
Gitelmanův syndrom provázený manifestní tetanií – kazuistika
Gitelman’s Syndrome Associated with Tetany – a Case Report
Tetany is a condition of increased neuromuscular excitability with variable clinical manifestations. Tetany is the most frequently associated with hypocalcaemia. Tetany is less frequently associated with Gitelman’s syndrome. It is a salt-losing renal tubulopathy that is characterized by hypokalaemia, hypomagnesaemia, metabolic alkalosis, hypocalciuria and hyperreninemic-hyperaldosteronism. This case study describes a 16-year-old girl with two undiagnosed episodes of normocalcaemic tetany. Following the third manifestation of tetany, a large battery of laboratory tests was performed and, based on these data, clinical diagnosis of Gitelman’s syndrome was made. Diagnosis of Gitelman’s syndrome was confirmed by gene-sequencing analysis.
Key words:
hypokalaemia – hypomagnesaemia – tetany – Gitelman’s syndrome
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
Z. Doležel 1; H. Ošlejšková 2; J. Papež 1; P. Hanáková 2
Authors‘ workplace:
LF MU a FN Brno
Pediatrická klinika
1; LF MU a FN Brno
Klinika dětské neurologie
2
Published in:
Cesk Slov Neurol N 2013; 76/109(5): 634-636
Category:
Case Report
Overview
Tetanie je stav zvýšené nervosvalové dráždivosti s variabilní klinickou manifestací a nejčastěji je asociována s hypokalcemií. K méně častým nozologickým jednotkám provázených tetanií patří Gitelmanův syndrom. Jedná se vrozené onemocnění, jehož podkladem je dysfunkce iontového transportního systému v distálním tubulu ledvin. Gitelmanův syndrom je charakterizován hypokalemií, hypomagnezemií, metabolickou alkalózou, hypokalcurií a hyperreninovým hyperaldosteronizmem. Je prezentován případ 16leté dívky, u které dvě epizody normokalcemické tetanie nebyly diagnosticky přesně určeny. Teprve při třetí manifestaci tetanie bylo provedeno širší spektrum laboratorních analýz, jejichž výsledky umožnily vyslovit podezření na Gitelmanův syndrom. Diagnóza onemocnění byla potvrzena molekulárněgenetickým vyšetřením.
Klíčová slova:
hypokalemie – hypomagnezemie – tetanie – Gitelmanův syndrom
Úvod
Buněčné membrány jsou pro většinu iontů/ molekul neprostupné, a mají tak rozhodující význam při udržování celulární integrity. Přitom se však mezi intracelulárním a extracelulárním kompartmentem odehrávají neustálé a fascinující procesy vzájemných výměn, které jsou nezbytné pro udržení homeostázy organizmu. Část z těchto výměn je realizována prostřednictvím iontových kanálů (IK), z nichž většina umožňuje pouze selektivní průnik iontů. Funkce IK je determinována geneticky. V případě mutací regulačních genů dochází k dysfunkci příslušného IK, což je provázeno mnohdy velmi rozmanitými fenotypickými projevy, které mohou přispívat i k opožděné diagnostice některých onemocnění.
Tetanie je klinicky charakterizována spazmy určitých svalových skupin (např. faciální, karpopedální, laryngální) s doprovázejícími paresteziemi. Nejčastější příčinou tetanie je snížení sérové koncentrace ionizované frakce vápníku, což vede k senzitizaci napěťově řízených sodíkových kanálů s následným ovlivněním frekvence akčních potenciálů. Vedle hypokalcemie je však nezbytné pamatovat také na některé jiné dyselektrolytemie, které mohou být provázeny tetanií.
Kazuistika
Pro akutní tonzilitidu provázenou horečkou byla u 16leté dívky zahájena praktickým dětským lékařem léčba perorálně podávaným antibiotikem penicilinového typu. Za 48 hod od iniciální dávky začalo děvče udávat měnlivou bolest v oblasti čela a pozitivní senzitivní symptomy parestezie (trnutí obličeje, jazyka a prstů obou rukou). Při klinickém vyšetření na dětském oddělení regionální nemocnice byly u dívky zaznamenány jen zarudlé a prosáklé patrové mandle a zadní stěna faryngu, dále submandibulární lymfadenopatie (uzliny velikosti 2 × 3 cm), ostatní nález byl normální. Z pomocných vyšetření byl u pacientky vyšetřen vzorek z kapilárního odběru krve (z iontů stanoveno: Na, K, Ca), kde byla jen mírně snížená hodnota draslíku. Dívka byla následně odeslána domů s doporučením další kontroly u svého dětského lékaře. Po třech týdnech se u děvčete z plného zdraví manifestovaly zcela identické subjektivní potíže. Při přijetí na dětské oddělení, kde proběhla také předchozí návštěva dívky, byla při fyzikálním vyšetření popisována pouze nepřesvědčivá pozitivita Chvostkova příznaku (Trousseauův příznak nebyl hodnocen), zbývající nález byl opět bez odchylek. Bez znalosti výsledků laboratorních vyšetření byla u dívky zahájena i.v. infuze s obsahem kalcia. Tato parenterální aplikace však byla ihned ukončena po obdržení biochemických hodnot sérové koncentrace celkového vápníku. Z ostatních iontů byly stanoveny ještě Na, Cl a K, opět byla přítomna mírná hypokalemie. Pacientka byla po krátké hospitalizaci propuštěna domů, doporučena jí byla přechodná perorální substituce draslíku. Třetí ataka obdobných subjektivních potíží dívky se projevila za čtyři týdny od epizody druhé a rodina vyhledala pomoc pracoviště autorů sdělení.
Rodinná anamnéza pacientky byla bez pozoruhodností. Dívka se narodila z první nekomplikované gravidity v termínu (porodní hmotnost 3 500 g, délka 49 cm), bez jakýchkoli perinatálních komplikací. Její duševní a pohybový vývoj byl bez odchylek. Děvče nebývalo častěji nemocné, nebylo pravidelně sledováno pro některé jiné onemocnění a nemělo žádnou trvalou farmakoterapii, vč. perorální antikoncepce (menzes od 13. roku života). Při přijetí na kliniku byly jedinými odchylkami fyzikálního vyšetření neúplně vyjádřený obraz „porodnické ruky“ na obou horních končetinách a intermitentní fascikulace jazyka. Při odborném neurologickém vyšetření byl prokázán obdobný nález, vč. pozitivního Trousseauova příznaku; nebyla shledána jiná ložisková symptomatika (vč. pyramidových jevů, bez poruchy chůze nebo změny svalové síly). Z pomocných vyšetření v den přijetí na kliniku byly u pacientky abnormální tyto nálezy: hypokalemie, hypomagnezemie, metabolická alkalóza; dále pak v jednorázovém vzorku moči změny koncentrace K, Ca a Mg. Následně bylo proto provedeno širší spektrum laboratorních analýz, jejichž výsledky umožnily vyslovit podezření, že dívka má renální tubulopatii označovanou Gitelmanův syndrom (GS). V tab. 1 je uvedena dynamika změn vybraných laboratorních ukazatelů pacientky.
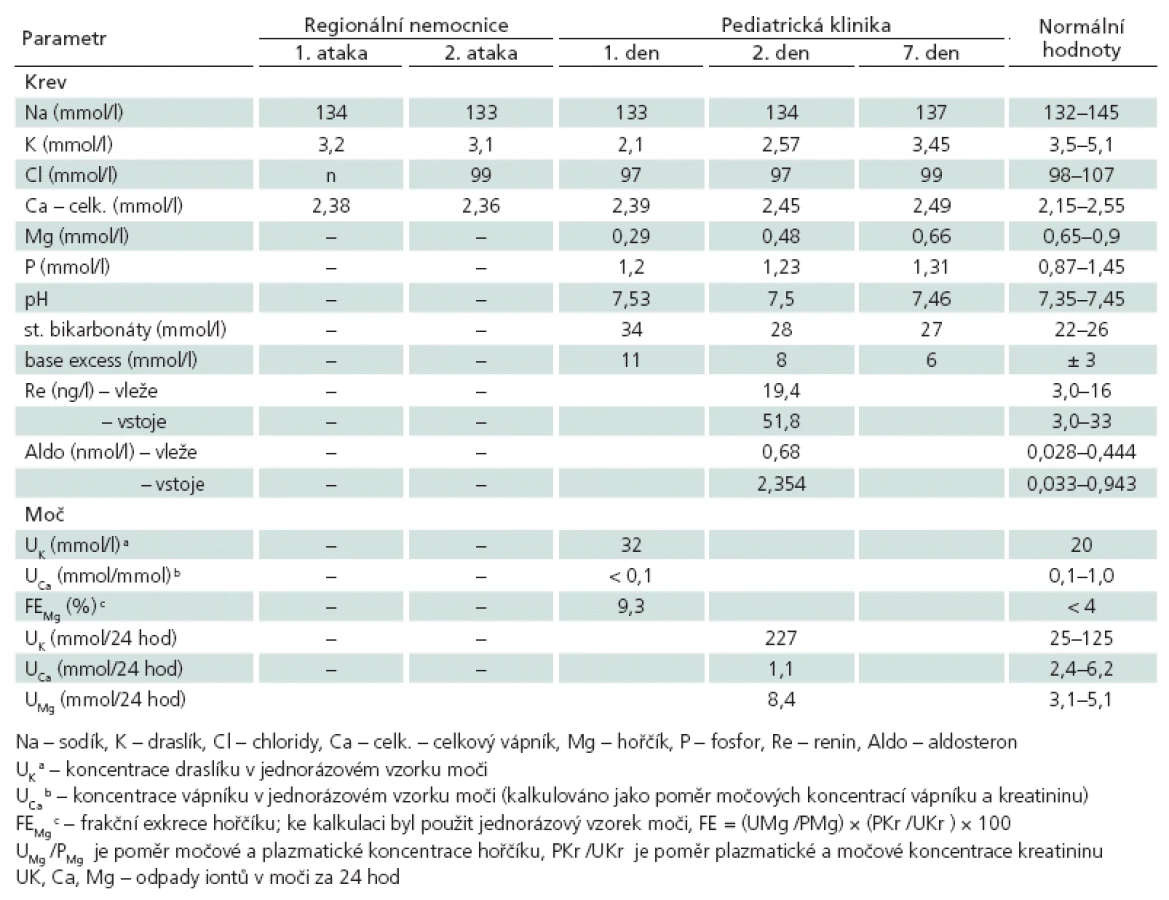
K preciznímu určení GS byl se souhlasem rodičů děvčete vzorek její DNA zaslán do zahraniční laboratoře (Dept. of Pediatrics, Philips University Marburg, Germany). Zde byla prokázána kombinovaná heterozygotní mutace (c.5 GT > AT a c.439 GT > AT) genu SLC12A3, a diagnóza GS tak byla přesvědčivě určena. Léčba GS byla u pacientky zahájena perorálním podáváním spironolaktonu a dále přípravků s vyšším obsahem hořčíku a chloridů. Při této terapii nedošlo u dívky v průběhu následujících 12 měsíců k žádné epizodě tetanie. Bylo však nezbytné postupně zvyšovat substituci hořčíkem a draslíkem, neboť jejich sérové koncentrace neustále oscilovaly při dolní hranici normálního rozmezí hodnot, a také zvýšit perorální příjem NaCl pro dosažení stabilizace sérových hodnot sodíku a chloridů.
Diskuze
GS (OMIM 263800), označovaný také jako familiální hypokalemická hypomagnezemie, je autozomálně recesivně dědičná renální tubulopatie. Bez ohledu na věk se přibližná prevalence GS uvádí 25 na 1 000 000 jedinců [1]. Podkladem GS je mutace genu SLC12A3, který kóduje thiazid ‑ senzitivní Na ‑ Cl kotransportní systém v distálním tubulu ledvin. Tento systém promiscue označovaný NCCT/ NCC se nachází v apikální membráně buněk distálního tubulu a výrazným způsobem se podílí na homeostáze Na, Cl a vody. V případě dysfunkce NCCT dochází k rozvoji GS, jehož charakteristickými klinickými příznaky jsou svalová slabost, snadná únava a projevy tetanie. U části pacientů však mohou být přítomny i další symptomy, jako jsou anorexie, cefalea, vertigo, neostré vidění nebo různé parézy [2]. Ojedinělá literární sdělení uvádějí případy dětí, u nichž dominující klinické projevy GS byly růstové opoždění nebo psychomotorická retardace [3,4].
Typickými laboratorními abnormitami GS jsou hypokalemie, hypomagnezemie, metabolická alkalóza, zvýšená sérová koncentrace reninu (Re) a aldosteronu (Ald) a hypokalciurie. Zvýšení Re a Aldo nebývá u nemocných s GS obvykle výrazné, každopádně toto zvýšení nedoprovází hypertenze. Je to důležitý symptom, který GS odlišuje od jiných geneticky podmíněných ledvinných tubulopatií, zejména pak Bartterova syndromu (BS) a Liddleova syndromu (LS). Podobně jako GS, také BS a LS z klinického hlediska charakterizuje svalová slabost a snadná únava, tetanie je však u BS a LS neobvyklá, u LS je navíc vždy přítomna hypertenze. Laboratorně je pro BS a LS typická výrazná hypokalemie a metabolická alkalóza, kalcemie je většinou normální. GS je entita, která je ve větším povědomí dětských nefrologů, jiným odbornostem věnujícím se dětskému věku nemusí být tato choroba všeobecně známa. Diagnóza GS proto může být nezřídka opožděná, zejména pak tehdy, pokud se nález i mírné a především opakované hypokalemie podcení a nejsou přesvědčivě vysvětleny její příčiny. Obdobné platí pro hypomagnezemii, zvláště pokud při manifestaci tetanie není vedle kalcemie stanovena také aktuální hodnota sérové koncentrace hořčíku [5,6]. Tak tomu bylo v případě i naší pacientky, která měla opakovaně prokazovánu hypokalemii, jejíž původ nebyl diagnosticky objasňován. Sérová koncentrace hořčíku a hodnoty acidobazické rovnováhy byly u dívky stanoveny teprve až při třetí epizodě jejích potíží. Pro precizní určení GS, ale i BS a LS je nezbytné molekulárněgenetické vyšetření, které je v současnosti dostupné také v ČR.
V léčbě GS se používají kalium šetřící diuretika, která blokují přítomný hyperaldosteronizmus. Dále je nezbytná vysoká dodávka NaCl a léků/ doplňků stravy s vysokým obsahem hořčíku a draslíku. Substituce elektrolytů je u některých pacientů s GS špatně tolerována, protože vysoký perorální přívod hořečnato ‑ draselných solí vyvolává značný dyskomfort GIT v podobě bolestí břicha, nadýmání a průjmů. Opakované parenterální aplikaci minerálů se tak někteří nemocní nevyhnou. Dyselektrolytemie u pacientů s GS je však nezbytné korigovat, neboť déletrvající hypokalemie může vést k poškození funkce ledvin nebo k závažným srdečním dysrytmiím; s chronickou hypomagnezemií je asociován rozvoj chondrokalcinózy [7 – 9]. Mimořádnou léčebnou pozornost zasluhují těhotné s GS [10]. I přes uvedenou léčebnou obtížnost je prognóza většiny jedinců s GS příznivá, nemocní mají mít zajištěno soustavné nefrologické sledování.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prof. MUDr. Zdeněk Doležel, CSc.
Pediatrická klinika
LF MU a FN Brno
Černopolní 9
625 00 Brno
e-mail: zdoleze@fnbrno.cz
Přijato k recenzi: 30. 1. 2013
Přijato do tisku: 20. 5. 2013
Sources
1. Vargas ‑ Poussou R, Dahan K, Venisse A, Riviera ‑ Munoz E, Debaix H, Grisart B et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol 2011; 22(4): 693 – 703.
2. Ueda K, Makita N, Kawarazaki H, Fujiwara T, Unuma S, Monkawa T et al. A novel compound heterozygous mutation of Gitelman’s syndrome in Japan, as diagnosed by an extraordinary response of the fractional excretion rate of chloride in the trichlormethiazide loading test. Intern Med 2012; 51(12): 1549 – 1553.
3. Skalova S, Neuman D, Lnenicka P, Stekrova J. Gitelman syndrome as a cause of psychomotor retardation in a toddler. Arab J Nephrol Transplant 2013; 6(1): 37 – 39.
4. Raza F, Sultan M, Qamar K, Jawad A, Jawa A. Gitelman syndrome manifesting in early childhood and leading to delayed puberty: a case report. J Med Case rep 2012; 6(1): 331.
5. Tammaro F, Bettinelli A, Cattarelli D, Cavazza A, Colombo C, Syrén ML et al. Early appearance of hypokalemia in Gitelman syndrome. Pediatr Nephrol 2010; 25(10): 2179 – 2182.
6. Fremont OT, Chan JC. Understanding Bartter syndrome and Gitelman syndrome. World J Pediatr 2012; 8(1): 25 – 30.
7. Cortesi C, Lava SA, Bettinelli A, Tammaro F, Giannini O, Caiata ‑ Zufferey M et al. Cardiac arrhythmias and rhabdomyolysis in Bartter ‑ Gitelman patients. Pediatr Nephrol 2010; 25(10): 2005 – 2008.
8. Rim PC, Keith MP. Chondrocalcinosis and hypomagnesemia in a 26‑year ‑ old woman. J Clin Rheumatol 2011; 17(6): 334 – 335.
9. Slovacek L. Gitelman’s syndrome: A hereditary disorder characterized by hypokalemia and hypomagnesaemia. Eur J Gen Med 2009; 6(2): 127 – 130.
10. Rušavý Z, Hudec A, Karbanová J, Korečko V, Janů R,Kališ V. Gitelman syndrome in pregnancy – a severe hypokalemia with favorable perinatal prognosis. Ceska Gynekol 2012; 77(5): 421 – 423.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 5
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Wilsonova nemoc
- Multiformní glioblastom – přehled nových poznatků o patogenezi, biomarkerech a perspektivách léčby
- Tumoriformní varianta roztroušené sklerózy – dvě kazuistiky
- Test 3F Dysartrický profil – normativní hodnoty řeči v češtině