
Souběžný výskyt mutace v genu ZNF9 (myotonická dystrofie typu 2) a v genu CLCN1 (myotonia congenita) v jedné rodině – kazuistika
Co-occurrence of the Gene ZNF9 Mutations (Myotonic Dystrophy Type 2) and Gene CLCN1 (Myotonia Congenita) in One Family – a Case Report
Myotonia is delayed skeletal muscle relaxation after voluntary contraction. It is associated with impaired chloride or, less frequently, sodium channels in the muscle membrane. The chloride channel defect may be caused by a primary mutation in the skeletal muscle chloride channel gene (CLCN1) or it is secondary as in myotonic dystrophy caused by post-transcription effect of accumulated ribonucleic acid, containing expanded triplets or tetraplets of the mutated gene, on the synthesis of chloride channels. Impaired muscle relaxation that occurs in childhood dominates in patients with congenital myotonia. Clinically, the myotonia is evident and is mitigated by moderate exercise (warmup phenomenon). On the contrary, muscle weakness and systemic symptoms are prominent in myotonic dystrophy, myotonia is is less apparent. Unlike the size of the expansion (myotonic dystrophy type 1), there is no evidence that homozygous state in myotonic dystrophy would be associated with poorer clinical course or an earlier onset of the disease. We present a rare case of a patient with decontraction disorder from 33 years of age. The objective neurological finding included mild pelvic girdle muscle weakness, significant action myotonia, percussion myotonia and positive warm-up phenomenon. Molecular genetic testing confirmed myotonic dystrophy type 2 with the expansion on both alleles of the ZNF9 gene, homozygous state was supported by examination of ancestors and their relatives. Concurrently, the patient is a carrier of a mutation in the semidominate CLCN1 gene that she acquired from her mother, a carrier of heterozygous mutations in the ZNF9 gene. Homozygous phenotype of the expansive autosomal dominant gene mutations (myotonic dystrophy) generally does not lead to more severe phenotype, this condition is probably due to a concurrent mutation in chloride channels.
Key words:
myotonia – myotonic dystrophy – congenital myotonia – molecular genetics
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
O. Parmová 1,2; S. Voháňka 1,2; L. Fajkusová 2,3; K. Stehlíková 3
Authors‘ workplace:
Neurologická klinika LF MU a FN Brno
1; CEITEC – Středoevropský technologický institut, MU, Brno
2; Centrum molekulární biologie a genové terapie IHOK, FN Brno
3
Published in:
Cesk Slov Neurol N 2013; 76/109(5): 648-651
Category:
Case Report
Overview
Myotonie je zpomalená relaxace kosterního svalstva po volní kontrakci. Je spojena s poruchou chloridových a méně často i natriových kanálů ve svalové membráně. Porucha chloridových kanálů je způsobena primárně mutací v genu pro kosterní svalový chloridový kanál (CLCN1) nebo je sekundární jako v případě myotonické dystrofie, kdy je způsobena posttranskripčním ovlivněním syntézy chloridových kanálů akumulovanou ribonukleovou kyselinou obsahující expandované triplety či tetraplety mutovaného genu. U nemocných s kongenitální myotonií dominuje porušená svalová relaxace, která je provází od dětství. Myotonie je klinicky velmi zřetelná, mírní se cvičením (warm ‑ up fenomén). Oproti tomu u myotonických dystrofií je dominantní svalová slabost a systémové příznaky, myotonie většinou není v popředí. Na rozdíl od velikosti expanze (myotonická dystrofie typ 1) neexistují důkazy, že homozygotní stav u myotonických dystrofií vede k horšímu klinickému průběhu či časnějšímu začátku onemocnění. Uvádíme ojedinělou kazuistiku pacientky, která má od 33 let potíže s dekontrakcí. V objektivním neurologickém nálezu je lehká slabost svalstva pletence pánevního, ale výrazná intenční myotonie, přítomnost myotonie perkusní a pozitivní warm ‑ up fenomén. Molekulárněgenetickým vyšetřením byla potvrzena myotonická dystrofie typu 2 s expanzí na obou alelách genu ZNF9, zároveň byl homozygotní stav podpořen vyšetřením předků a jejich příbuzných. Současně je pacientka nositelkou semidominantní mutace v genu CLCN1, kterou získala od své matky, nositelky heterozygotní mutace v genu ZNF9. Homozygotní stav u expanzivních autozomálně dominantních genových mutací (myotonická dystrofie) obecně nevede k těžšímu fenotypu, ten je u pacientky způsoben patrně současnou mutací v chloridovém kanálu.
Klíčová slova:
myotonie – myotonická dystrofie – kongenitální myotonie – molekulární genetika
Použité zkratky
CLCN1 gen pro kosterní svalový chloridový iontový kanál 1 (ClC1)
DMPK proteinkináza pro myotonickou dystrofii
MCB kongenitální myotonie, Beckerův typ
MCT kongenitální myotonie, Thomsenův typ
MD myotonická dystrofie
PCR polymerázová řetězová reakce
RNA ribonukleová kyselina
SB Southern Blot
ZNF9 protein s motivem zinkových prstů 9
Úvod
Myotonie je zvýšená excitabilita svalových vláken charakterizovaná zpomalenou relaxací kosterního svalstva po volní kontrakci. Je spojena s poruchou chloridových a méně často i natriových napětím řízených kanálů ve svalové membráně. Porucha chloridových kanálů je primárně způsobena mutací v genu pro chloridový kanál (CLCN1) nebo je sekundární jako v případě myotonické dystrofie, kdy je způsobena posttranskripčním ovlivněním syntézy chloridových kanálů akumulovanou RNA obsahující abnormální triplety či tetraplety genu DPMK, resp. ZNF9. Myotonia congenita je způsobena mutací v genu pro chloridový kanál (CLCN1), který je lokalizován na dlouhém raménku 7. chromozomu (7q35). Podle typu dědičnosti se tradičně rozděluje na Thomsenovu (dominantní) a Beckerovu (recesivní) variantu. CLCN1 mutace jsou rozesety po celém genu a nevykazují žádné zvláštní umístění pro mutace vedoucí k MCB nebo MCT [1]. Většina mutací vede k výskytu autozomálně recesivní formy, existují však mutace, které jsou spojené s oběma typy dědičnosti – tzv. semidominantní [2]. Molekulární mechanizmus tohoto komplexního chování zůstává nejasný, může jít o variabilní penetranci, neúplnou dominanci, rozdílnou alelickou expresi a nižší klinickou expresi u žen [1]. U nemocných s kongenitální myotonií dominuje porušená svalová relaxace, která je provází od dětství. Myotonie je klinicky velmi zřetelná, objevuje se po první kontrakci po odpočinku a mírní se opakováním pohybu (warm ‑ up fenomén). U pacientů s recesivní formou je tíže myotonie větší s občasným výskytem svalové slabosti, která se taktéž projevuje po období klidu. Muskulatura je u pacientů často hypertrofická.
Myotonická dystrofie má autozomálně dominantní typ dědičnosti. Molekulárněgenetickým vyšetřením se u myotonické dystrofie typu 1 (MD1) prokazuje CTG trinukleotidová expanze v genu DMPK na 19. chromozomu, u myotonické dystrofie typu 2 (MD2) se prokazuje CCTG tetranukleotidová expanze v genu ZNF9 na chromozomu 3. U myotonických dystrofií je prominentní svalová slabost a systémové příznaky (katarakta, poruchy srdečního rytmu a kardiomyopatie, endokrinní poruchy aj.). Myotonie většinou není v popředí klinického obrazu nemoci. U MD1 tíže fenotypu do určité míry koreluje s velikosti CTG expanze, u MD2 tato závislost chybí. Také neexistují důkazy, že homozygotní stav u myotonických dystrofií vede k horšímu klinickému průběhu či časnějšímu začátku [3].
Kazuistika
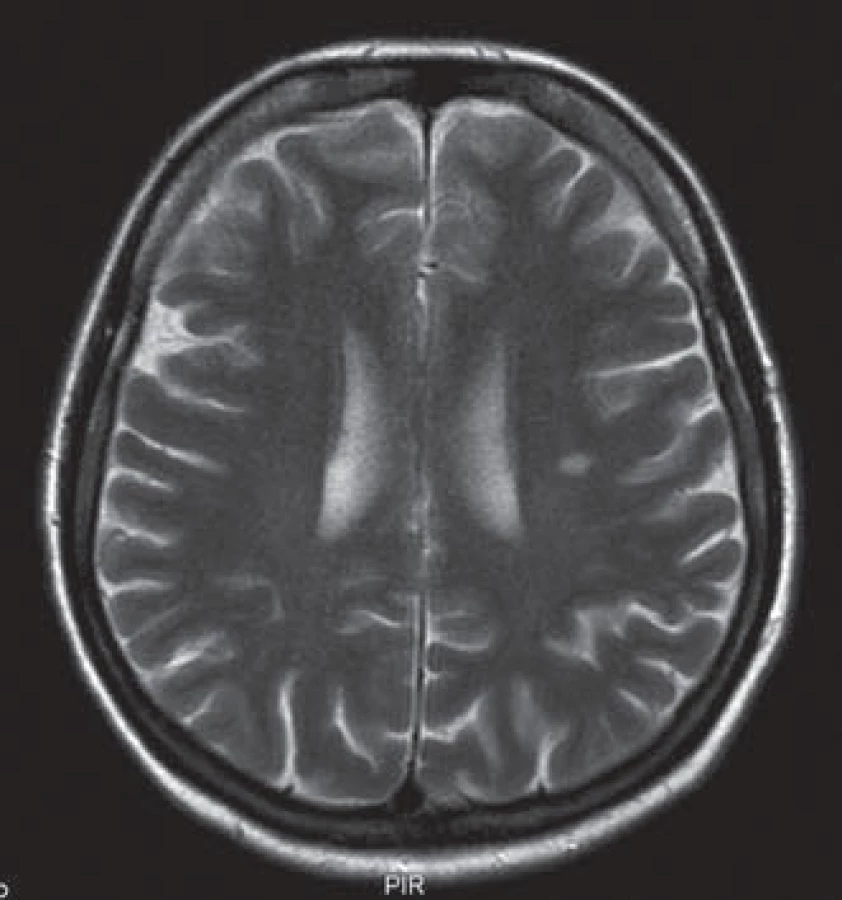
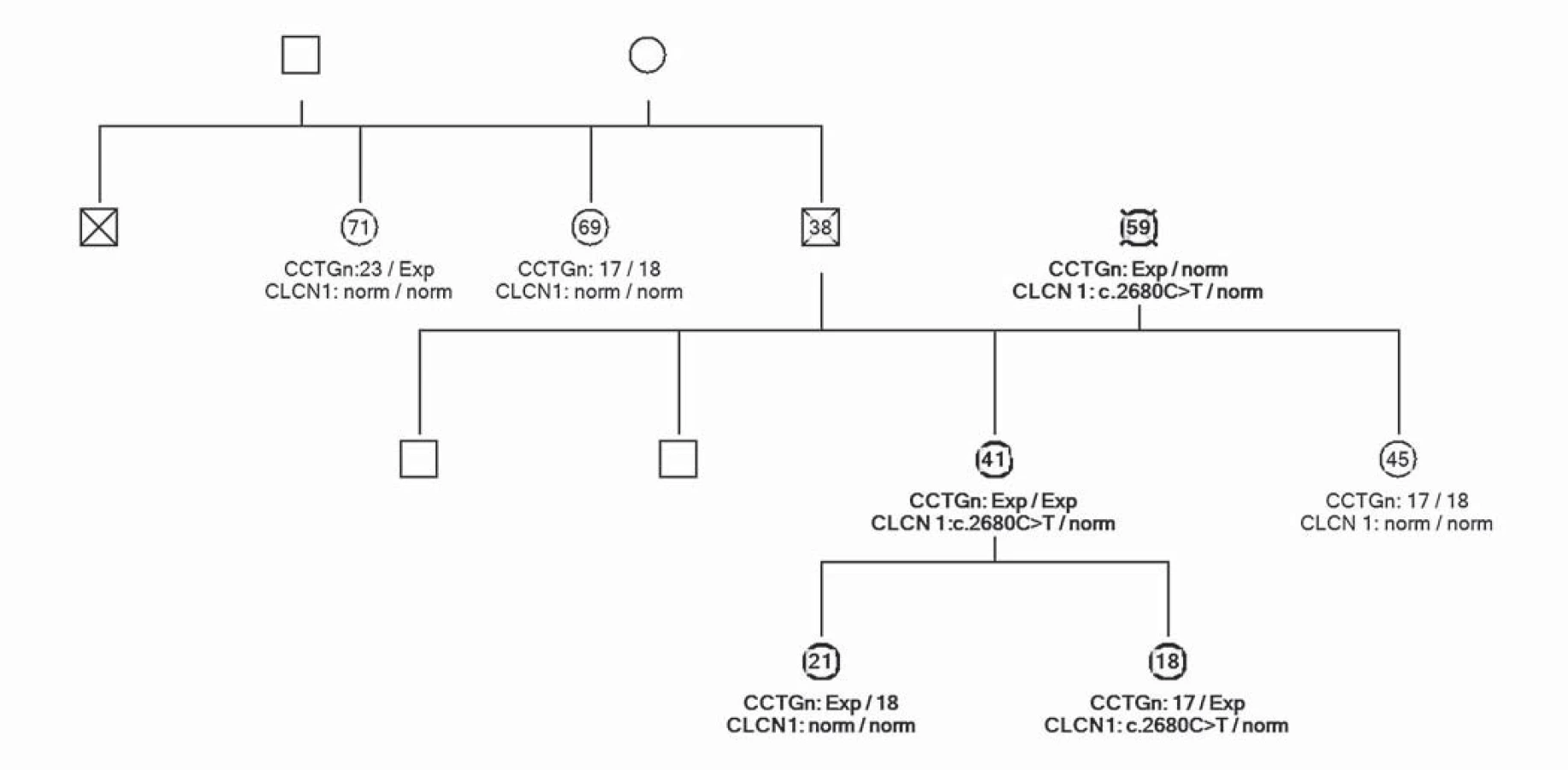
Uvádíme ojedinělou kazuistiku pacientky a její rodiny, u které byl zjištěn současný výskyt myotonické dystrofie a kongenitální myotonie. Pacientka ve věku 41 let má od 33 let potíže s dekontrakcí. V objektivním neurologickém nálezu je lehká slabost svalstva pletence pánevního, která se objevila před dvěma lety. Pacientka má však výraznou intenční myotonii a myotonii perkusní, je také pozitivní warm ‑ up fenomén. Molekulárněgenetickým vyšetřením Repeat ‑ Primed PCR (RP PCR) a Souther Blot (SB) byla potvrzena MD2 s expanzí na obou alelách postiženého genu. Vyšetřené hladiny kreatinkinázy (5,6 µkat/ l) a myoglobinu (110,7 µg/ l) jsou zvýšené. Na EMG vyšetření byly nalezeny myotonické výboje. Na MR mozku je přítomno hypersignální ložisko v bílé hmotě (obr. 1). Strukturální a signálové změny především v bílé hmotě mozku se typicky vyskytují u pacientů s myotonickou dystrofii, ale jsou samy o sobě nespecifické. Kardiologické vyšetření neprokázalo poruchy srdečního rytmu či kardiomyopatii. Taktéž oční vyšetření bylo bez průkazu katarakty. Z důvodu přítomnosti výrazné poruchy dekontrakce byla provedena i analýza genu CLCN1 (PCR ‑ sekvenace exonů a přilehlých intronových oblastí). Molekulárněgenetickým vyšetřením bylo potvrzeno, že pacientka je zároveň nositelkou semidominantní mutace c.2680C>T, p.(Arg894*) ve 23. exonu genu CLCN1. Tuto mutaci získala od své matky. Homozygotní stav mutace v genu ZNF9 byl podpořen vyšetřením rodinných příslušníků. Rodokmen rodiny zobrazuje přenos mutací těchto chorob v rodině (obr. 2). Matka pacientky je nositelkou heterozygotní mutace v genu ZNF9, má klinické projevy odpovídající MD2 (kořenovou slabost svalstva pletence pánevního od 38 let věku, intenční myotonii a kataraktu od 55 let), myotonické výboje v jehlové EMG a zvýšenou hladinu kreatinkinázy v séru. Vyšetření otce nebylo možné, jelikož zemřel v mladém věku a byl zpopelněn. Abychom potvrdili homozygotní expanzi genu ZNF9 u pacientky, provedli jsme vyšetření jeho sourozenců a nalezli mutaci v genu ZNF9 u jedné z jeho sester, která je ale ve věku 70 let kompletně asymptomatická. Taktéž jsme vyšetřili sourozence pacientky. U její sestry nebyla zjištěna mutace v genu ZNF9 ani v genu CLCN1. Její dva bratři doposud vyšetřeni nebyli. V genetickém vyšetřování jsme pokračovali i u dětí pacientky. Obě dcery jsou presymptomatickými nositelkami heterozygotní mutace v genu ZNF9, mladší dcera je však zároveň i nositelkou mutace pro kongenitální myotonii.
Diskuze
Myotonická dystrofie je považována za autozomálně dominantní chorobu bez rozdílu ve fenotypu mezi heterozygotním a homozygotním stavem. Homozygotní stav je velmi vzácný a byl doposud popsán jen u několika pacientů [4]. Na rozdíl od MD1, kde tíže fenotypu do určité míry koreluje s velikosti CTG expanze, tato či jiné závislosti u MD2 chybí. V literatuře byla popsána rodina s výskytem myotonické dystrofie typu 2, kde u třech sourozenců byla zjištěna homozygotní mutace [3]. I když jeden ze sourozenců byl vážněji klinicky postižen, klinický průběh symptomů všech tří homozygotů byl v rozmezí klinického průběhu heterozygotů. Ani velikost expanze a jiná vyšetření neprokázaly žádné rozdíly mezi heterozygotním a homozygotním stavem. Homozygotní stav se může dokonce klinicky projevit pouze výskytem katarakty jako jediného symptomu nemoci [5]. Homozygotní stav u expanzivních autozomálně dominantních genových mutací (MD1, MD2) nemění fenotyp nemoci ve srovnání s heterozygotním stavem. Lze tedy říci, že u pacientky výskyt mutace na obou alelách genu pro myotonickou dystrofii nevede k těžšímu fenotypu. Ten je způsoben patrně současnou mutací v chloridovém kanálu, pro co svědčí dominantní myotonické projevy.
Výskyt kongenitální myotonie je popisován různě od 1 : 10 000 až 1 : 100 000. Doposud je identifikováno okolo 200 mutací v genu CLCN1 a toto číslo se neustále zvyšuje, což dokládá vysoký stupeň alelické heterogenity spojené s touto chorobou [6]. U pacientky byla zjištěna mutace p.(Arg894*). Tato mutace patří k jedné z nejčastějších mutací v genu CLCN1 v české populaci [7] a může mít autozomálně dominantní nebo recesivní charakter. U pacientky výskyt semidominantní mutace v genu CLCN1 pravděpodobně přispěl, jak bylo uvedeno, k výraznějšímu klinickému postižení.
Současný výskyt mutace v genu ZNF9 a v genu CLCN1 je ojedinělý, ale možný případ [8]. Tento stav byl například popsán u jedné rodiny, kde koexistence obou mutací zvýšila závažnost myotonie u některých členů rodiny [9]. Zároveň to přispělo k výrazné variabilitě v myotonickém fenotypu sledované rodiny. V jiném případě nosiči mutace chloridového kanálu měli kromě větší myotonie i výraznější svalové bolesti [10]. V uvedené kazuistice je myotonie u pacientky taktéž výraznější než u ostatních členů rodiny. Také je zajímavý fenotyp sestry otce pacientky, kterou jsme vyšetřovali v rámci ověření homozygotního stavu pacientky při nemožnosti vyšetřit otce. Vzhledem k věku 72 let jde zatím o dlouhodobě kompletně asymptomatickou nosičku mutace v genu ZNF9 s normální nejen svalovou silou a nepřítomností katarakty a myotonie, ale i s normální hodnotou CK a normálním EMG nálezem.
Závěr
Uvedli jsme kazuistiku pacientky s klinicky výraznou intenční myotonií, u níž jsme molekulárněgenetickým vyšetřením zjistili koexistenci mutací v genu pro myotonickou dystrofii 2 a v genu pro kongenitální myotonii.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Olesja Parmová
Neurologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: olesja.parmova@fnbrno.cz
Přijato k recenzi: 24. 4. 2013
Přijato do tisku: 31. 5. 2013
Sources
1. Clinical and genetic investigations of patients with myotonia congenita in Northern Norway [on-line]. Available from URL: munin.uit.no/ bitstream/ handle/ 10037/ 3993/ thesis.pdf?sequence=2.
2. Dunø M, Colding ‑ Jørgensen E, Grunnet M, Jespersen T, Vissing J, Schwartz M. Difference in allelic expression of the CLCN1 gene and the possible influence on the myotonia congenita phenotype. Eur J Hum Genet 2004; 12(9): 738 – 743.
3. Schoser BG, Kress W, Walter MC, Halliger ‑ Keller B,Lochmüller H, Ricker K. Homozygosity for CCTG mutation in myotonic dystrophy type 2. Brain 2004; 127(8): 1868 – 1877.
4. Cerghet M, Tapos D, Serajee FJ, Mahbubul Huq AH. Homozygous myotonic dystrophy with craniosynostosis. J Child Neurol 2008; 23(8): 930 – 933.
5. Martorell L, Illa I, Rosell J, Benitez J, Sedano MJ, Baiget M. Homozygous myotonic dystrophy: clinical and molecular studies of three unrelated cases. J Med Genet 1996; 33(9): 783 – 785.
6. The human gene mutation database [on-line]. Available from URL: www.hgmd.cf.ac.uk/ ac/ gene.php?gene=CLCN1.
7. Sedlackova J, Vohanka S, Hermanova M, Vondracek P, Fajkusova L. Analysis of the CLCN1 gene in Czech patients with myotonia congenita. Neuromuscul Disord 2009; 19 : 645.
8. Neuromuscular disease center [on-line]. Available from URL: neuromuscular.wustl.edu/ mother/ activity.html.
9. Sun C, Van Ghelue M, Tranebjærg L, Thyssen F, Nilssen Ø, Torbergsen T. Myotonia congenita and myotonic dystrophy in the same family: coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin Genet 2011; 80(6): 574 – 580.
10. Ursu SF, Alekov A, Mao NH, Jurkat ‑ Rott K. ClC1chloride channel in myotonic dystrophy type 2 and ClC1 splicing in vitro. Acta Myol 2012; 31(2): 144 – 153.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 5
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Wilsonova nemoc
- Multiformní glioblastom – přehled nových poznatků o patogenezi, biomarkerech a perspektivách léčby
- Tumoriformní varianta roztroušené sklerózy – dvě kazuistiky
- Test 3F Dysartrický profil – normativní hodnoty řeči v češtině