
Flekční cervikální myelopatie (Hirayamova choroba) – skutečnost, nebo mýtus? Dvě kazuistiky
Flexion Cervical Myelopathy (Hirayama Disease) – Reality or Myth? Two Case Reports
Cervical flexion myelopathy (Hirayama disease) is a rare disease of the cervical spine. It is characterized by progressive muscular weakness and atrophy of the distal upper limb (brachioradialis muscle is spared), predominantly affecting male adolescents between 15 and 25 years of age. There is no sensory or deep tendon reflexes involvement. Cervical MRI images show local cord atrophy, T1-weighted images show widened lateral epidural space on flexion on that is hyperintense on T2-weighted, especially contrast-enhanced, images. The existence of the Hirayama disease is disputed by many authors. We present two patients with clinical symptoms of this disease and summarize facts about the diagnosis and treatment of Hirayama disease.
Key words:
Hirayama disease – cervical myelopathy – segmental spinal muscular atrophy – amyotrophy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
Z. Kadaňka Jr; B. Adamová
Authors‘ workplace:
Neurologická klinika LF MU a FN Brno
Published in:
Cesk Slov Neurol N 2014; 77/110(3): 362-367
Category:
Case Report
Overview
Flekční cervikální myelopatie (Hirayamova choroba) je vzácné onemocnění krční míchy. Manifestuje se asymetrickou, velmi pomalu progredující atrofií a slabostí svalů ruky a předloktí (s ušetřením m. brachioradialis) a postihuje většinou mladé muže mezi 15 a 25 lety. Nebývají přítomny změny reflexologické ani poruchy senzitivity. Na MR krční páteře je typicky popisována lokální atrofie míchy, ve flexi v T1 vážených obrazech dochází k rozšíření zadního epidurálního prostoru, který je hyperintenzní v T2 vážených obrazech a zvýrazňuje se po aplikaci kontrastní látky. Existence choroby samotné je mnohými autory zpochybňována. Autoři popisují kazuistiky dvou pacientů, které svým průběhem připomínaly tuto chorobu, a shrnují dosavadní poznatky o diagnostice a léčbě této sporné nozologické jednotky.
Klíčová slova:
Hirayamova choroba – cervikální myelopatie – segmentální spinální muskulární atrofie – amyotrofie
Úvod
Flekční cervikální myelopatie (Hirayamova choroba) je vzácné onemocnění krční míchy (synonyma: monomelická amyotrofie – MMA, juvenilní asymetrická segmentální spinální muskulární atrofie – JASSMA, juvenilní distální amyotrofie horní končetiny – JMADUE). Choroba byla poprvé popsána v roce 1959 [1]. Jde o onemocnění, jehož existence bývá zpochybňována [2]. Vyskytuje se zejména v asijských zemích, ale byly popsány případy i v Evropě a Severní Americe [3 – 6]. Postihuje nejčastěji mladé muže mezi 15 a 25 lety [7].
Onemocnění má tyto základní charakteristiky:
- svalové atrofie jsou omezeny na svaly předloktí a ruky (mimo m. brachio ‑ radialis),
- výskyt je obvykle sporadický, vzácně familiární,
- vyskytuje se zejména u mladých mužů (15 – 25 let) – většinou jednostranně,
- není porucha čití nebo změna reflexů na končetinách,
- pomalá progrese (roky),
- přítomna ztráta uchycení mezi zadní částí durálního vaku a laminami (v oblasti dolní krční páteře) a charakteristický obraz na MR (zvětšení zadního epidurálního prostoru ve flexi krční páteře).
Nejčastěji uváděná patogenetická hypotéza je mechanické a ischemické poškození cervikálních spinálních motoneuronů [8,9], ale poukazuje se rovněž na neurodegenerativní či autoimunitní příčinu [3]. Histopatologické nálezy ukazují na změnu pružnosti tkáně durálního vaku jako základní příčinu vedoucí k opakovaným míšním kompresím a myelopatii [10]. Při pitvě bývá zjišťována atrofie, nekróza a glióza v předních rozích míšních C5 – Th1 (obzvláště v C7 – 8), ale bez poškození intra ‑ i extramedulárních cév [11]. Léčba Hirayamovy choroby je pouze empirická. Na základě postulované hypotézy o patogenezi choroby a jejím průběhu a vzhledem k tomu, že v prvních letech onemocnění progreduje, se doporučuje intermitentní nošení měkkého límce po dobu 3 – 4 roků [12]. V některých případech se zvažuje i operační léčba, která je však stále považována za kontroverzní. Uvádíme kazuistiky dvou mladých pacientů, jejichž příznaky byly velmi podobné tomuto onemocnění.
Kazuistika 1
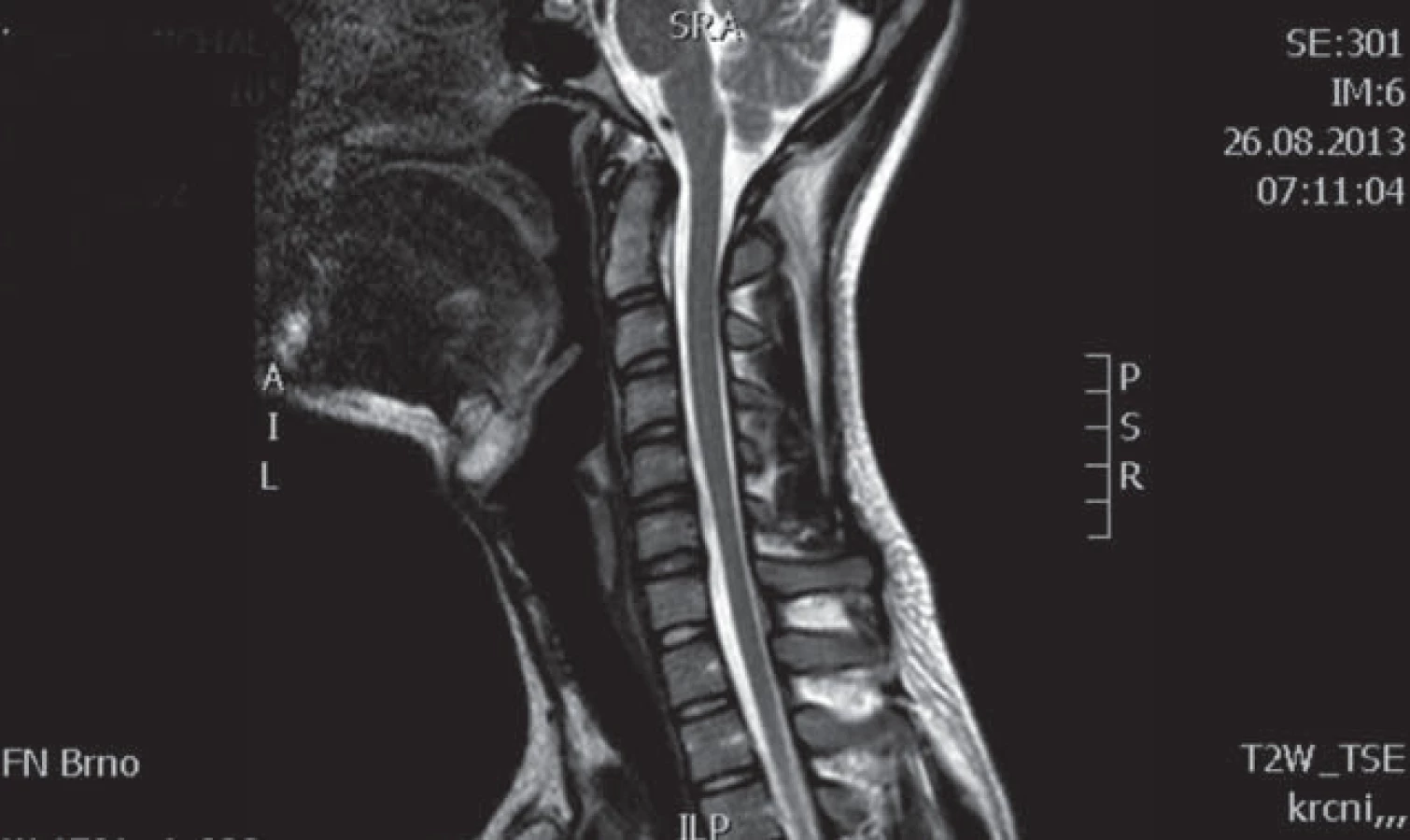
Muž, 37 let, od roku 1996 (od 20 let věku) pozoruje hubnutí svalstva a neobratnost levé horní končetiny s maximem akrálně. Rozvoj potíží byl pozvolný a progrese v dalších letech velmi pomalá. Pracuje jako technik u počítače, léky trvale žádné neužívá. Pacient je bývalý kuřák (přestal kouřit v roce 2008). V anamnéze neuvádí úrazy páteře, nemá další subjektivní problémy jako bolesti hlavy, bolesti páteře, parestezie končetin a jiné poruchy senzitivity. EMG provedené již v roce 1997 vykazovalo známky subakutní až chronické motorické axonopatie v myotomech C7 – Th1 vlevo s maximem v distribuci kořene C8. MR vyšetření krční páteře bylo provedeno v roce 1999 s nálezem diskrétní spondylózy C5 – 6, drobné paramediální pravostranné protruze C6 – 7a náznakem myelopatického ložiska (tehdy však ložisko nebylo radiologem zaznamenáno). Od roku 1999 již pacient nikde nebyl neurologicky vyšetřován ani sledován, až v roce 2009 se znovu rozhodl navštívit neurologa. Na kontrolním EMG vyšetření z HKK byl nález subakutní až chronické motorické axonopatie ve svalech myotomů C7 – 8 vlevo, bez průkazu bloku vedení motorických vláken. EMG z DKK bylo jen lehce abnormní, resp. byla přítomna lehká prolongace latencí F ‑ vln n. tibialis a n. peroneus, subjektivně pacient nemá žádné potíže z dolních končetin. Klinicky dominuje atrofie a paréza svalů inervovaných z myotomu C7 – 8 vpravo, bez poruchy senzitivity, jinak je ložiskový neurologický nález v normě. Bylo provedeno genetické vyšetření, které nesvědčí pro HNPP (tomakulozní neuropatie), Charcot ‑ Marie ‑ Tooth typ Ia, hereditární motorické neuropatie (mutace genu kódujícího protein HSP 22 a HSP 27). Celá baterie testů včetně likvorologického vyšetření nepodporovala diagnózu zánětlivého onemocnění nervového systému. Antigangliozidové protilátky anti‑GM 1, 2, 3, GD1A, GD1B, GQ1B byly v séru negativní. MR brachiální plexu je bez patologického nálezu. Na základě průběhu a klinického obrazu onemocnění vzniklo podezření na flekční cervikální myelopatii. Provedené kontrolní MR krční páteře prokázalo lokální míšní atrofii ve výši meziobratlového prostoru C5/ 6 a lehčí rozšíření zadního epidurálního prostoru při flexi (obr. 1 – 3). Diferenciálně diagnosticky by se však mohlo jednat o HMN5 (distální SMA s postižením horních končetin, lokus 7p15). Stav pacienta zůstává prakticky stacionární, objektivně je pozorována jen zcela mírná progrese atrofií.
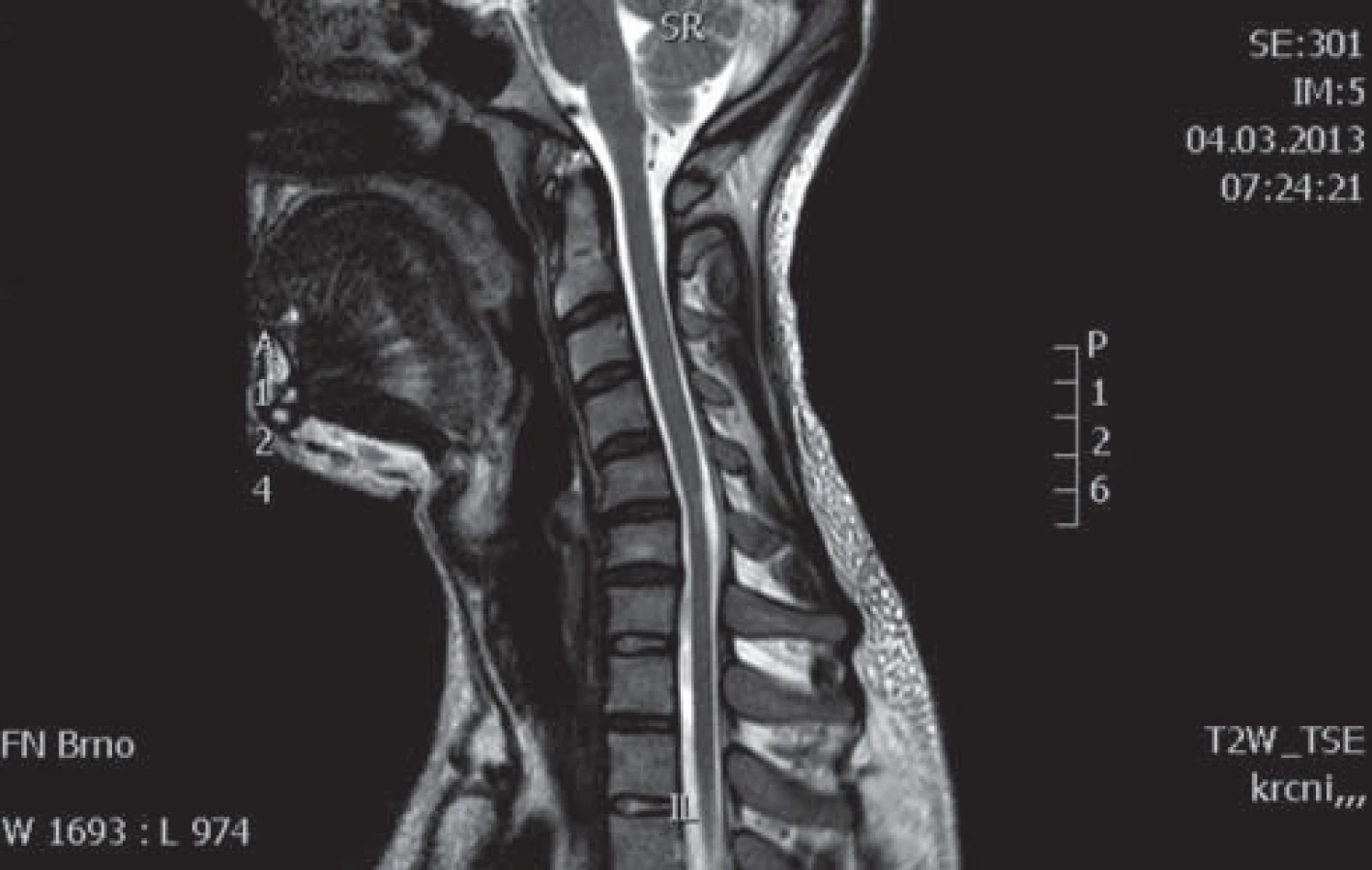
Kazuistika 2
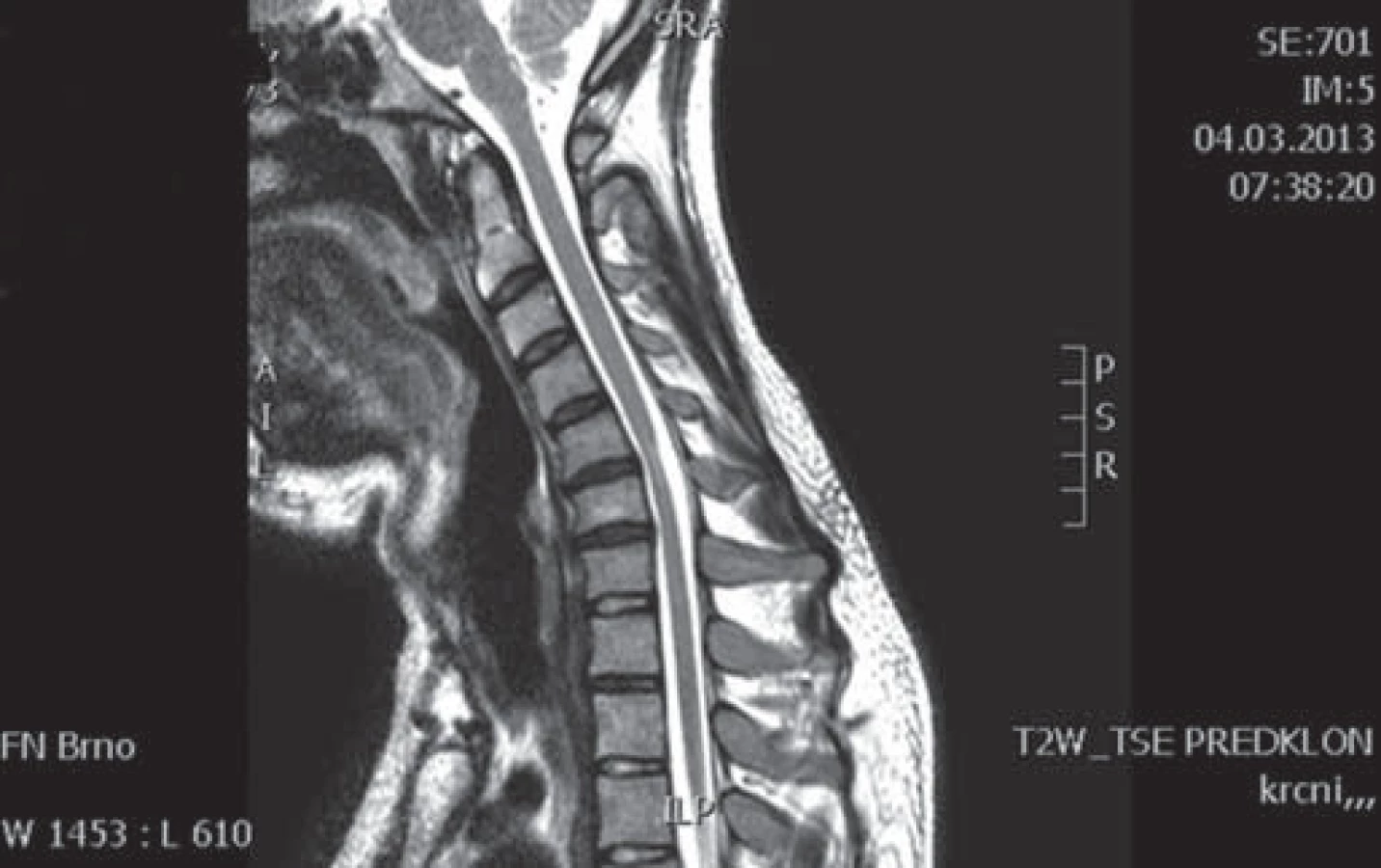
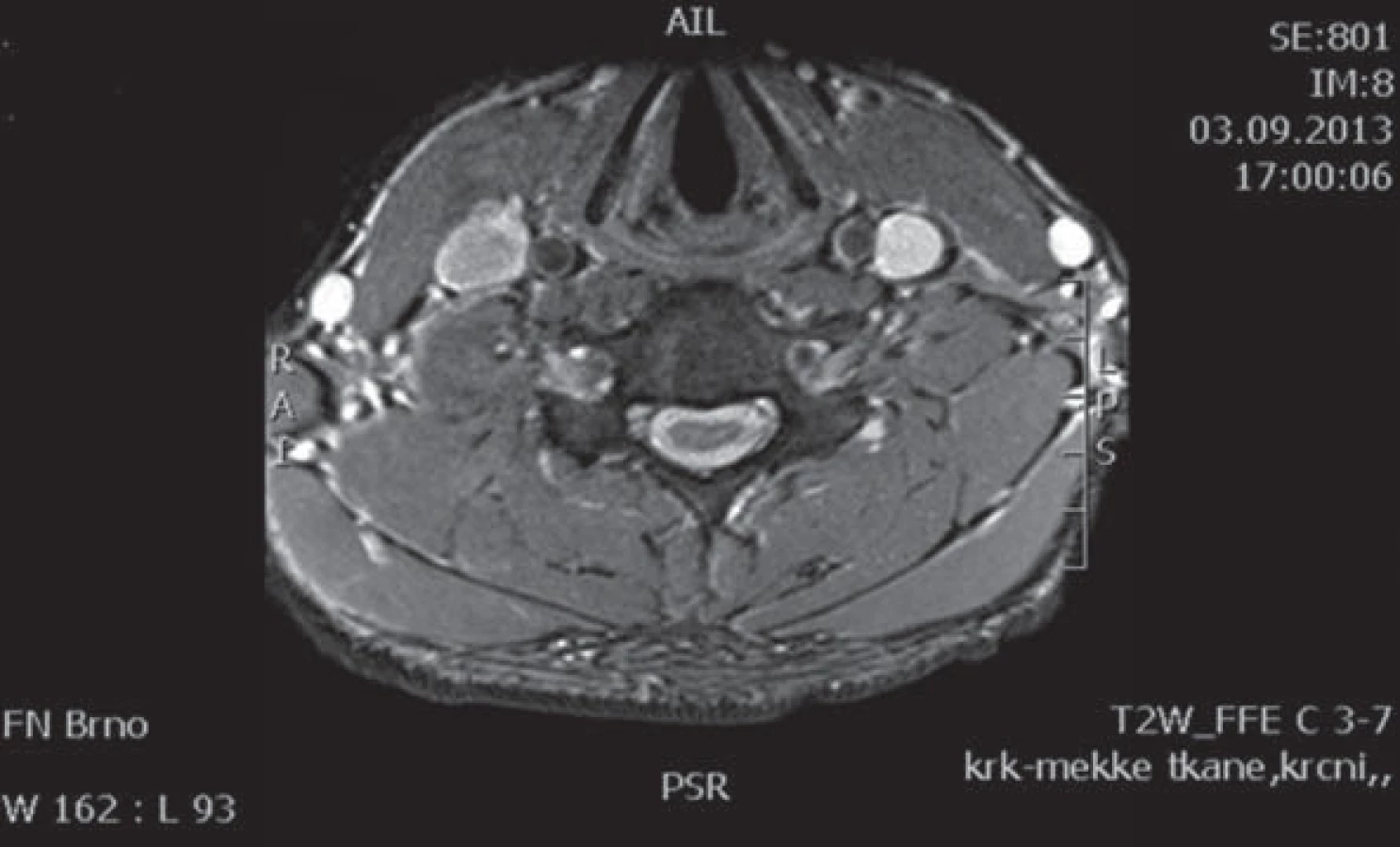
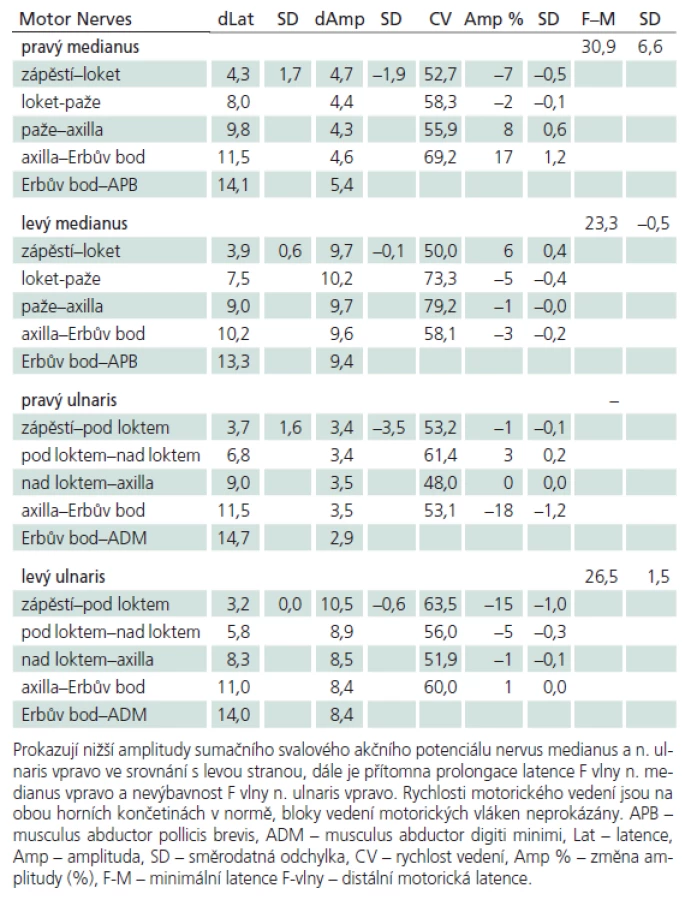
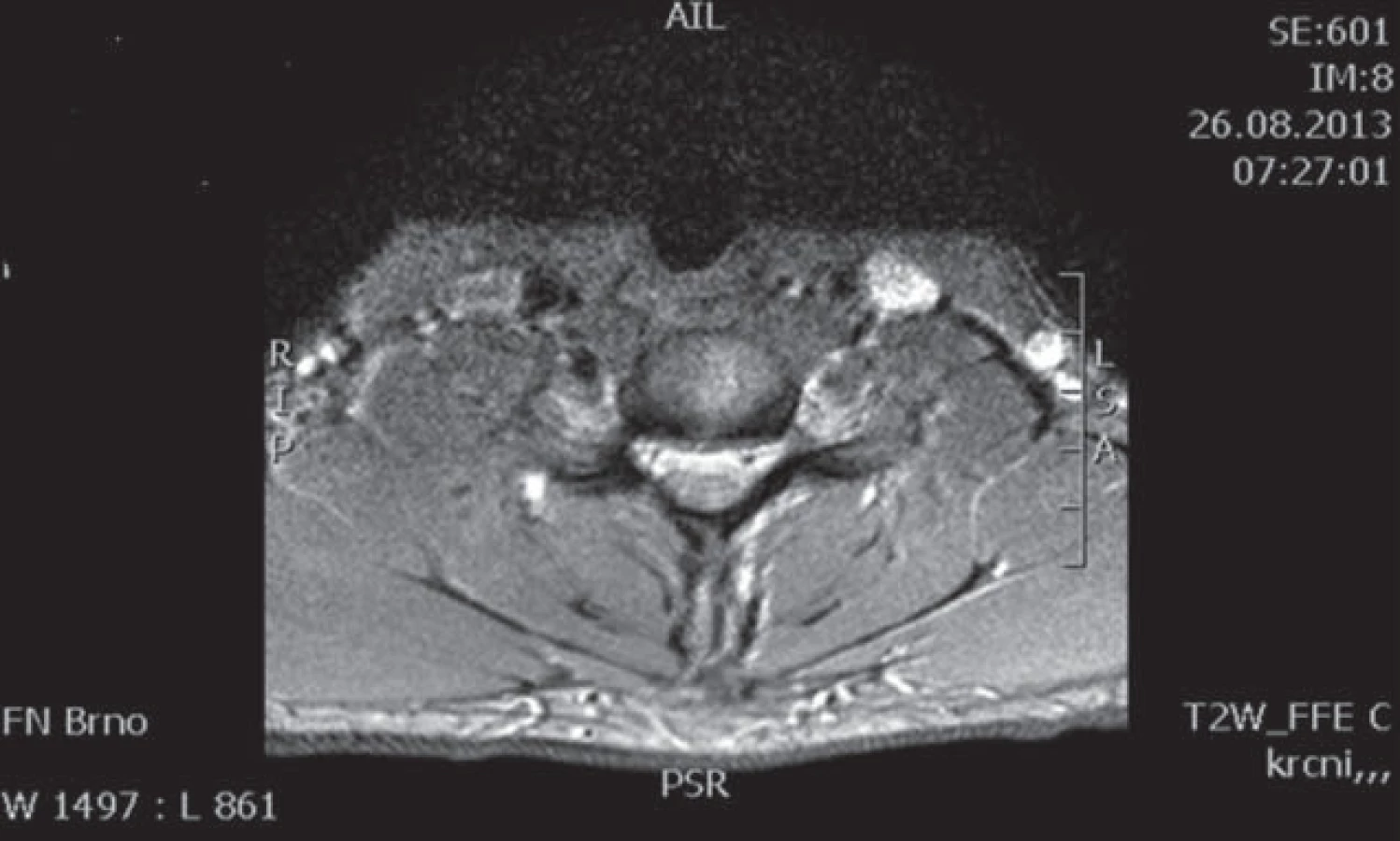
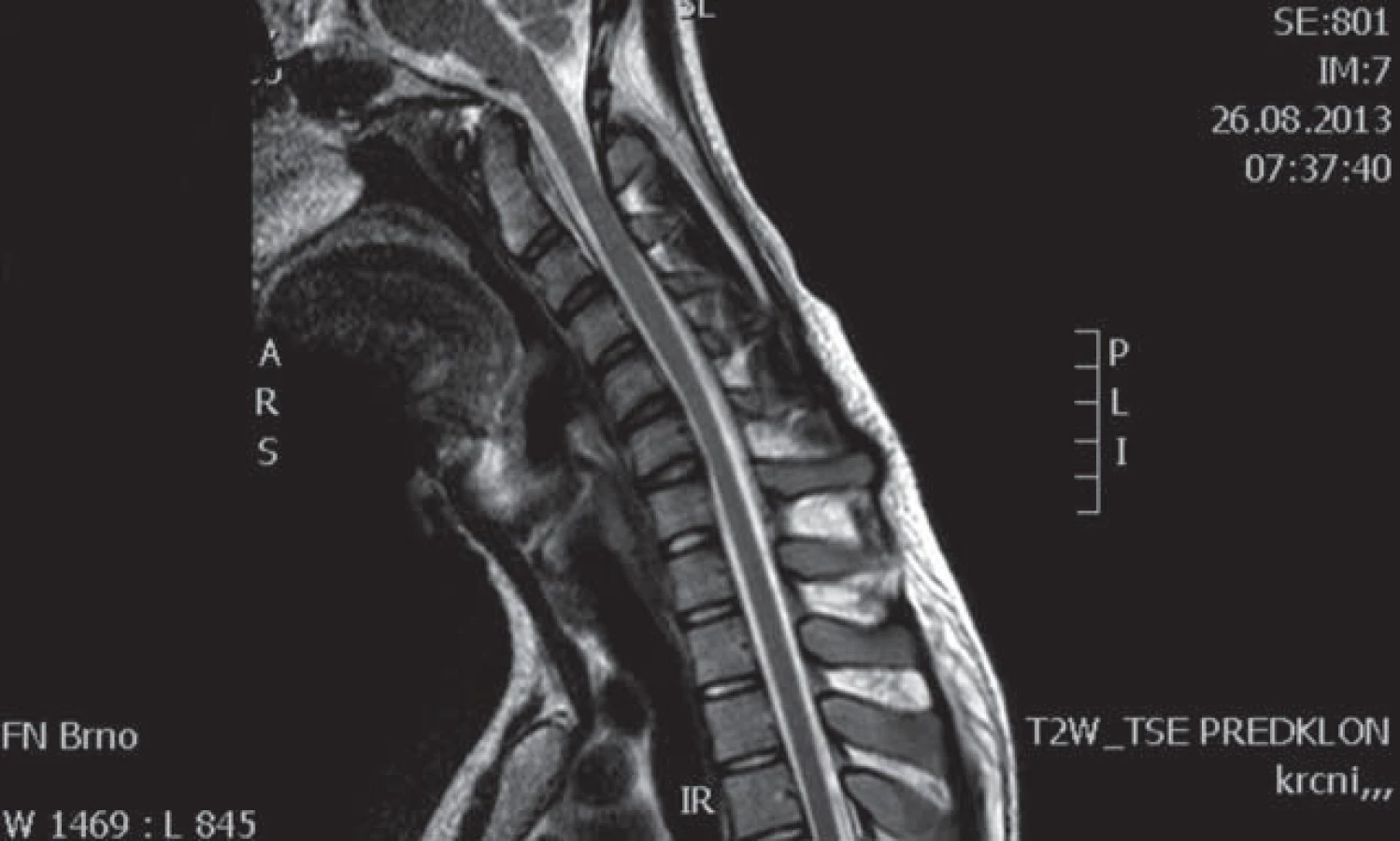
Muž, 21 let, vysokoškolský student, dosud zdravý, v anamnéze bez traumatu krční páteře. Od 17 let věku se u něj postupně pomalu rozvíjela neobratnost a slabost pravé horní končetiny. Nebyla přítomna bolest ani jiné senzitivní příznaky. Výše uvedené potíže progredovaly velmi pomalu tři roky, poslední rok je stav stacionární. Ve 20 letech podstoupil deliberaci n. ulnaris v lokti vpravo, která neměla klinický efekt. Operace byla provedena patrně na základě chybné interpretace potíží a EMG nálezu, ve kterém bylo vysloveno podezření na fokální lézi n. ulnaris v lokti (dominovalo postižení ulnarisových svalů). Pro nejasnou diagnózu byl pacient odeslán k vyšetření na naši kliniku. V objektivním neurologickém obraze dominovala hypotrofie akrálních svalů na PHK, byla přítomna periferní paréza s oslabením svalů inervovaných z myotomů C8 – Th1 (svalová síla dle MRC scale: m. abductor pollicis brevis 3, m. abductor digiti minimi 3, m. extensor indicis 3, m. brachioradialis 5). Nebyl přítomen senzitivní deficit na PHK. Objektivní neurologický nález na LHK a obou dolních končetinách byl v normě. EMG vyšetření prokazovalo známky čistě motorické axonální subakutní až chronické neuropatie v myotomech C8 – Th1 vpravo. EMG z LHK a obou DKK bylo v normě, bez známek periferně neurogenní léze, bloky vedení motorických vláken nebyly prokázány, stimulace na HKK byla provedena až do Erbova bodu (tab. 1). Magnetická rezonance krční páteře vykazovala známky atrofie míchy v etážích C6 – 7, přičemž mícha byla bez známek patologického signálu (obr. 4, 5). Při flexi krční páteře v místě atrofie míchy byl vrchol kyfotického ohybu a docházelo k rozšíření dorzálního arachnoidálního a epidurálního prostoru (obr. 6). Lumbální punkci pacient odmítl. Na základě klinického obrazu, EMG nálezu a MR krční páteře je zvažována diagnóza Hirayamovy choroby. Pacientovi byla doporučena pouze režimová opatření s vyvarováním se úrazů krční páteře, klinicky nedochází k progresi neurologického deficitu.
Diskuze
Hirayamova choroba je vzácné onemocnění, v literatuře bylo dosud popsáno zhruba 200 případů, zejména v asijských zemích [13]. Nejčastěji se uvádí tato etiopatogeneze nemoci: při flexi krční páteře dochází k abnormálnímu ventrálnímu posunu zadní části durálního vaku a sekundární míšní kompresi. Tento patologický posun je vysvětlován disproporcí mezi růstem páteřního kanálu (větší růst) a jeho obsahu (menší růst) zejména během růstového spurtu, který bývá rychlejší u mužů než u žen [14], tím dochází k relativnímu „poklesu durálního vaku“, resp. ke ztrátě jeho uchycení. Jiný mechanizmus předpokládá, že tento posun je způsoben zahuštěním a změnou počtu elastických vláken durálního vaku, což vede ke ztrátě elasticity vaku a též k mechanickému míšnímu postižení [10]. Další autoři předpokládají ztrátu „ukotvení“ mezi zadní částí dury a ligamentum flavum způsobenou snížením počtu epidurálních ligament [15]. Výše popsané pojetí patogeneze bývá ale zpochybňováno z následujících důvodů: nevysvětluje, proč udávaná dynamická komprese krční míchy poškozuje exkluzivně motoneurony a nepostihuje dlouhé dráhy, proč je neprogresivní a tak často přísně jednostranná. V jedné studii např. nebyl zjištěn rozdíl předozadního rozměru míchy u nemocných a kontrol, což by ukazovalo na to, že se jedná o primární degeneraci motoneuronů [16]. Podobně zpochybňuje flekční mechanizmus jako příčinu amyotrofie další práce, ve které mírnou redukci páteřního kanálu ve flexi našli jak u nemocných, tak i u pěti zdravých kontrol. Předozadní rozměr míchy byl stejný u pacientů jako u kontrol a u žádného z nemocných nedošlo k úplné obliteraci zadního subarachnoidálního prostoru [2]. Hirayamovu chorobu někteří považují spíše za formu MND (Motor Neuron Disease) [2]. Ostatní práce však oproti kontrolám a nemocným s amyotrofickou laterální sklerózou nacházejí u Hirayamovy choroby posun dury ventrálně a míšní kompresi [17]. Na MR krční páteře se u nich charakteristicky zadní epidurální prostor zvětšuje ve flexi a je možno pozorovat zvyšující se intenzitu signálu tohoto prostoru na T2 vážených obrazech [18]. Je též prokázáno, že extenze krční páteře signifikantně zvyšuje míru komprese míchy ve srovnání se standardním postavením při CT či NMR vyšetření [19]. Epidurální prostor je vyplněn zmoženými cévami venózního plexu. Uniformní enhancement tohoto epidurálního prostoru se pak ještě zvýrazní po aplikaci kontrastní látky [9]. U některých jedinců s tímto postižením se však patologický posun durálního vaku krční míchy ventrálně nezjistil, což bývá vysvětlováno tím, že postupem času (tedy v pozdějším věku nemocných) již laxita vaku plen mizí a typický MR nález se neprokáže. Chybění posunu durálního vaku ventrálně v pozdních a neprogresivních stadiích choroby ukazuje na to, že dynamická komprese má patogenetický význam v počátku choroby [20]. Diferenciální diagnostika Hirayamovy choroby je široká. Mezi další příčiny jednostranné amyotrofie horní končetiny patří amyotrofická laterální skleróza, a to zejména v počátečních stadiích onemocnění, kdy postižení může být výrazně lateralizováno, dále zánětlivé či kompresivní postižení krční míchy a kořenů, brachiální neuritida (amyotrofická neuralgie brachiálního plexu), multifokální motorická neuropatie anebo spinální svalová atrofie. Spinální svalová atrofie (SMA) je heterogenní skupina geneticky podmíněných chorob s různou distribucí postižení čistě dolních motoneuronů s pomalou progresí. Přes předpokládaný hereditární základ se však objevují i zcela sporadické případy, i když lze těžko v těchto případech vyloučit autozomálně recesivní dědičnost. Zvláštní formou je dominantně hereditární distální SMA, u které se prokázalo, že vzniká na podkladě mutace genu glycyl ‑ tRNA syntetázy (HMN V), a která má predilekci postižení na horních končetinách. Kolem roku 1983 byly některé případy asymetrických atrofií na HKK označovány jako chronická asymetrická spinální svalová atrofie [21]. Je možné, že v některých případech šlo o Hirayamovu chorobu. Je též nutno vyloučit i kompresivní mononeuropatie – zvláště lézi n. medianus a ulnaris.
Vzhledem k vzácnému výskytu bývají klinické a radiologické nálezy u Hirayamovy choroby popisovány na jednotlivých případech, souhrnnější obraz o nálezech podává jedna studie, která zahrnuje skupinu 11 nemocných s tímto postižením. Bylo zjištěno, že 10 pacientů mělo méně než 25 let, jeden nemocný měl 53 let. Všichni pacienti měli šikmou amyotrofii předloktí (ušetřený m. brachioradialis), 36 % chladovou parézu, 27 % mělo fascikulace. EMG vykazovalo chronickou denervaci v myotomech C7 – Th1, u 82 % pacientů byla atrofie dolní části krční míchy, asymetrické oploštění míchy bylo přítomno ve 100 %, míšní hyperintenzity u 18 %, v 90 % byl zachycen ventrální posun dury a ztráta úponů durálního vaku. Léčba límcem zpomalila progresi ve většině případů [22]. Rovněž Hirayama popisuje, že léčba choroby v progresivním stadiu měkkým límcem se jeví jako účinná [20]. Nicméně přirozený průběh nemoci není spolehlivě dokumentován.
Oběma našim pacientům byla doporučena terapeuticky pouze režimová opatření s vyvarováním se přetěžování a poranění krční páteře. Rovněž z retrospektivních studií vyplývá, že většina nemocných je léčena konzervativně [23].
Jelikož patofyziologie cervikální flekční myelopatie není zcela objasněna, tak i v literatuře doporučovaná chirurgická léčba zůstává kontroverzní. Přední dekomprese a fúze bývá užívána jako metoda první volby [24]. Podle některých autorů duroplastika a laminoplastika může být užitečná v časné fázi nemoci, šlachově svalový transfer ve fázi pozdní. Někteří chirurgové provádějí laminektomii a duroplastiku bez spinální fúze a popisují zástavu progrese nemoci i po dvou letech po operaci – tyto chirurgické metody ovšem nejsou v této indikaci obecně přijímány [25]. Je také otázka, zda zástava progrese choroby byla na podkladě operace či je dána přirozeným vývojem nemoci.
Závěr
Hirayamova choroba (flekční cervikální myelopatie) je jednotka, která je sporná a bývá mnohými autory zpochybňována, což je dáno zejména nejasností v etiopatogenezi choroby. Měli bychom na její existenci pomýšlet u mladých jedinců (zejména mužů) s asymetrickou atrofií ruky a předloktí, která nevykazuje známky výraznější progrese. U těchto pacientů je indikována MR krční páteře ve flexi, při níž jsou změny nejvýraznější a v případě jasné komprese masou v zadním epidurálním prostoru přichází v úvahu i operační řešení. Nicméně stále jde o jednotku, která dosud nebyla definitivně zařazena mezi onemocnění krční páteře a míchy a jejíž léčba je pouze empirická.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Zdeněk Kadaňka jr
Neurologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: zdenek.kadanka@fnbrno.cz
Přijato k recenzi: 10. 12. 2013
Přijato do tisku: 20. 2. 2014
Sources
1. Hirayama K, Toyokura Y, Tsubaki T. Juvenile muscular atrophy of unilateral upper extremity: a new clinical entity. Psychiatr Neurol Jpn 1959; 61 : 2190–2197.
2. Schröder R, Keller E, Flacke S, Schmidt S, Pohl C,Klockgether T et al. MRI findings in Hirayama disease: flexion‑induced cervical myelopathy or intrinsic motor neurone disease? J Neurol 1999; 246(11): 1069 – 1074.
3. Kang JS, Jochem ‑ Gawehn S, Laufs H, Ferbert A, Vieregge P, Ziemann U. Hirayama disease in Germany: case reports and review of the literature. Nervenarzt 2011; 82(10): 1264 – 1272. doi: 10.1007/ s00115 - 011 - 3320 - 9.
4. Ghosh PS, Moodley M, Friedman NR, Rothner AD, Ghosh D. Hirayama disease in children from North America. J Child Neurol 2011; 26(12): 1542 – 1547. doi: 10.1177/ 0883073811409226.
5. Finsterer J, Löscher W, Wanschitz J, Baumann M, Quasthoff S, Grisold W. Hirayama disease in Austria. Joint Bone Spine 2013; 80(5): 503 – 507. doi: 10.1016/ j.jbspin.2012.10.013.
6. Dejobert M, Geffray A, Delpierre C, Chassande B, Larrieu E, Magni C. Hirayama disease: three cases. Diagn Interv Imaging 2013; 94(3): 319 – 323. doi: 10.1016/ j.diii.2012.10.008.
7. Tashiro K, Kikuchi S, Itoyama Y, Tokumaru Y, Sobue G, Mukai E et al. Nationwide survey of juvenile muscular atrophy of distal upper extremities (Hirayama disease) in Japan. Amyotroph Later Scler 2006; 7(1): 38 – 45.
8. Hirayama K. Non ‑ progressive juvenile spinal atrophy od the distal upper limb (Hirayama disease). In: De Jong JM (ed). Handbook of clinical neurology. Amsterdam: The Netherlands: Elsevier 1991; 15 : 107 – 120.
9. Chen CJ, Chen CM, Wu CL, Ro LS, Chen ST, Lee Th.Hiarayama disease: MR diagnosis. AJNR Am J Neuroradiol 1998; 19(2): 365 – 368.
10. Konno S, Goto S, Murakami M, Mochizuki M, Motegi H, Moriya H. Juvenile amyotrophy of the upper extremity: pathologic fidings of the dura mater and surgical management. Spine 1997; 22(5): 486 – 492.
11. Hirayama K, Tomonaga M, Kitano K, Yamada T, Kojima S, Arai K. Focal cervical poliopathy causing juvenile muscular atrophy of distal upper extremity: a pathological study. J Neurol Neurosurg Psychiatry 1987; 50(3): 285 – 290.
12. Tokumaru Y, Hirayama K. Cervical collar therapy for juvenile of juvenile muscular atrophy of distal upper extremity (Hirayama disease): results from 38 cases. Rinsho Shinkeigaku 2001; 41(4 – 5): 173 – 178.
13. Vargas MC, Castillo M. Magnetic resonance imaging in Hirayama Disease. J Radiol Case Rep 2011; 5(3): 17 – 23. doi: 10.3941/ jrcr.v5i3.602.
14. Huang YC, Ro LS, Chang HS, Chen CM, Wu YR, Lee JD et al. A clinical study of Hirayama disease in Taiwan. Muscle Nerve 2008; 37(5): 576 – 582. doi: 10.1002/ mus.20980.
15. Shinomiya K, Dawson J, Spengler DM, Konrad P, Blumenkopf B. An analysis of the posterior epidural ligament role on the cervical spinal cord. Spine 1996; 21(18): 2081 – 2088.
16. Willeit J, Kiechl S, Kiechl ‑ Kohlendorfer U, Golaszewski S, Peer S, Poewe W. Juvenile asymmetric segmental spinal muscular atrophy (Hirayama’s disease): three cases without evidence of „flexion myelopathy“. Acta Neurol Scand 2001; 104(5): 320 – 322.
17. Pradhan S, Gupta RK. Magnetic resonance imaging in juvenile asymmetric segmental spinal muscular atrophy. J Neurol Sci 1997; 146(2): 133 – 138.
18. Hirayama K, Tokumaru Y. Cervical dural sac and spinal cord in juvenile muscular atrophy of distal upper extremity. Neurology 2000; 54(10): 1922 – 1926.
19. Bartlett RJ, Rowland Hill CA, Rigby AS, Chandrasekaran S, Narayanamurthy H. MRI of the cervical spine with neck extension: is it useful? Br J Radiol 2012; 85(1016): 1044 – 1051. doi: 10.1259/ bjr/ 94315429.
20. Hirayama K. Juvenile muscular atrophy of unilateral upper extremity (Hiarayama disease) – half century progress and establishment its discovery. Brain Nerve 2008; 60(1): 17 – 29.
21. Harding AE, Bradbury PG, Murray NM. Chronic asymmetric spinal muscular atrophy. J Neurol Sci 1983; 59(1): 69 – 83.
22. Hassan KM, Sahni H, Jah A. Clinical and radiological profile of Hirayama disease: a flexion myelopathy due to tight cervical dural canal amenable to collar therapy. Ann Indian Acad Neurol 2012; 15(2): 106 – 112. doi: 10.4103/ 0972 – 2327.94993.
23. Agundez M, Rouco I, Barcena J, Mateos B, Barredo J, Zarranz JJ. Hirayama disease: Is surgery an option? Neurologia 2013. doi: 10.1016/ j.nrl.2013.05.005.
24. Watanabe K, Hasegawa K, Hirano T, Endo N, Yamazaki K, Homma T. Anterior spinal decompression and fusion for cervical flexion myelopathy in young patients. J Neurosurg Spine 2005; 3(2): 86 – 91.
25. Arrese I, Rivas JJ, Esteban J, Ramos A, Lobato RD. A case of Hirayama disease treated with laminectomy and duraplasty without spinal fusion. Neurochirurgia 2009; 20(6): 555 – 558.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2014 Issue 3
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Funkční poruchy hybnosti
- Přehled méně častých primárních bolestí hlavy
- Neurologické hypotézy u panické poruchy
- Neuromodulace sakrálních nervů při řešení inkontinence stolice