
A Novel Mutation in the GIGYF2 Gene in a Patient with Parkinson’s Disease
Nová mutace v genu GIGYF2 u pacienta s Parkinsonovou chorobou
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Authors:
J. Necpál 1; R. Vodička 2;
p. Kaňovský 3
Authors‘ workplace:
Department of Neurology, Hospital Zvolen, Slovak Republic
1; Department of Medical Genetics, Faculty of Medicine and Dentistry, Palacký University and University Hospital Olomouc, Czech Republic
2; Department of Neurology, Faculty of Medicine and Dentistry, Palacký University and University Hospital Olomouc, Czech Republic
3
Published in:
Cesk Slov Neurol N 2017; 80(6): 719-721
Category:
Letter to Editor
doi:
https://doi.org/10.14735/amcsnn2017719
Overview
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Dear editor,
Although Parkinson’s disease (PD) is mainly a sporadic disorder, there is a demonstrable familial occurrence in up to 30% of cases [1]. Thanks to the improving of molecular genetics technologies, the list of identifiable genetic mutations related to genetic forms of PD is ever broader. In general, these are labelled with the abbreviation PARK with a number reflecting the chronological order in which they were discovered (to date PARK 1 – 23) [2]. Although we know the gene loci of all currently known PARK diseases, we do not yet know all of their causative genes.
We describe a 59-year-old woman with a history of 7 years of anxious depressive disorder, insomnia and personality disorder with paranoid behaviour (documented as a depressive type of schizoaffective disorder), treated with various types of antidepressants and antipsychotics, including clonazepam, cinolazepam, mirtazapine, venlafaxine, alprazolam and risperidone. She is currently taking olanzapine, venlafaxine and alprazolam. Her psychological assessment showed cognitive decline and organicity, without significant evidence of psychosis. She was examined for the first time at the age of 57, when she complained of left hand clumsiness and nonspecific leg weakness when walking, occurring for about 2 years, short-term memory loss, urinary frequency and orthostatic problems. Initially, we suggested a neuroleptic-induced parkinsonism. However, the patient referred to a history of adult-onset parkinsonism in more family members on her mother’s side, some with clear benefit after levodopa treatment (Fig. 1). On examination, there was a hypokinetic-rigid syndrome, more on the left side, with slight postural tremor of the left hand, hypomimia, seboroic dermatitis and positive Babinski sign on the left. Magnetic resonance imaging of the brain displayed slightly asymmetric frontoparietal atrophy (more on the left) and incipient leukoencephalopathy. Electroencephalography showed theta activity with sporadic delta dysrhythmia on the right side. Dopamine transporter single-photon emission computed tomography (DaT-SPECT) confirmed the reduced uptake of presynaptical dopaminergic neurons, thus a neurodegenerative parkinsonism. A slit-lamp examination for the presence of a Kayser-Fleischer ring was negative. The patient was extensively tested neurologically and neuropsychologically (Tab. 1). She started to take rasagiline, with no subjective improvement. A levodopa trial was theninitiated with improvement of her parkinsonism at a dose of 750 mg daily. Owing tosuspicion of having a familial form of parkinsonism, we conducted genetic testing.

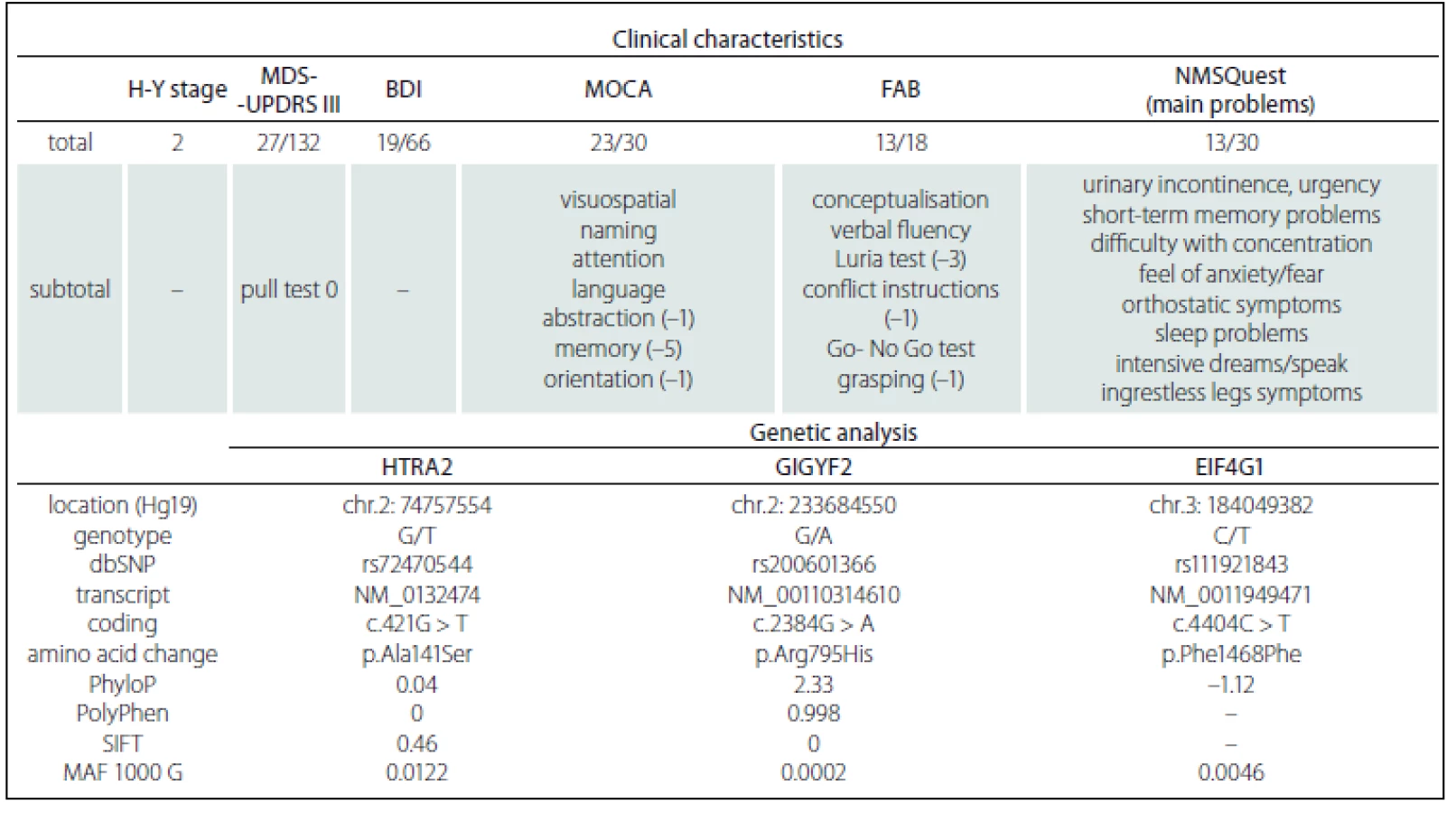
The patient’s DNA was isolated from peripheral blood. The amplicon library was in-silico-, designed by Ion AmpliSeq Designer (Thermo Fisher Scientific, Waltham,USA) for the ADH1C, EIF4G1, FBXO7, GBA + GBAP1, GIGYF2, HTRA2, LRRK2, MAPT, PARK2, PARK7, PINK1, PLA2G6, SNCA, UCHL1 and VPS35 genes. In total 617 amplicons covered 92.59% of the coding DNA sequence, including 100 bp into the introns and both 5’ and 3’ UTRs of the particular gene regions. After filtering the variants, two missense and one synonymous rare mutation were found (Tab. 1). The most important mutation was found in exon 21 in a coiled coil domain of the GIGYF2 gene on chromosome 2 (c.2384G>A; p.Arg795His), which has not been published until now. Since the majority of symptomatic family members including the patient’s brother have died, the patient is childless and cooperation with her is quite difficult, extensive genetic family testing is, unfortunately, not possible.
In 2002, Pankratz et al identified, by way of whole-genome linkage analysis, a locus for a new genetic form of PD [3]. The locus on chromosome 2 (2q36 – 37) corresponds with the 18-cM interval between the microsatellite markers D2S396 and D2S338, which contains 73 potential candidate genes. In this region, the gene encoding a protein with 1,299 amino acids, named GIGYF2 (Grb10-Interacting GYF protein 2), was found. Both known homologues, GIGYF1 and GIGYF2, interact by their GYF domain with the proline-rich region of Grb10 adaptor protein [4,5]. Although it is hypothesized that GYF-domain-containing proteins take part in mRNA splicing or other steps of mRNA processing, the exact function of the GIGYF2 protein is not yet known [4]. Lautier et al. [4] have studied all 27 exons of the GIGYF2 gene in 249 PD patients with at least one affected first-degree relative in comparison to 227 control subjects. In the PD group there were seven heterozygous mutations in 12 unrelated identified cases. These mutations were consistent with autosomal dominant adult onset levodopa-responsive PD with incomplete penetrance. The GIGYF2 gene was then added to the candidate genes for a monogenic form of PD, later designated as PARK 11. Nichols et al. carefully examined a sample of 566 familial PD cases with nearly 1,500 affected individuals for these seven variants. Additionally, another three new mutations were discovered. All these variants were found in only six PD families, and the authors believe they are not responsible for the heredity of PD at the 2q36 – 37 locus [5].
Haplotype analysis of the GIGYF2 gene proved that mutations are not associated with a higher risk of sporadic PD [6]. In an animal study, abrogation of GIGYF2 function in zebrafish did not lead to a loss of diencephalic dopaminergic cells, suggesting that this gene is probably not required for dopaminergic differentiation of cells [7]. Afterwards, another study which questioned the role of the GIGYF2 gene in causal relation to familial PD and its association with PARK 11 disease started to appear [8,9]. Therefore in the contemporary literature, the mutations in the GIGYF2 gene no longer appear as a cause of PARK 11 disease [10].
We report here a patient with possible autosomal dominant PD with incomplete penetrance with genetically confirmed mutation in the GIGYF2 gene. Unfortunately, the patient’s symptomatic and asymptomatic relatives were not clinically or genetically tested. In our patient, psychiatric manifestation (depression, paranoid behaviour and cognitive decline) preceded her motor symptoms by some years. They more likely represent associated symptoms in PD rather than simple coexistence of a schizoaffective disorder with PD. Psychiatric symptoms in carriers of the GIGYF2 mutation have thus far not been reported. As previously mentioned, the causality of GIGYF2 mutation in PARK 11 disease has been disproved [10]. However, there is a need to answer two principal questions: 1. What other gene mutation could be responsible for the developing of the disease? 2. What is a result of GIGYF2 mutation in our patient, if it is not pathogenic? Therefore, we believe that further studies to solve this problem are necessary.
Acknowledgements
We thank the patient for allowing us to report her clinical data and Michal Patarák, MD, for his technical support.
Funding
This study was funded by grant MZ-NV15-32715A.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Ján Necpál, MD
Department of Neurology
Hospital Zvolen
Kuzmányho nábrežie 28
960 01 Zvolen Slovakia
e-mail: necpal.neuro@gmail.com
Accepted for review: 7. 7. 2017
Accepted for print: 25. 10. 2017
Sources
1. Payami H, Zareparsi S. Genetic epidemiologyof Parkinson’s disease. J Geriatr Psychiatry Neurol 1998;11(2):98 – 106. doi: 10.1177/ 089198879801100207.
2. Parkinson disease 23, autosomal recessive early-onset; PARK23. OMIM [accessed 2017 Jul 7]. Available from URL: https:/ / omim.org/ entry/ 616840.
3. Pankratz N, Nichols WC, Uniacke SK, et al. Genome screen to identify susceptibility genes for Parkinson disease in sample without parkin mutations. Am J HumGenet 2002;71(1):124 – 35. doi: 10.1086/ 341282.
4. Lautier C, Goldwurm S, Dürr A, et al. Mutations in the GIGYF2 (TNRC15) gene at the PARK 11 locus in familial Parkinson disease. Am J Hum Genet 2008;82(4):822 – 33. doi: 10.1016/ j.ajhg.2008.01.015.
5. Nichols WC, Kissell DK, Pankratz N, et al. Variation in GIGYF2 is not associated with Parkinson disease. Neurology 2009;72(22):1886 – 92. doi: 10.1212/ 01.wnl.0000346517.98982.1b.
6. Sutherland GT, Siebert GA, Newman JR, et al. Haplotype analysis of the PARK 11 gene, GIGYF2, in sporadic Parkinson’s disease. Mov Disord 2009;24(3):449 – 52. doi: 10.1002/ mds.22427.
7. Guella I, Pistocchi A, Asselta R, et al. Mutational screening and zebrafish functional analysis of GIGYF2as a Parkinson-disease gene. Neurobiol Aging 2011;32(11): 1994 – 2005. doi: 10.1016/ j.neurobiolaging.2009.12.016.
8. Samaranch L, Lorenzo E, Pastor MA, et al. Analysis of the GIGYF2 gene in familial and sporadic Parkinson disease on the Spanish population. Eur J Neurol 2010; 17(2):321 – 5. doi: 10.1111/ j.1468-1331.2009.02812.x.
9. Li L, Funayama M, Tomiyama H, et al. No evidence for pathologic role of GIGYF2 mutation in Parkinson disease in Japanese patients. Neurosci Lett 2010;479(3):245 – 8. doi: 10.1016/ j.neulet.2010.05.071.
10. Kalinderi K, Bostantjopoulou S, Fidani L. The genetic background of Parkinson’s disease: current progress and future prospects. Acta Neurol Scand 2016;134(5):314 – 26. doi: 10.1111/ ane.12563.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2017 Issue 6
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Brief Test of Verbal Memory Using the Sentence in Alzheimer Disease
- State-of-the-Art MRI Techniques for Multiple Sclerosis
- H-reflex and Its Role in EMG Laboratory and Clinical Practice
- When to Operate on Temporal Bone Fractures?