
Neurosarkoidóza u muže středního věku – kazuistika
Neurosarcoidosis in a Middle-aged Man – a Case Report
Sarcoidosis is a multisystem granulomatous disease of unknown origin that rarely affects the nervous system. Granulomatous process may affect all structures of the central and peripheral nervous system. Neurosarcoidosis is associated with bad prognosis and the treatment should be started at early stages of the disease. We present a case of a middle-aged man with sarcoidosis of the peripheral lung lymph nodes and lung parenchyma. After 10 years of follow-up, the disease progressed with cardiac and neurological involvement. The peripheral nervous system was affected with peripheral neuropathy associated with sarcoidosis; other reasons for peripheral neuropathy were excluded. Treatment with corticosteroids led to improvements in neurological symptoms. At present, the patient is on a maintenance dose of corticosteroids, shows low activity of lung disease and neurological findings have been stable throughout the follow-up period. A brief literature review of the latest diagnostic and treatment options in patients with neurosarcoidosis is presented, including the first experience with novel biological treatments of this rare, very serious disease.
Key words:
sarcoidosis – neurosarcoidosis – involvement of central nervous system – peripheral neuropathy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
M. Žurková 1; P. Otruba 2; V. Lošťáková 1; Z. Tudos 3; V. Kolek 1; E. Kriegová 4
:
Klinika plicních nemocí a tuberkulózy LF UP a FN Olomouc
1; Neurologická klinika LF UP a FN Olomouc
2; Radiologická klinika LF UP a FN Olomouc
3; Ústav imunologie, LF UP v Olomouci
4
:
Cesk Slov Neurol N 2016; 79/112(2): 225-230
:
Case Report
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amcsnn2016225
Grantová podpora: IGA MZ ČR NT11117.
Sarkoidóza je multisystémové onemocnění granulomatózního charakteru neznámé etiologie, které velmi vzácně postihuje nervový systém. Granulomatózní proces může postihovat všechny struktury centrální i periferní nervové soustavy. Neurosarkoidóza má špatnou prognózu a léčbu je nutno zahájit v časných stadiích. Uvádíme kazuistiku muže středního věku s diagnostikovanou sarkoidózou nitrohrudních uzlin a plicního parenchymu, u kterého s časovou latencí 10 let došlo k rozvoji kardiálního a neurologického postižení. Bylo diagnostikováno postižení periferního nervového systému charakteru periferní neuropatie a symptomatika zadních míšních provazců asociované se základním onemocněním sarkoidózou. Byla nasazena léčba kortikoidy, která vedla ke zmírnění neuropatických obtíží. Pacient je v současnosti na udržovací dávce kortikoidů, vykazuje nízkou aktivitu plicního onemocnění a neurologický nález se během doby sledování nezměnil. Sdělení obsahuje přehled základních diagnostických a léčebných algoritmů u pacientů se stanovenou diagnózou neurosarkoidózy, vč. prvních zkušeností s novými biologickými preparáty při léčbě tohoto raritního, ale velmi závažného onemocnění.
Klíčová slova:
sarkoidóza – neurosarkoidóza – postižení centrálního nervového systému – periferní neuropatie
Úvod
Sarkoidóza je multisystémové granulomatózní onemocnění neznámé etiologie. V České republice je incidence tohoto onemocnění 3,1/ 100 000 obyvatel a prevalence mezi 70 – 80 případy/ 100 000 obyvatel [1,2]. Přestože se hovoří o systémovém onemocnění, velmi často se sarkoidóza projevuje ve formě klinicky limitované pouze na plíce [3]. U přibližně 30 – 50 % případů se však setkáváme současně s mimohrudním postižením [3], z toho klinicky rozpoznatelné postižení nervové soustavy je patrné u méně než 5 % případů [4]. V české populaci je výskyt neurosarkoidózy ještě raritnější, postihuje méně než 1 % pacientů se sarkoidózou [5,6].
Diagnostika neurosarkoidózy je velmi složitá. Důvodem je velmi rozmanitá neurologická manifestace sarkoidózy a skutečnost, že toto onemocnění může imitovat řadu dalších nemocí postihujících neurologický systém. Při diagnostických rozpacích je možno provést i biopsii mening a mozku, senzitivního periferního nervu nebo také epidermální biopsii s kvantitativní analýzou k potvrzení neuropatie malých vláken. Přesto je diagnostika neurosarkoidózy většinou založena na průkazu sarkoidních granulomů v jiných tkáních než neurologických biop-siích, dále na charakteristických syndromech, funkčním vyšetření, zobrazovacích technikách a vyloučení jiných chronických neuropatií. Proto je nezbytně nutné znát syndromy asociované s neurosarkoidózou, což může pomoci včas diagnostikovat toto velmi raritní, ale velmi závažné onemocnění se špatnou prognózu. Léčbu je nutno zahájit v časných stadiích. Současná protizánětlivá event. imunosupresivní léčba je empirická a vyžaduje důkladné zvážení klinických symptomů, postižení dalších orgánů, aktivity onemocnění a dalších klinických a paraklinických parametrů. Preparáty na bázi moderní biologicky cílené léčby nabízí pro pacienty s neurosarkoidózou nové možnosti, nepatří však zatím mezi standardní léčebné postupy.
V našem sdělení popisujeme případ muže středního věku s diagnostikovanou plicní sarkoidózou s hyperkalciurií, u kterého s časovou latencí 10 let došlo k rozvoji srdečního a neurologického postižení. Dále je uveden přehled základních údajů o diagnostice a léčbě neurosarkoidózy.
Kazuistika
Prezentujeme případ 57letého muže, který je sledován na Plicní klinice LF UP a FN Olomouc od roku 2004 pro recidivující nodózní erytém a zvýšenou hladinu enzymu konvertujícího angiotenzin v séru (SACE; 84,7 U/ l). Cytologickým vyšetřením bronchoalveolární tekutiny byla zjištěna lymfocytární alveolitida (11 %; poměr CD4+/ CD8+ lymfocytů byl 1,23) a současně mikromorfologicky prokázán epiteloidní granulom. Na vysoce rozlišovacím CT (HRCT) plic byly popsány dvě drobné buly v pravém hrotu a nečetné nodulace na periferii plicního parenchymu. Stav nemocného byl veden jako rentgenologické stadium 0. Standardně byl proveden skríning očního postižení a sběr moče na odpad vápníku za 24 hod, vše s negativním výsledkem. Pacient splňoval kritéria pro diagnózu sarkoidózy [1,3].
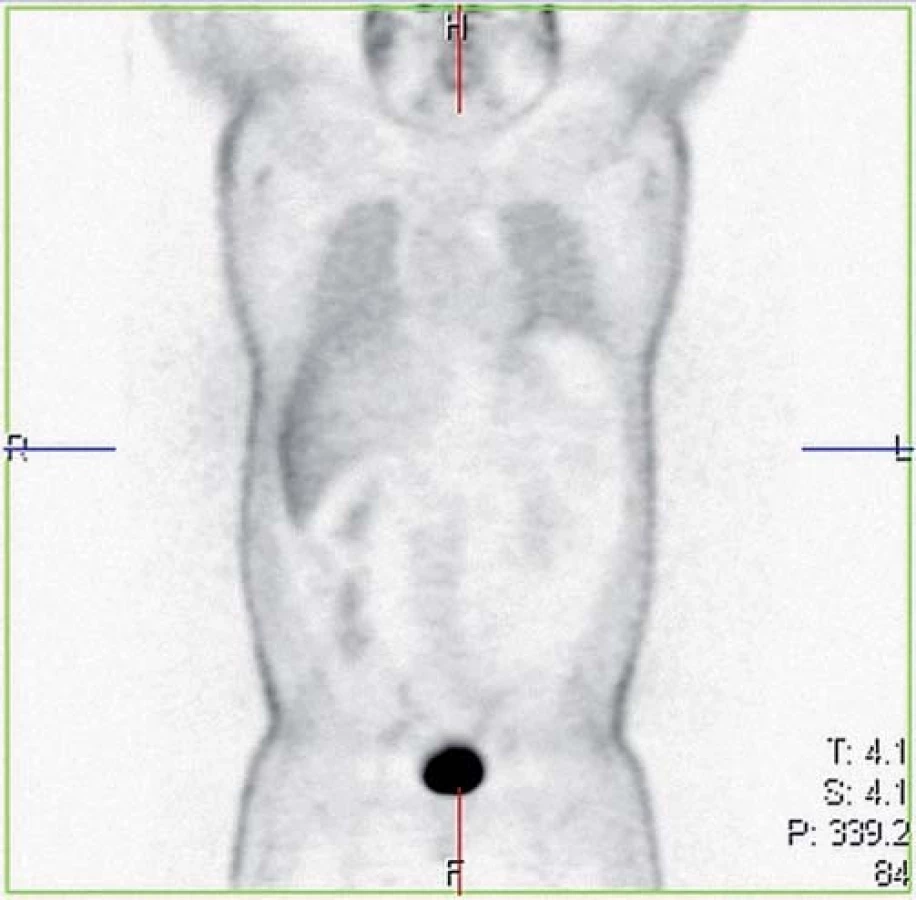
V září 2007 byla zjištěna hyperkalciurie a následně zahájena kortikoterapie v iniciální dávce 40 mg/ den prednizonu s postupným poklesem. V lednu 2009 byl pacient opět vyšetřen pro nově zjištěné infiltráty v podkoží předloktí rukou, bolesti kloubů a svalů a mravenčení v dolních končetinách a bylo indikováno oční, kožní a neurologické vyšetření vč. PET/ CT k vyloučení možného latentního mimoplicního postižení. U nemocného byly opakovaně prováděny biopsie z míst kožních změn a histologicky z biopsie kůže paže pravé horní končetiny byl potvrzen atypický lipom a lobulární panikulitida a nejspíše netuberkulózní varianta erythema induratum. Kožní postižení sarkoidózou nikdy doloženo nebylo. Hyperkalciurie nebyla prokázána, kontrolní oční a neurologické vyšetření byla bez průkazu patologie. HRCT hrudníku prokazoval stacionární nález dvou emfyzematózních bul v pravém hrotu a nečetné drobné nodulace na periferii plicního parenchymu bilaterálně (obr. 1). Bylo doplněno celotělové PET/ CT (obr. 2), kde nebyla prokázána aktivita sarkoidózy. Pacient byl nadále sledován bez léčby kortikoidy.


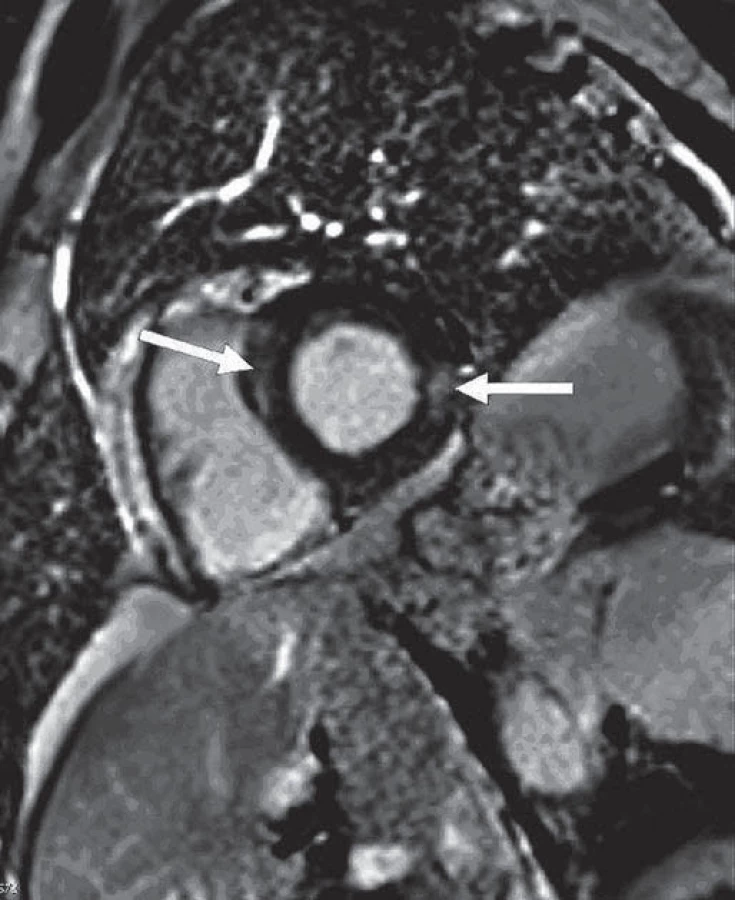
V lednu 2014 bylo u pacienta provedeno neurologické vyšetření pro symptomy periferní neuropatie. V objektivním nálezu byla zjištěna hyporeflexie šlachově-okosticových reflexů L2 – S2, nebyly shledány známky svalové hypotrofie, nebylo zjištěno snížení svalové síly na horních nebo dolních končetinách. Pacient udával dysestezie s typickou lokalizací na akrech DKK ponožkovitého charakteru, termické a vibrační modality nebyly postiženy. Bylo doplněno elektromyografické (EMG) vyšetření, při kterém byla prokázána chronická axonální převážně senzitivní neuropatie dolních končetin lehkého stupně. Parametry senzitivního vedení n. suralis a n. peroneus superficialis byly zpomaleny na hodnoty 35 – 39 m/ s. Byla registrována změna velikosti sumačního senzitivního akčního potenciálu. Naopak motorické kmenové vedení zůstalo zachováno při normálních parametrech motorického vedení, pouze s lehkou redukcí amplitudy motorického akčního potenciálu. V jehlové technice nebyly zaznamenány akutní denervační potenciály, při hodnocení jednotlivých motorických akčních jednotek byla registrována lehce vyšší amplituda při normální délce trvání. Klinické podezření na spinální ataxii bylo verifikováno provedením somatosenzitivních evokovaných potenciálů stimulací n. medianus a n. tibialis oboustranně. Při stimulaci n. tibialis bylo oboustranně registrováno prodloužení latencí kortikálního komplexu při normálních parametrech periferního vedení a při vyšetření somatosenzitivních evokovaných potenciálů při stimulaci n. medianus. Analýza likvoru nebyla provedena z důvodu nesouhlasu pacienta s invazivním vyšetřením. V rámci standardního algoritmu při nově se vyskytujícím mimoplicním postižení, příp. při zhoršení klinického stavu, byla indikována bronchoskopie s bronchoalveolární laváží (BAL) a transbronchiální biopsie (TBB). Cytologie BAL tekutiny prokázala nález lymfocytární alveolitidy (23 %), poměr CD4+/ CD8+ byl 1,05 a histologie z TBB bez průkazu granulomu. Současně bylo prokázáno postižení srdce sarkoidózou pomocí techniky magnetické rezonance (MR) (obr. 3). Vzhledem k nově prokázanému mimoplicnímu postižení nervového a srdečního systému bylo indikováno opětovné zahájení kortikoterapie, byl nasazen prednizon v iniciální dávce 60 mg/ den. Do léčby byly nasazeny vitamíny skupiny B (milgamma 3 × 1) a pregabalin (lyrica 150 mg/ den).
Kontrolní HRCT plic v únoru 2014 popisuje jemné retikulace a opacity mléčného skla, vícečetné drobné nodulace s maximem subpleurálně. V červenci 2014 podstoupil pacient opakovaně komplexní neurologická vyšetření, při kterých byly dominující příznaky postižení periferního nervového systému charakteru periferní senzomotorické neuropatie a postižení charakteru spinální ataxie lehkého stupně. Klinicky byly zjištěny známky zadně provazcového postižení, které bylo potvrzeno pomocí vyšetření somatosenzitivních evokovaných potenciálů (SSEP n. medianus a n. tibialis). Vyšetření míchy pomocí MR nebylo provedeno pro malou výtěžnost u daného typu postižení. Opětovně byl bez známek svalové hypotrofie, přetrvává hyporeflexie reflexů L2 – S2 oboustranně, jsou přítomny známky poruchy vibračního čití při testování kalibrovanou ladičkou. Při testování stoje se objevuje nejistota v Rombergově zkoušce. Bylo pokračováno v terapii prednizonem a vitamíny skupiny B (milgamma 3 × 1) a pregabalin (lyrica 150 mg/ den).
V září 2015 bylo provedeno kontrolní MR srdce a echokardiografické vyšetření, nálezy byly bez známek progrese postižení srdce sarkoidózou. Holter EKG při kontrolním vyšetření indikován nebyl. Kontrolní HRCT plic popisuje regresi minimálních jemných retikulací a opacit mléčného skla, jinak přetrvává nález drobných nodulací s maximem subpleurálně. Hodnoty biomarkerů byly jen s lehkou elevací SACE, sILR2 a neopterin byly v normě. Pacient je v současnosti ve stabilizovaném stavu na udržovací malé dávce 5 mg prednizonu denně, vykazuje nízkou aktivitu plicního onemocnění a neurologický nález se během doby sledování nezměnil, jsou přítomny známky senzomotorické neuropatie a spinální ataxie.
Diagnostika neurosarkoidózy
Granulomatózní proces u neurosarkoidózy může postihovat všechny struktury centrální i periferní nervové soustavy. Nejčastěji se u neurosarkoidózy setkáváme s periferními neuropatiemi, ojedinělé jsou případy s postižením centrálního nervového systému [4,5,7], v literatuře se popisují i případy postižení mozku nebo míchy [4,5,7]. Postižení hlavových nervů, např. n. facialis (VII.), se někdy kombinuje s uveitidou a s infiltrací především příušních žláz („febris uveoparotidea“ popsaná Heerfordtem) [8]. S postižením centrální nervové soustavy souvisí i atrofie optického nervu s následnou možnou amaurózou nebo se při hypofyzární lokalizaci může vyskytnout diabetes insipidus. Popisuje se neuropatie malých vláken („small fibre neuropathy“) [4,7]. Periferní neuropatie se může kombinovat se svalovým postižením [9]. U řady pacientů s neurosarkoidózou se setkáváme s celkovou slabostí a únavou, které jsou u některých nemocných těžko řešitelným problémem. V diferenciální diagnostice neurosarkoidózy je nutno vyloučit tuberkulózu a jiné granulomatózy a mykotické infekce. V širší diferenciální diagnostice se zaměřujeme na autoimunitní onemocnění centrální a periferní nervové soustavy demyelinizačního charakteru (roztroušená skleróza mozkomíšní, chronická zánětlivá demyelinizační polyneuropatie) a vylučujeme onemocnění charakteru izolovaných vaskulitid CNS nebo raritní postižení při syndromu získané imunodeficience.
Z důvodu vzácného výskytu a velmi rozmanité klinické manifestace je diagnostika neurosarkoidózy velmi složitá. Většinou se neurosarkoidóza vyskytuje v průběhu onemocnění zjištěného v jiné lokalizaci. Zcela vzácně je neurosarkoidóza prvním projevem sarkoidózy. U pacientů s neurosarkoidózou lze objektivizovat postižení hlavových nervů (n. VII), které se může kombinovat s uveitidou a s infiltrací především příušních žláz („febris uveoparotidea“) [2,4,5,8]. K průkazu postižení jednotlivých struktur centrální i periferní nervové soustavy, příp. postižení mozku nebo míchy granulomatózním procesem využíváme řadu zobrazovacích metod. Při podezření na postižení centrální nervové soustavy (CNS) sarkoidózou se uplatňuje MR, elektromyografie (EMG) a vyšetření multimodálních evokovaných potenciálů. Přínosné může být vyšetření mozkomíšního moku, ve kterém se prokazuje zvýšený počet lymfocytů. Při imunofenotypizaci lymfocytů z likvoru je charakteristický zvýšený poměr CD4+/ CD8+ (tzv. imunoregulační index), v likvoru bývají i vyšší hladiny lysozymu a SACE. Při diagnostických rozpacích je možno provést i biopsii mening a mozku. Dále je možno v indikovaných případech provést biopsii senzitivního periferního nervu nebo také epidermální biopsii s kvantitativní analýzou k potvrzení neuropatie malých vláken. Ve většině případů je však diagnostika neurosarkoidózy založena na průkazu sarkoidních granulomů v jiných tkáních než neurologických biopsiích a dále na charakteristických syndromech a zobrazovacích technikách [7,10]. Jednotlivé typy postižení nervového systému a symptomů u neurosarkoidózy jsou uvedeny v tab. 1.
![Typy postižení neurosarkoidózou a její symptomy (upraveno dle Agnihotri et al [7].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/21ee786d8dfdfef0bd769f5b76d32574.png)
Léčba neurosarkoidózy
Současná léčba je přísně individuální dle vyhodnocení rozsahu a tíže orgánového postižení a posouzení aktivity onemocnění (neaktivní, stabilizované, progredující) [7,10,11]. Doporučuje se začít s léčbou neurosarkoidózy v časných stadiích. Vzhledem ke skutečnosti, že se jedná o systémové onemocnění, které většinou postihuje kromě neurologického systému také jiné orgány, je nutno spolupracovat se specialisty z jiných oborů. Konzultován by měl být pneumolog. Přehled současných léčebných možností a dávkování u neurosarkoidózy jsou uvedeny v tab. 2.

Léčba neurosarkoidózy vychází z doporučených konsenzů a je zde indikována léčba kortikoidy [4,8]. Dávky a délka trvání léčby jsou individuální a záleží na tíži onemocnění a léčebné odpovědi. Nemocní s parézou n. facialis nebo aseptickou meningitidou by měli být léčeni prednizonem v iniciální dávce 0,5 mg/ kg/ den po dobu dvou týdnů s následným snižováním dávek po dobu jednoho roku. U nemocných s myopatií či neuropatií je délka léčby vyššími dávkami (0,5 – 1,0 mg/ kg/ den) čtyři týdny. Pokud jsou postiženy meningy nebo jsou známky encefalopatie či symptomatického hydrocefalu, je dávkování kortikoidů vyšší – prednizon 1 – 1,5 mg/ kg/ denně po čtyři týdny s následným snižováním dávek po dobu jednoho roku. Nemocní, kteří se horší při terapii kortikoidy nebo u kterých jsou tyto léky kontraindikovány, mohou profitovat z alternativní terapie jako např. metotrexát, mykofenolát mofetil, azathioprin, cyklofosfamid, cyklosporin, chlorambucil a purinová analoga – kladribin [4,8,12]. Dále jsou využívány hydroxychlorochin, talidomid jako steroid-šetřící léky, či jako dodatková léčba u nemocných s vysokodávkovanými kortikoidy, kteří jsou rezistentní na léčbu samotnými kortikoidy. U nemocných se sarkoidózou CNS bývá zcela výjimečně indikována i chirurgická léčba, a to jen v případě perzistentních či zvětšujících se lézí. Dále jen ve speciálních případech, jako je refrakterní onemocnění na léčbu kortikoidy či selhání léčby kortikoidy, bývá indikována radioterapie CNS [4,8,10]. V průběhu radioterapie je doporučeno pokračovat v imunosupresivní léčbě, ale v nižších dávkách.
V nedávné době byly popsány další léčebné možnosti u neurosarkoidózy s využitím léků blokujících zánětlivý cytokin tumor nektorizující faktor TNF-α podílející se na patogenezi sarkoidózy, a to zejména u pacientů refrakterních na léčbu kortikoidy. U těchto nemocných je indikována biologická léčba TNF-α inhibitory infliximabem nebo rituximabem [13]. Nedávná práce prokázala také možnost úspěšného použití etanerceptu u pacientky s neurosarkoidózou refrakterní na kortikosteroidy, cyklofosfamid, alemtuzumab, azathioprin, mykofenolát mofetil i rituximab [14]. Výhodou použití etanerceptu, který inhibuje vazbu TNF-α na povrchové buněčné receptory, je skutečnost, že se na rozdíl od infliximabu nebo rituximabu nevytváří neutralizační protilátky. Preparáty na bázi moderní biologicky cílené léčby zatím nepatří mezi standardní léčebné postupy.
Diskuze
Postižení granulomatózním zánětem se může velmi vzácně vztahovat i na nervový systém [1 – 3]. Nejčastěji se u neurosarkoidózy setkáváme s periferními neuropatiemi, ojedinělé jsou případy s postižením centrálního nervového systému [4,5]. Z důvodu vzácného výskytu a velmi rozmanité klinické manifestace je diagnostika neurosarkoidózy velmi složitá. Celosvětově je udáván výskyt klinicky rozpoznatelného postižení nervové soustavy sarkoidózou u 5 % všech případů [3]. Její výskyt v české populaci je však velmi raritní a v průběhu let 2007 – 2014 byla na Plicní klinice FN Olomouc ve spolupráci s Neurologickou klinikou FN Olomouc diagnostikována neurosarkoidóza pouze u dvou pacientů (1 % případů) [6]. První případ je muž prezentovaný v kazuistickém sdělení a druhým případem byla žena s postižením centrálního nervového systému. Společným rysem u obou případů bylo současné postižení plicní formou sarkoidózy. Tento nález je ve shodě s popisovanými případy neurosarkoidózy, která se u většiny pacientů nachází v kombinaci s plicním postižením (generalizovaná forma) a velmi zřídka jako izolované postižení nervového systému [4,5]. U našeho pacienta bylo kromě neurosarkoidózy diagnostikováno i kardiální postižení. Jedná se o raritní kombinaci kardiálního, neurologického a plicního postižení, kdy v literatuře jsou uváděny pouze jednotlivé kazuistiky takto klinicky systémově manifestního onemocnění. Tato vysoce urgentní forma sarkoidózy, která ohrožuje život, vyžadovala nasazení léčby kortikoidy, jež vedla ke zmírnění obtíží neuropatického charakteru. Pacient je v současnosti na udržovací dávce kortikoidů, vykazuje nízkou aktivitu plicního onemocnění a neurologický nález se během doby sledování nezměnil.
Závěr
Neurosarkoidóza je velmi vzácné závažné onemocnění, které má velmi rozmanitý klinický průběh. Léčbu je nutné indikovat již v časných stadiích, v současnosti jsou lékem první volby kortikosteroidy. V nedávné době se objevily první práce prokazující léčebný efekt biologických preparátů blokujících zánětlivý cytokin TNF-α, jenž se podílí na patogenezi sarkoidózy. Tyto preparáty nabízí nové možnosti léčby neurosarkoidózy. Onemocnění jednoznačně vyžaduje mezioborovou spolupráci, jak bylo na našem případě doloženo.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Dr. Eva Kriegová
Ústav imunologie
LF UP v Olomouci
Hněvotínská 3
Olomouc
e-mail: eva.kriegova@email.cz
Přijato k recenzi: 1. 10. 2015
Přijato do tisku: 28. 12. 2015
Sources
1. Loštáková V, Kolek V, Vašáková M. Sarkoidóza – doporučený postup diagnostiky, terapie a sledování vývoje onemocnění. Standardy ČPFS – Sekce pro intersticiální plicní procesy. Dostupné z URL: http:/ / pneumologie.cz/ odborne/ doc/ Sarkoidoza.doc.
2. Kolek V. Sarkoidóza: známé a neznámé. Praha: Grada 1998.
3. American Thoracic Society, European Respiratory Society and World Association of Sarcoidosis and Other Granulomatous Disorders. Joint Statement on sarcoidosis. Am J Respir Crit Care Med 1999;160(2):736 – 55.
4. Nozaki K, Judson MA. Neurosarcoidosis: clinical manifestations, diagnosis and treatment. Presse Med 2012;41(2):e331 – 48. doi: 10.1016/ j.lpm.2011.12.017.
5. Kolek V, Pěničková V, Heřman M, et al. Neurosarcoidosis. Evaluation of new diagnostic methods in early diagnosis and treatment. Cas Lek Ces 1996;135(5):141 – 4.
6. Zurková M, Kolek V, Tomankova T, et al. Extrapulmonary involvement in patients with sarcoidosis and comparison of routine laboratory and clinical data to pulmonary involvement. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2014;158(4):613 – 20. doi: 10.5507/ bp.2014.026.
7. Agnihotri SP, Singhal T, Stern BJ, et al. Neurosarcoidosis. Semin Neurol 2014;34(4):386 – 94. doi: 10.1055/ s-0034-1390387.
8. Heerfordt CF. „Über eine „Febris uveo-parotidea subchronica“ an der Glandula parotis und der Uvea des Auges lokalisiert und häufug mit Paresen cerebrospinaler Nerven kompliziert. Albrecht Von Graefes Arch Klin Exp Ophthalmol 1909;70 : 254 – 73.
9. Zámecník J, Ambler Z, Ehler E, et al. Granulomatous myopathy in patients with sarcoidosis and myasthenia gravis. Cesk Patol 2006;42(4):175 – 81.
10. Bagnato F, Stern BJ. Neurosarcoidosis: diagnosis, therapy and biomarkers. Expert Rev Neurother 2015;15(5):533 – 48. doi: 10.1586/ 14737175.2015.1037288.
11. Hoyle JC, Jablonski C, Newton HB. Neurosarcoidosis: clinical review of a disorder with challenging in patient presentations and diagnostic considerations. Neurohospitalist 2014;4(2):94 – 101. doi: 10.1177/ 1941874413519447.
12. Tana C, Wegener S, Borys E, et al. Challenges in the diagnosis and treatment of neurosarcoidosis. Ann Med 2015;47(7):576 – 91. doi: 10.3109/ 07853890.2015.1093164.
13. Doty JD, Mazur JE, Judson MA. Treatment of sarcoidosis with infliximab. Chest 2005;127(3):1064 – 71.
14. Marques I, Giovannonni G, Marta M. Mononeuritis multiplex as the first presentation of refractory sarcoidosis responsive to etanercept. BMC Neurol 2014;14 : 237. doi: 10.1186/ s12883-014
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2016 Issue 2
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Ramsay-Hunt Syndrome – a Rare Manifestation of Relatively Frequent Condition
- Transient Ischemic Attack and Minor Stroke Management
- Neurosarcoidosis in a Middle-aged Man – a Case Report
- Autonomic Dysfunction and its Diagnostic Tools in Multiple Sclerosis