values correspond to the normal range with regard to the body size [23], as well as to the 22nd week of fetal development [24]. The fiber type analysis, performed according to the standard Ca-ATPase reaction at pH 9.4, did not reveal any distinct populations of muscle fibers, while in a normal developing muscle histochemical fiber types are recognizable after the 18th to 19th week of gestation [24]. Either a complete uniformity or only small fluctuations in the enzyme activity (Fig. 1b) could be seen in both muscles examined: reactivity to developmental myosin heavy chain (dMHC) was uniformly positive in all myofibers (Fig. 1c). Uniform myofibers without fiber type differentiation were also identified after staining with the antibody to the fast MHC (MHCf) and to mitochondrial dehydrogenases. Early differentiation of the muscle fibers was identified through reactivity to slow MHC (MHCs) (Fig. 1d). Histological examination of the esophagus, stomach, and intestine showed clear stratification of the wall into the mucous membrane (epithel, lamina propria, muscularis mucosae), submucosa, muscularis propria, and serous membrane.
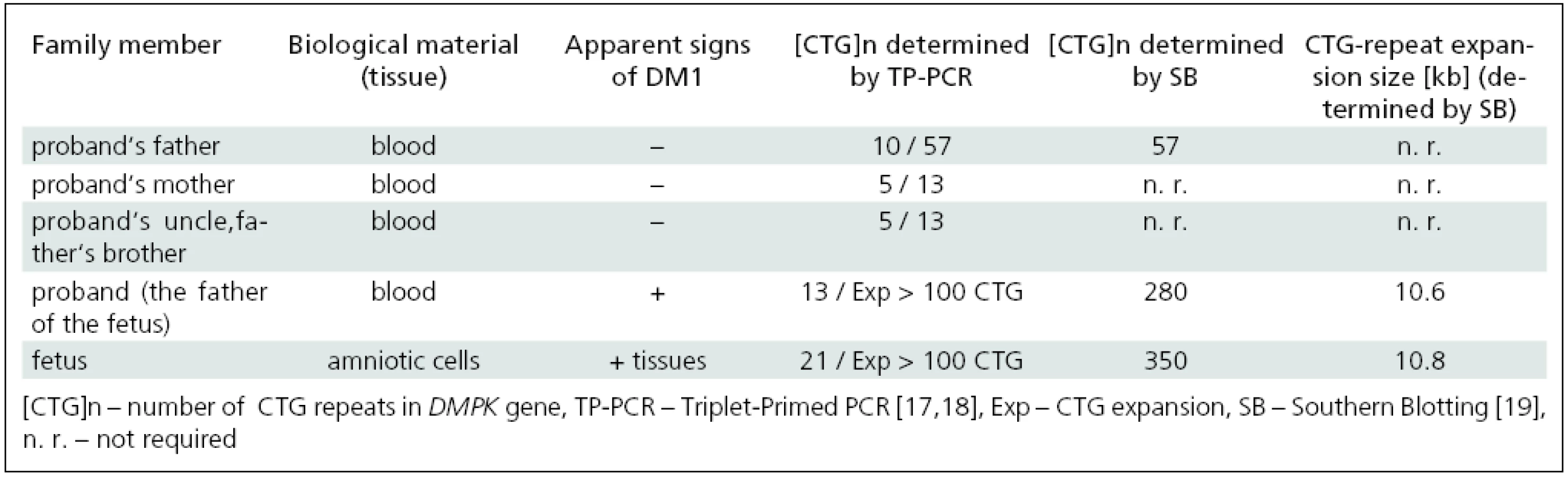

Mutation analysis of the DMPK gene in the affected family; confirmation of MD1 in the fetus
Mutation analysis of the DMPK gene performed by TP-PCR in accessible members of the affected family revealed CTG expansions in leukocytes of the father and grandfather as well as in amniotic cells of the fetus (Tab. 1). This confirmed the diagnosis of MD1 in symptomatic members of the family (the fetus and its father) and in the still asymptomatic grandfather. The mother had, in line with her non-DM status, no expansion of the DMPK gene. These results reveal a rare case of paternal transmission of the disease in a family.
Determination of the CTG-repeat size in the fetus and members of the affected family by Southern blotting
Determination of expanded allele sizes larger than about 100 CTG is not possible with TP-PCR [17–18,20]. Therefore, Southern blot analysis (SB) was used to further confirm MD1 diagnosis and to evaluate the size of large expansions (especially those associated with CDM). SB proved CTG expansions in the DMPK gene in the fetus and its father, 350 and 280 CTGs, respectively, and clearly demonstrated both clinical and genetic anticipation of the disease in the three generations studied (Tab. 1).
CUGexp ribonuclear foci in fetal tissue
Neither the DMPK gene mutation analysis nor the presence of CUGexp ribonuclear foci were studied in situ in fetal tissue. Therefore, we attempted to visualize these foci in individual cell types using the RNA-FISH method in combination with high-resolution fluorescence microscopy.
Using this method, CUGexp ribonuclear foci were identified in all muscle tissues studied: in numerous myonuclei of cross-striated muscles (vastus lateralis and gastrocnemius) (Fig. 2a), nuclei of smooth muscle cells of muscularis externa (propria) of the esophagus, stomach and intestines (Fig. 2b), and vascular smooth muscle (not shown). In addition, CUGexp foci were found in some nuclei of Schwann cells and ganglion cells in the intrinsic (Auerbach) ganglionic plexus (not shown); the combined FISH and immunofluorescence staining (IFL-FISH) of nerve fibers (NF-H neurofilaments) showed an intimate spatial proximity of the Schwann nuclei with the nerve fibers, thus confirming the identity of the Schwann cell (Fig. 2c). The foci were also present in epithelial cells of the esophagus (Fig. 2d).

These results provide evidence that transcription of the DMPK gene and nuclear retention of the expanded transcripts take place in all cell types studied, even in a 22-week fetus with congenital MD1.
Expression of MBNL1 and MBNL2 proteins in fetal tissues and their co-localization with CUGexp foci
The immunofluorescence (IFL) showed expression of MBNL1 protein in all the tissues examined. Reactivity with the MBNL1 antibody in the cross-striated muscle was mostly confined to the nuclei and appeared as foci or granules (Fig. 3a); sporadically, the signals were observed also as groups of granules in the sarcoplasm. A combined immunofluorescence and fluorescence in situ hybridization (IFL-FISH) clearly demonstrated co-localization of the MBNL1 protein with the CUGexp RNA in intranuclear foci in smooth muscle cells (Fig. 3b) as well as in skeletal muscle fibres (Fig. 3c). In addition, some nuclei contained MBNL1 deposits but no or single CUGexp foci (Fig. 3d). These findings indicate that nucleoplasmic MBNL1 is only partially sequestered into the CUGexp nuclear foci (Fig. 3d). MBNL1 expression was also found in fetal smooth muscle cells of the esophagus, intestines, Schwann cells, and in capillary endothelial cells.

The MBNL2 protein was detected in both nuclei and cytoplasm of the fetal tissue studied. IFL-FISH demonstrates co-localization with CUGexp in ribonuclear foci as well as complete separation of CUGexp from MBNL2 reactivity in skeletal muscle fibers (Fig. 4).

In summary, these results show that CUGexp RNA is retained as ribonuclear foci even at the fetal stage of the development. The MBNL proteins co-localize and are sequestered in the foci.
Discussion
In this paper we present the first in situ molecular genetic and histopathological analysis of human fetal tissue of a 22-week fetus from a family affected by myotonic dystrophy (MD1), where a paternal path of transmission was established. The uniform reactivity of the fetal muscle fibres to mitochondrial dehydrogenases differs from that described by Dubowitz [25] who found a small proportion of large dark fibers in a human (21–23 weeks of gestation) triceps. Retarded fiber type differentiation suggesting muscle immaturity in 30–37 week old fetuses with the congenital form of DM (CDM) was described by Farkas-Bargeton et al [26] and in infants with congenital and infantile myotonic dystrophy by Saghal et al [27], Iannaccone et al [28], and Karpati et al [29]. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation were also established in human myotonic dystrophy myoblasts [16].
First, we speculated that we were presented with congenital MD1 (CDM) resulting from a rare paternal transmission [30]. CTG expansion in the fetus and its father was 350 and 280 CTG, respectively. Even though the fetus has a larger expansion than its father, the size of CTG expansion was small in comparison to the CTG expansions usually associated with the CDM form (>1,000 CTG). With only slightly delayed differentiation of the skeletal muscles and without clinical manifestations that are usually associated with the severe congenital form (decreased fetal movements, polyhydroamnion and ultrasound findings before a termination of pregnancy), diagnosis of this form of MD1 was difficult. Therefore, this case could not be classified as typical CDM. We provide in situ molecular genetic evidence that the fetal tissue was affected by myotonic dystrophy but the actual phenotypic expression of the disease could not be predicted. On the other hand, by definition, a condition existing at birth or before birth is congenital – congenital disorder, or congenital disease.
Our results address several aspects of MD1 molecular pathogenesis: the current model of MD1 and MD2 pathogenesis involves sequestration of MBNL1 into CUGexp or CCUGexp ribonuclear foci, respectively, and loss of MBNL1 function in the nucleoplasm. The relationship between CUGexp foci and MBNL1 proteins was studied on cell lines and skeletal muscles of adult DM patients [21, and the citation therein]; however, at the fetal stage of the development, histopathological changes and nuclear retention of CUGexp transcripts were only described in vitro, in myoblast cell lines with about 2300 CTGs, derived from three fetuses suffering from CDM of various severity [16]; the authors demonstrated a defect in the proliferation and fusion of myogenic precursor cells in the skeletal muscle that resulted in delayed formation and growth of secondary myotubes.
The present demonstration of CUGexp ribonuclear foci in fetal cell types provides evidence that transcription of the expanded DMPK gene and nuclear retention of its transcripts also take place at the fetal stage of the MD1 development.
Different role of MBNL1 and MBNL2 in the pathogenesis of DM was illustrated by transgenic and MBNL1 knockout models of MD1 and mice that have reduced expression of MBNL2. Misregulated splicing of some target pre-mRNAs in a transgenic model that express CUGexp RNA was closely reproduced by deficiency of MBNL1 but not MBNL2 [31]. The results point to a pivotal role of MBNL1 in the pathogenesis of DM. The MBNL1 sequestration causes loss of the MBNL1 function leading to an increase in fetal forms of mRNAs. How would this play in the pathogenic mechanism in the fetal MD1 where both target mRNAs and MBNL1 proteins are predominantly in fetal forms?
Holt et al [21] found that MBNL2 levels decrease during muscle development in vivo and in vitro, while MBNL1 levels remain unchanged. MBNL2 declines during muscle fiber or myotube formation in culture but decline in MBNL2 with fetal development occurs during muscle maturation (between 20th and 31st week) after initial fiber formation and activation of muscle-specific genes had occurred [16]. Positive reactivity to MBNL2 protein in the fetal tissue gives evidence that the reduced MBNL2 expression observed in vivo during muscle maturation did not begin at the 22 weeks old fetus studied.
Sarcoplasmic deposits/traces of MBNL proteins may stand for remnants after relocalization from the cytoplasm to the nucleus. In the study by Miller et al [6], endogenous MBNL1 was mainly cytoplasmic in mouse myoblasts but it was distributed more evenly in the same type of human cells. In both, wild-type and MD1 transgenic mice, relocalization of the MBNL1 protein was observed between days 2 and 22 postnatally [31]. In cultured human MD1 myoblasts, relocalization occurred with both MBNL1 and MBNL2, and these proteins were always co-distributed [21]. Alternative splicing of MBNL exon 5 undergoes an early developmental transition in the developing heart and inclusion or skipping of the exon regulates subcellular localization of the proteins. Growing evidence suggests that MBNL proteins have important cytoplasmic functions, too. MBNL2 has been shown to regulate localization of α3 integrin mRNA to sites of focal adhesion for localized protein production; and, under stress conditions, MBNL2 has been shown to relocalize stress granules in the cytoplasm (reviewed by Terenzi and Ladd 2010 [32]).
On the other hand, the results presented here suggest that migration and redistribution of MBNL1 and MBNL2 proteins from the sarcoplasm to the nuclei occurs during fetal development. A comparison of the presented findings with non-affected age-matched fetuses would be necessary to assess migration and distribution of MBNL1 and 2 in fetal tissues.
The presence of DMPK transcript with expanded CUG repeat in the studied (non-muscle) fetal tissue may theoretically result in distinctive histopathological changes in MD1 patients, and thus contribute to the generation of the MD1 phenotype. For example, it would be useful to answer the question whether the presence of CUGexp nuclear foci and sequestration of the MBNL1 protein in fetal smooth muscle cells of the intestines, Schwann cells and neurons in intramural nerve plexuses is related to impaired motility of the gastrointestinal tract in MD1 patients [33–35]. Interestingly, Southern Blot analysis [36] showed that the size of CTGexp repeats varies in differentiated tissues (somatic mosaicism) and raised another question as to whether there was any relationship between the degree of expansion in various tissues and multisystemic involvement in MD1. In support of this, muscleblind proteins were shown to bind to CUGexp transcripts in a manner proportional to the size of the expanded repeat [6].
However, it is not known whether the fetal expression pattern of CUGexp RNA and MBNL proteins is preserved up to the mature state of the tissues. Therefore, more concrete conclusions on the pathological consequences and biological outcome of the findings presented here cannot be drawn. Nevertheless, it is evident that sequestration of the MBNL1 protein begins during fetal development, both in the muscles and non-muscle tissues studied.
RNDr. Martin Falk, Ph.D.
Institute of Biophysics
Academy of Sciences of the Czech Republic
Kralovopolska St. 135
612 65 Brno
e-mail: falk@ibp.cz
Accepted for review: 23. 2. 2012
Accepted for print: 10. 5. 2012
Acknowledgment
This research was supported by the grants: NS/9877-4 (IGA MH CR), IAA500040802 (GA of ASCR), OPVK CZ.1.07/2.3.00/30.0030, P302/12/G157 and P302/10/1022 (GACR), COST LD12039 (MEYS CR), EU COST MP1002 Nano-IBCT, and from the CR contribution to JINR Dubna (the Grant of the Government Plenipotentiary and the Grant 3+3). Monoclonal antibody MB1a was supplied by Professor Glenn Morris, RJAH Orthopaedic Hospital, Oswestry, UK.
Sources
1. Hamshere MG, Harley H, Harper P, Brook JD, Brookfield JF. Myotonic dystrophy: the correlation of (CTG) repeat length in leukocytes with age at onset is significant only for patients with small expansions. J Med Genet 1999; 36(1): 59–61.
2. Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3´ end of a transcript encoding a protein kinase family member. Cell 1992; 68(4): 799–808.
3. Fu YH, Pizzuti A, Fenwick RG jr, King J, Rajnarayan S, Dunne PW et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992; 255(5049): 1256–1258.
4. Davis BM, McCurrach ME, Taneja K, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3´ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci 1997; 94(14): 7388–7393.
5. Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 1995; 128(6): 995–1002.
6. Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 2000; 19(17): 4439–4448.
7. Fardaei M, Larkin K, Brook JD, Hamshere MG. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res 2001; 29(13): 2766–2771.
8. Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet 2002; 11(7): 805–814.
9. Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet 2006; 15(2): R162–R169.
10. Timchenko LT. Myotonic dystrophy: the role of RNA CUG triplet repeats. Am J Hum Genet 1999; 64(2): 360–364.
11. Timchenko LT, Caskey CT. Triplet repeat disorders: discussion of molecular mechanisms. Cell Mol Life Sci 1999; 55(11): 1432–1447.
12. Day JW, Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord 2005; 15(1): 5–16.
13. Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation 2006; 74(2–3): 65–80.
14. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind protein and deregulated alternative splicing in neurons. Hum Mol Genet 2004; 13(24): 3079–3088.
15. Mankodi A, Lin X, Blaxall BC, Swanson MS, Thornton CA. Nuclear RNA foci in the heart in myotonic dystrophy. Circ Res 2005; 97(11): 1152–1155.
16. Furling D, Coiffier L, Mouly V, Barbet JP, St Guily JL, Taneja K et al. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet 2001; 10(19): 2079–2087.
17. Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR et al. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet 1996; 33(12): 1022–1026.
18. Falk M, Vojtíšková M, Lukáš Z, Kroupová I, Froster U. Simple procedure for automatic detection of unstable alleles in the myotonic dystrophy and Huntington’s disease loci. Genet Test 2006; 10(2): 83–95.
19. Guida M, Marger RS, Papp AC, Snyder PJ, Sedra MS, Kissel JT et al. A molecular protocol for diagnosing myotonic dystrophy. Clin Chem 1995; 41(1): 69–72.
20. Dubowitz V, Sewry CA. Muscle biopsy. A practical approach. 3rd ed. London: Elsevier Saunders 2007.
21. Holt I, Jacquemin V, Fardaei M, Sewry CA, Butler-Browne GS, Furling D et al. Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle. Am J Pathol 2009; 174(1): 216–227.
22. Holt I, Mittal S, Furling D, Butler-Browne GS, Brook JD, Morris GE. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells 2007; 12(9): 1035–1048.
23. Stickland NC. Muscle development in the human fetus as exemplified by m. sartorius: a quantitative study. J Anat 1981; 132 (Pt 4): 557–579.
24. Carpenter S, Karpati G. Pathology of skeletal muscle. 2nd ed. Oxford: Oxford University Press 2001.
25. Dubowitz V. Enzyme histochemistry of skeletal muscle. J Neurol Neurosurg Psychiatry 1965; 28(6): 519–524.
26. Farkas-Bargeton E, Barbet JP, Dancea S, Wehrle R, Checouri A, Dulac O. Immaturity of muscle fibers in the congenital form of myotonic dystrophy: its consequences and its origin. J Neurol Sci 1988; 83(2–3): 145–159.
27. Sahgal V, Bernes S, Sahgal S, Lischwey, Subramani V. Skeletal muscle in preterm infants with congenital myotonic dystrophy. J Neurol Sci 1983; 59(1): 47–55.
28. Iannaccone ST, Bove KE, Vogler C, Azzarelli B, Muller J. Muscle maturation delay in infantile myotonic dystrophy. Arch Pathol Lab Med 1986; 110(5): 405–411.
29. Karpati G, Carpenter S, Watters GV, Eisen AA, Andermann F. Infantile myotonic dystrophy. Histochemical and electron microscopic features in skeletal muscle. Neurology 1973; 23(10): 1066–1077.
30. Anand KM, Biradar VM, Panicker JN, Nampoothiri S. Congenital myotonic dystrophy with asymptomatic mother. Indian Pediatr 2011; 48(7): 565–567.
31. Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 2006; 15(13): 2087–2097.
32. Terenzi F, Ladd AN. Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol 2010; 7(1): 43–55.
33. Bellini M, Biagi S, Stasi C, Costa F, Mumolo MG, Ricchiuti A et al. Gastrointestinal manifestations in myotonic muscular dystrophy. World J Gastroenterol 2006; 12(12): 1821–1828.
34. Casasa JM, Isnard RM, Ojanguren I, Castellví A,Blanco JA, López P. Intestinal myopathy in Steinert’s disease. Cir Pediatr 2003; 16(3): 149–151.
35. Lecointe-Besancon I, Leroy F, Devroede G, Chevrollier M, Lebeurier F, Congard P et al. A comparative study of esophageal and anorectal motility in myotonic dystrophy. Dig Dis Sci 1999; 44(6): 1090–1099.
36. Kinoshita M, Takahashi R, Hasegawa T, Komori T, Nagasawa R, Hirose K et al. (CTG)n expansions in various tissues from a myotonic dystrophy patient. Muscle Nerve 1996; 19(2): 240–242.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2012 Issue 6
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- A Global Epidemic of Multiple Sclerosis?
- Cortical Pathology in Multiple Sclerosis – Morphology, Immunopathology and Clinical Context
- Structure of Care in Neurorehabilitation
- Endovascular Treatment of an Ischemic Cerebrovascular Event